A Hepatitis B Virus-Derived Peptide Exerts an Anticancer Effect via TNF/iNOS-producing Dendritic Cells in Tumor-Bearing Mouse Model



Abstract
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Poly6 Treatment Leads to Tip-DC Development from DCs in an IFN-I-Dependent Manner by Evoking Mitochondrial ROS-Mediated Cytosolic Release of Oxidized Mitochondrial DNA
2.2. Poly6 Exerts Anticancer Effects in Mouse Models in an IFN-I Dependent Manner
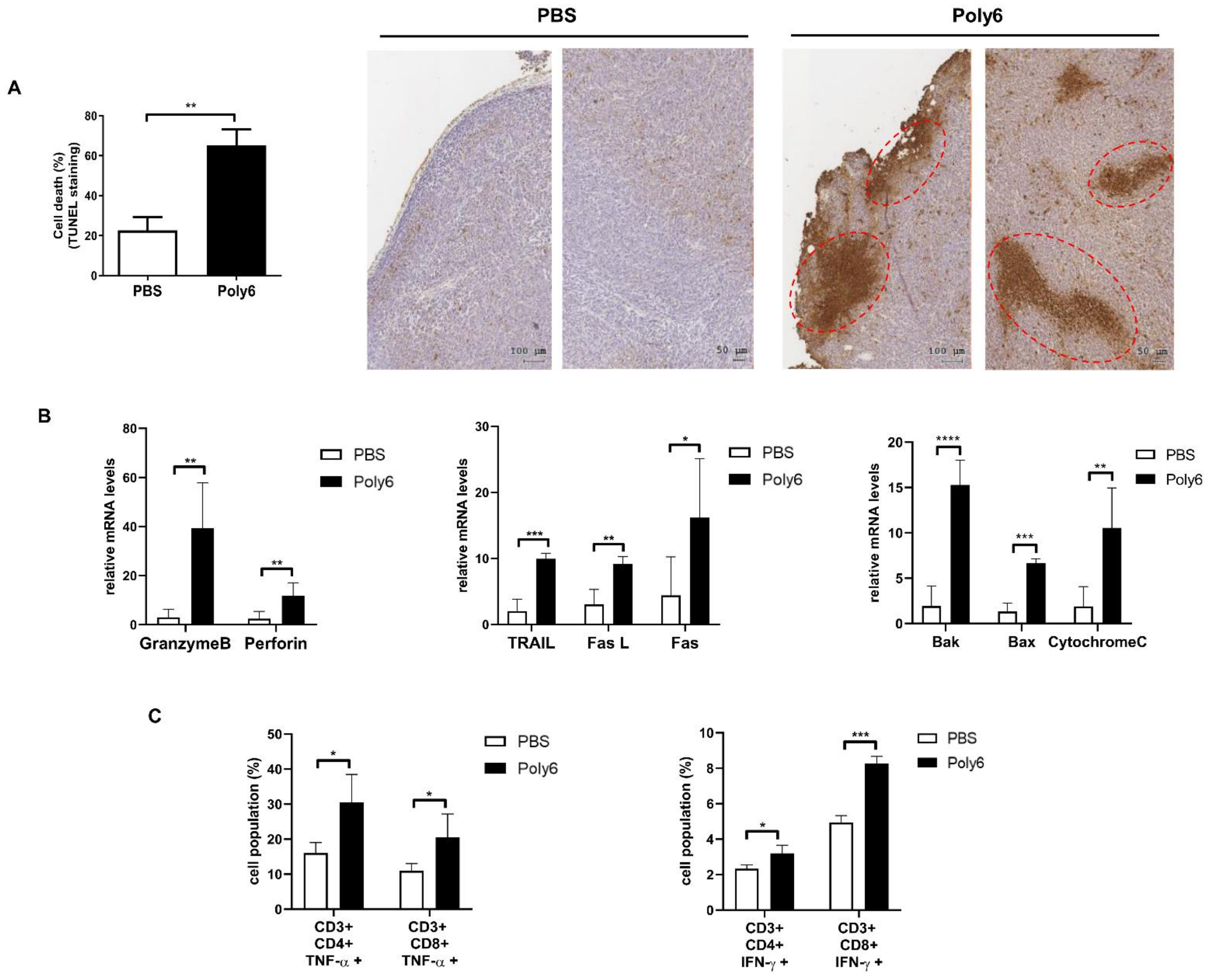
2.3. Poly6 Exerts Anticancer Effects via Induction of Apoptotic Tumor Cell Death in the Tumor Microenvironment Primarily by Activating CD8 T Cell-Mediated CTL Response
2.4. Poly6 Induces Generation of Tip-DCs and CD40 Activation of DCs
2.5. Poly6 Leads to Direct Oncolytic Activity of Tip-DCs in an NO-Dependent Manner
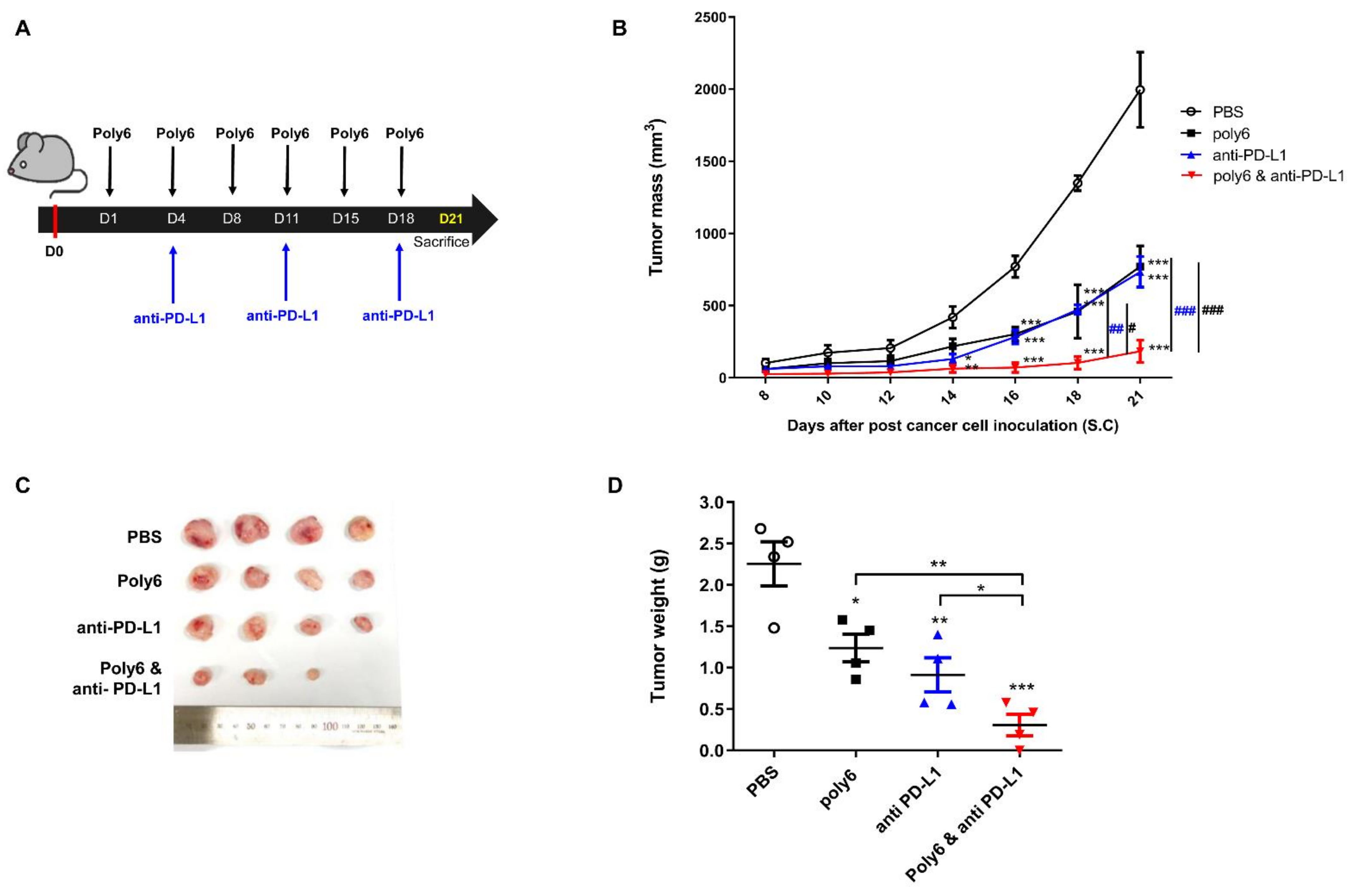
2.6. Combination of Poly6 with Anti-PD-L1 Ab Treatment Exerts an Enhanced Anticancer Effect in Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cells and Cell Culture
4.3. Tumorigenesis Studies
4.4. Histopathological Study
4.5. Flow Cytometry
4.6. Analysis of mRNA by Real-Time PCR
4.7. Western Blot
4.8. Immunofluorescence
4.9. Cytotoxicity
4.9.1. Direct Cell Cytotoxicity
4.9.2. Cell-Mediated Cytotoxicity
4.10. Cytokine and Nitrate Assay
4.11. Dissociation of Tumor, Lymph Nodes, and Spleen
4.12. Measurement and Quantification of Oxidative DNA Damage
4.13. Detection of Mitochondrial ROS and Cytosolic Mitochondrial DNA
4.13.1. Mitochondrial ROS
4.13.2. Cytosolic Mitochondrial DNA
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017, 3, 250–261. [Google Scholar] [CrossRef]
- Schuster, M.; Nechansky, A.; Kircheis, R. Cancer immunotherapy. Biotechnol. J. Healthc. Nutr. Technol. 2006, 1, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Overcoming obstacles to the effective immunotherapy of human cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 12643–12644. [Google Scholar] [CrossRef] [PubMed]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Marigo, I.; Dolcetti, L.; Serafini, P.; Zanovello, P.; Bronte, V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol. Rev. 2008, 222, 162–179. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Intratumoral dendritic cells in the anti-tumor immune response. Cell. Mol. Immunol. 2015, 12, 387–390. [Google Scholar] [CrossRef]
- Amigorena, S.; Savina, A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr. Opin. Immunol. 2010, 22, 109–117. [Google Scholar] [CrossRef]
- Ballestrero, A.; Boy, D.; Moran, E.; Cirmena, G.; Brossart, P.; Nencioni, A. Immunotherapy with dendritic cells for cancer. Adv. Drug Deliv. Rev. 2008, 60, 173–183. [Google Scholar] [CrossRef]
- Datta, M.; Coussens, L.M.; Nishikawa, H.; Hodi, F.S.; Jain, R.K. Reprogramming the tumor microenvironment to improve immunotherapy: Emerging strategies and combination therapies. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 165–174. [Google Scholar] [CrossRef]
- Shi, C.; Liu, T.; Guo, Z.; Zhuang, R.; Zhang, X.; Chen, X. Reprogramming tumor-associated macrophages by nanoparticle-based reactive oxygen species photogeneration. Nano Lett. 2018, 18, 7330–7342. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Porta, C.; Morlacchi, S.; Banfi, S.; Strauss, L.; Rimoldi, M.; Totaro, M.G.; Riboldi, E. Origin and functions of tumor-associated myeloid cells (TAMCs). Cancer Microenviron. 2012, 5, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Bian, K.; Ghassemi, F.; Sotolongo, A.; Siu, A.; Shauger, L.; Kots, A.; Murad, F. NOS-2 signaling and cancer therapy. Iubmb Life 2012, 64, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Marigo, I.; Zilio, S.; Desantis, G.; Mlecnik, B.; Agnellini, A.H.; Ugel, S.; Sasso, M.S.; Qualls, J.E.; Kratochvill, F.; Zanovello, P. T cell cancer therapy requires CD40-CD40L activation of tumor necrosis factor and inducible nitric-oxide-synthase-producing dendritic cells. Cancer Cell 2016, 30, 377–390. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.-Y.; Choi, Y.-M.; Kim, B.-J. HBV polymerase-derived peptide exerts an anti-HIV-1 effect by inhibiting the acetylation of viral integrase. Biochem. Biophys. Res. Commun. 2018, 501, 541–546. [Google Scholar] [CrossRef]
- Dresing, P.; Borkens, S.; Kocur, M.; Kropp, S.; Scheu, S. A fluorescence reporter model defines “Tip-DCs” as the cellular source of interferon β in murine listeriosis. PLoS ONE 2010, 5, e15567. [Google Scholar] [CrossRef]
- Ali, S.; Mann-Nüttel, R.; Schulze, A.; Richter, L.; Alferink, J.; Scheu, S. Sources of type I interferons in infectious immunity: Plasmacytoid dendritic cells not always in the driver’s Seat. Front. Immunol. 2019, 10, 778. [Google Scholar] [CrossRef]
- Auwerx, J.; Li, T.Y. A conserved role of CBP/p300 in mitochondrial stress response and longevity. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Zaini, M.A.; Müller, C.; de Jong, T.V.; Ackermann, T.; Hartleben, G.; Kortman, G.; Gührs, K.-H.; Fusetti, F.; Krämer, O.H.; Guryev, V. A p300 and SIRT1 regulated acetylation switch of C/EBPα controls mitochondrial function. Cell Rep. 2018, 22, 497–511. [Google Scholar] [CrossRef]
- Choi, Y.-M.; Kim, H.; Lee, S.-A.; Lee, S.-Y.; Kim, B.-J. A telomerase-derived peptide exerts an anti-hepatitis B virus effect via mitochondrial DNA stress-dependent Type I interferon production. Front. Immunol. 2020, 11, 652. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Kim, B.-R.; Kim, B.-J.; Kook, Y.-H.; Kim, B.-J. Mycobacterium abscessus infection leads to enhanced production of type 1 interferon and NLRP3 inflammasome activation in murine macrophages via mitochondrial oxidative stress. PLoS Pathog. 2020, 16, e1008294. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Shin, J.B.; Park, B.C.; Lee, J.-W.; Byun, S.W.; Jang, N.-Y.; Kim, Y.J.; Kim, Y.; Kim, Y.K.; Cho, N.-H. Application of radially grown ZnO nanowires on poly-L-lactide microfibers complexed with a tumor antigen for cancer immunotherapy. Nanoscale 2019, 11, 4591–4600. [Google Scholar] [CrossRef]
- Duan, X.; Chan, C.; Lin, W. Nanoparticle-mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angew. Chem. Int. Ed. 2019, 58, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Fauskanger, M.; Haabeth, O.A.W.; Skjeldal, F.M.; Bogen, B.; Tveita, A.A. Tumor killing by CD4+ T cells is mediated via induction of inducible nitric oxide synthase-dependent macrophage cytotoxicity. Front. Immunol. 2018, 9, 1684. [Google Scholar] [CrossRef]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef]
- Thwe, P.M.; Amiel, E. The role of nitric oxide in metabolic regulation of dendritic cell immune function. Cancer Lett. 2018, 412, 236–242. [Google Scholar] [CrossRef]
- Lee, S.-A.; Kim, H.; Won, Y.-S.; Seok, S.-H.; Na, Y.; Shin, H.-B.; Inn, K.-S.; Kim, B.-J. Male-specific hepatitis B virus large surface protein variant W4P potentiates tumorigenicity and induces gender disparity. Mol. Cancer 2015, 14, 23. [Google Scholar] [CrossRef]
- Fraszczak, J.; Trad, M.; Janikashvili, N.; Cathelin, D.; Lakomy, D.; Granci, V.; Morizot, A.; Audia, S.; Micheau, O.; Lagrost, L. Peroxynitrite-dependent killing of cancer cells and presentation of released tumor antigens by activated dendritic cells. J. Immunol. 2010, 184, 1876–1884. [Google Scholar] [CrossRef]
- Szabó, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef]
- Singh, M.; Vianden, C.; Cantwell, M.J.; Dai, Z.; Xiao, Z.; Sharma, M.; Khong, H.; Jaiswal, A.R.; Faak, F.; Hailemichael, Y. Intratumoral CD40 activation and checkpoint blockade induces T cell-mediated eradication of melanoma in the brain. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Movahedi, K.; Bosschaerts, T.; VandenDriessche, T.; Chuah, M.K.; Hérin, M.; Acosta-Sanchez, A.; Ma, L.; Moser, M.; Van Ginderachter, J.A. IL-10 dampens TNF/inducible nitric oxide synthase-producing dendritic cell-mediated pathogenicity during parasitic infection. J. Immunol. 2009, 182, 1107–1118. [Google Scholar] [CrossRef]
- Serbina, N.V.; Salazar-Mather, T.P.; Biron, C.A.; Kuziel, W.A.; Pamer, E.G. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 2003, 19, 59–70. [Google Scholar] [CrossRef]
- Galmozzi, A.; Mitro, N.; Ferrari, A.; Gers, E.; Gilardi, F.; Godio, C.; Cermenati, G.; Gualerzi, A.; Donetti, E.; Rotili, D. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 2013, 62, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Shi, W.; Li, Z.; Liu, H. Activation of mPTP-dependent mitochondrial apoptosis pathway by a novel pan HDAC inhibitor resminostat in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2016, 477, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jin, L.; Chen, X.; Yuan, Y.; Zuo, Y.; Miao, Y.; Feng, Q.; Zhang, H.; Huang, F.; Guo, T. USP12 translocation maintains interferon antiviral efficacy by inhibiting CBP acetyltransferase activity. PLoS Pathog. 2020, 16, e1008215. [Google Scholar] [CrossRef] [PubMed]
- Melroe, G.T.; Silva, L.; Schaffer, P.A.; Knipe, D.M. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN-β induction. Virology 2007, 360, 305–321. [Google Scholar] [CrossRef]
- Schoenberger, S.P.; Toes, R.E.; Van Der Voort, E.I.; Offringa, R.; Melief, C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- Clarke, S.R.M. The critical role of CD40/CD40L in the CD4-dependent generation of CD8+ T cell immunity. J. Leukoc. Biol. 2000, 67, 607–614. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Hunziker, L.; Zinkernagel, R.M.; Storni, T.; Kopf, M. Maintenance of memory CTL responses by T helper cells and CD40-CD40 ligand: Antibodies provide the key. Eur. J. Immunol. 2004, 34, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Li, Y.; Long, K.B. Cancer immunotherapy: Activating innate and adaptive immunity through CD40 agonists. Expert Rev. Anticancer Ther. 2017, 17, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Glennie, M.J. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. 2013, 19, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016, 381, 244–251. [Google Scholar] [CrossRef]
- Yao, X.; Wu, J.; Lin, M.; Sun, W.; He, X.; Gowda, C.; Bolland, S.; Long, C.A.; Wang, R.; Su, X.-z. Increased CD40 expression enhances early STING-mediated type I interferon response and host survival in a rodent malaria model. PLoS Pathog. 2016, 12, e1005930. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Yang, S.-B.; Choi, Y.-M.; Oh, S.-J.; Kim, B.-J.; Kook, Y.-H.; Kim, B.-J. Heat-killed Mycobacterium paragordonae therapy exerts an anti-cancer immune response via enhanced immune cell mediated oncolytic activity in xenograft mice model. Cancer Lett. 2020, 472, 142–150. [Google Scholar] [CrossRef]
- Wu, T.-C.; Xu, K.; Banchereau, R.; Marches, F.; Chun, I.Y.; Martinek, J.; Anguiano, E.; Pedroza-Gonzalez, A.; Snipes, G.J.; O’Shaughnessy, J. Reprogramming tumor-infiltrating dendritic cells for CD103+ CD8+ mucosal T-cell differentiation and breast cancer rejection. Cancer Immunol. Res. 2014, 2, 487–500. [Google Scholar] [CrossRef]
- Zhang, D.; Shi, R.; Xiang, W.; Kang, X.; Tang, B.; Li, C.; Gao, L.; Zhang, X.; Zhang, L.; Dai, R. The Agpat4/LPA axis in colorectal cancer cells regulates antitumor responses via p38/p65 signaling in macrophages. Signal. Transduct. Target. Ther. 2020, 5, 1–13. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.-B.; Lee, M.-H.; Kim, B.-R.; Choi, Y.-M.; Kim, B.-J. A Hepatitis B Virus-Derived Peptide Exerts an Anticancer Effect via TNF/iNOS-producing Dendritic Cells in Tumor-Bearing Mouse Model. Cancers 2021, 13, 407. https://doi.org/10.3390/cancers13030407
Yang S-B, Lee M-H, Kim B-R, Choi Y-M, Kim B-J. A Hepatitis B Virus-Derived Peptide Exerts an Anticancer Effect via TNF/iNOS-producing Dendritic Cells in Tumor-Bearing Mouse Model. Cancers. 2021; 13(3):407. https://doi.org/10.3390/cancers13030407
Chicago/Turabian StyleYang, Soo-Bin, Mi-Hyun Lee, Bo-Ram Kim, Yu-Min Choi, and Bum-Joon Kim. 2021. "A Hepatitis B Virus-Derived Peptide Exerts an Anticancer Effect via TNF/iNOS-producing Dendritic Cells in Tumor-Bearing Mouse Model" Cancers 13, no. 3: 407. https://doi.org/10.3390/cancers13030407
APA StyleYang, S.-B., Lee, M.-H., Kim, B.-R., Choi, Y.-M., & Kim, B.-J. (2021). A Hepatitis B Virus-Derived Peptide Exerts an Anticancer Effect via TNF/iNOS-producing Dendritic Cells in Tumor-Bearing Mouse Model. Cancers, 13(3), 407. https://doi.org/10.3390/cancers13030407