Immune Resistance in Lung Adenocarcinoma


Simple Summary
Abstract
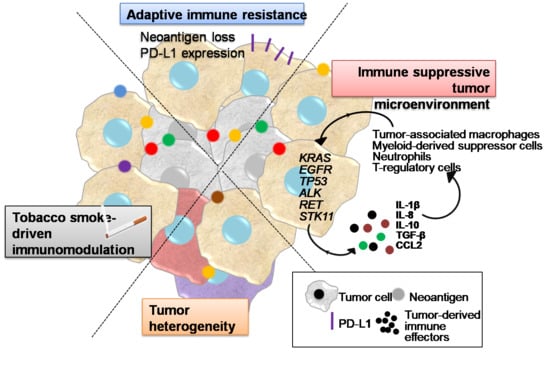
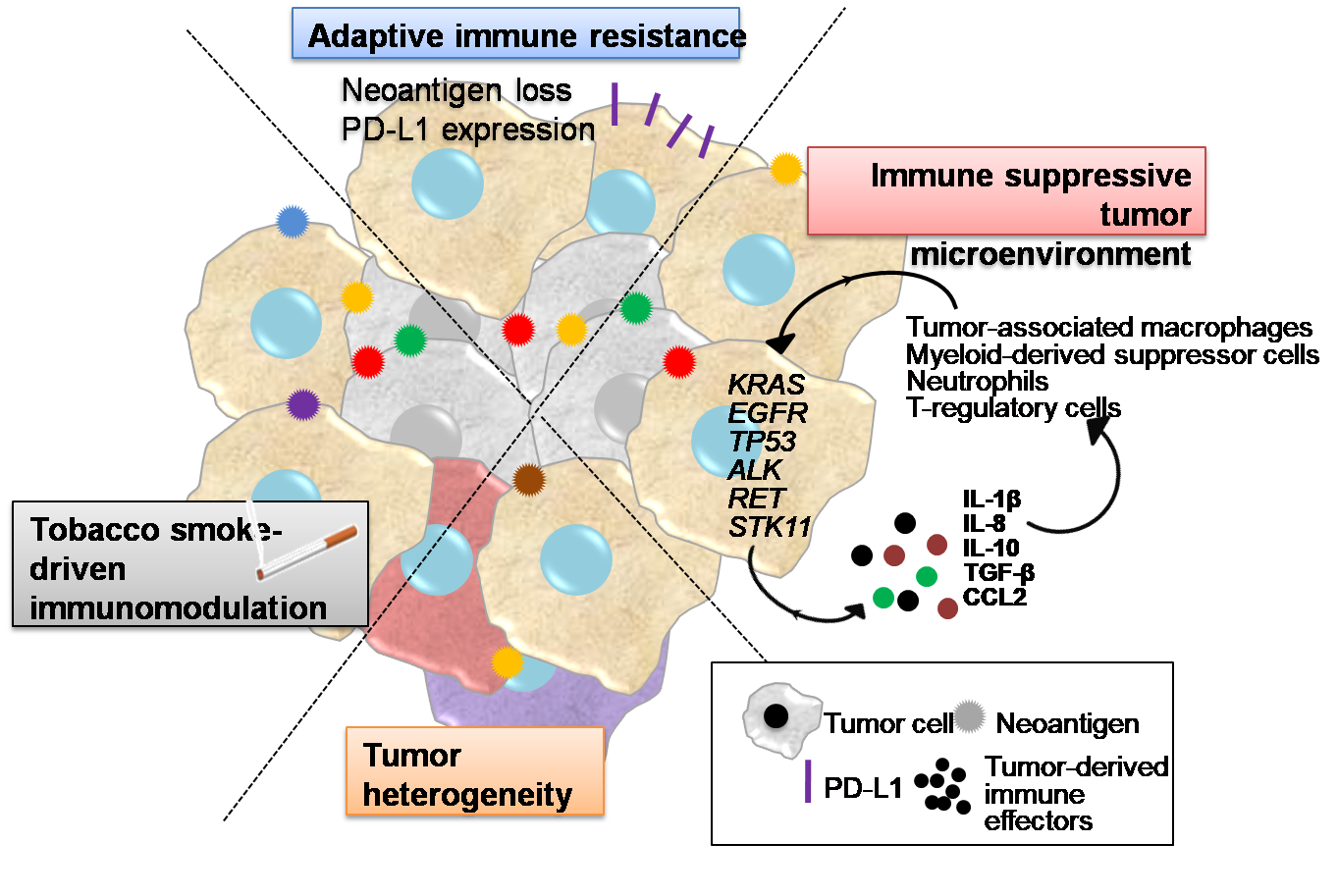
1. Introduction
2. Tobacco Smoke Immunomodulatory Effects
3. Tumor Heterogeneity
4. Inflammatory Interactions of LUAD Cells with the Tumor Microenvironment Mediating Immune Escape
5. Adaptive Immune Resistance
6. Acquired Immune Resistance
7. Drug Resistance
8. Strategies to Overcome Resistance
9. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ward, E.M.; Smith, R.; Jemal, A. Annual number of lung cancer deaths potentially avertable by screening in the United States. Cancer 2013, 119, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Dempke, W.C.; Suto, T.; Reck, M. Targeted therapies for non-small cell lung cancer. Lung Cancer 2010, 67, 257–274. [Google Scholar] [CrossRef]
- Shea, M.; Costa, D.B.; Rangachari, D. Management of advanced non-small cell lung cancers with known mutations or rearrangements: Latest evidence and treatment approaches. Ther. Adv. Respir. Dis. 2016, 10, 113–129. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, L.L.; Chen, J.H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target Ther. 2019, 4, 61. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef]
- Drilon, A.; Wang, L.; Hasanovic, A.; Suehara, Y.; Lipson, D.; Stephens, P.; Ross, J.; Miller, V.; Ginsberg, M.; Zakowski, M.F.; et al. Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013, 3, 630–635. [Google Scholar] [CrossRef]
- Lin, J.J.; Shaw, A.T. Resisting Resistance: Targeted Therapies in Lung Cancer. Trends Cancer 2016, 2, 350–364. [Google Scholar] [CrossRef]
- Hames, M.L.; Chen, H.; Iams, W.; Aston, J.; Lovly, C.M.; Horn, L. Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung cancer. Lung Cancer 2016, 92, 29–34. [Google Scholar] [CrossRef]
- Dias Carvalho, P.; Machado, A.L.; Martins, F.; Seruca, R.; Velho, S. Targeting the Tumor Microenvironment: An Unexplored Strategy for Mutant KRAS Tumors. Cancers 2019, 11, 2010. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Li, W.A.; Mooney, D.J.; Dranoff, G. Advances in Therapeutic Cancer Vaccines. Adv. Immunol. 2016, 130, 191–249. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Rosenberg, S.A. Decade in review-cancer immunotherapy: Entering the mainstream of cancer treatment. Nat. Rev. Clin. Oncol. 2014, 11, 630–632. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Hall, R.D.; Gray, J.E.; Chiappori, A.A. Beyond the standard of care: A review of novel immunotherapy trials for the treatment of lung cancer. Cancer Control 2013, 20, 22–31. [Google Scholar] [CrossRef]
- Bonavida, B.; Chouaib, S. Resistance to anticancer immunity in cancer patients: Potential strategies to reverse resistance. Ann. Oncol. 2017, 28, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Dias Carvalho, P.; Guimaraes, C.F.; Cardoso, A.P.; Mendonca, S.; Costa, A.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Agalioti, T.; Giannou, A.D.; Krontira, A.C.; Kanellakis, N.I.; Kati, D.; Vreka, M.; Pepe, M.; Spella, M.; Lilis, I.; Zazara, D.E.; et al. Mutant KRAS promotes malignant pleural effusion formation. Nat. Commun. 2017, 8, 15205. [Google Scholar] [CrossRef] [PubMed]
- Lilis, I.; Ntaliarda, G.; Papaleonidopoulos, V.; Giotopoulou, G.A.; Oplopoiou, M.; Marazioti, A.; Spella, M.; Marwitz, S.; Goldmann, T.; Bravou, V.; et al. Interleukin-1beta provided by KIT-competent mast cells is required for KRAS-mutant lung adenocarcinoma. Oncoimmunology 2019, 8, 1593802. [Google Scholar] [CrossRef]
- Marazioti, A.; Lilis, I.; Vreka, M.; Apostolopoulou, H.; Kalogeropoulou, A.; Giopanou, I.; Giotopoulou, G.A.; Krontira, A.C.; Iliopoulou, M.; Kanellakis, N.I.; et al. Myeloid-derived interleukin-1beta drives oncogenic KRAS-NF-kappaBeta addiction in malignant pleural effusion. Nat. Commun. 2018, 9, 672. [Google Scholar] [CrossRef]
- Giotopoulou, G.A.; Stathopoulos, G.T. Effects of Inhaled Tobacco Smoke on the Pulmonary Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1225, 53–69. [Google Scholar] [CrossRef]
- Engels, E.A. Inflammation in the development of lung cancer: Epidemiological evidence. Expert Rev. Anticancer. Ther. 2008, 8, 605–615. [Google Scholar] [CrossRef]
- Kode, A.; Yang, S.R.; Rahman, I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir. Res. 2006, 7, 132. [Google Scholar] [CrossRef]
- Reynolds, P.R.; Kasteler, S.D.; Schmitt, R.E.; Hoidal, J.R. Receptor for advanced glycation end-products signals through Ras during tobacco smoke-induced pulmonary inflammation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 411–418. [Google Scholar] [CrossRef]
- Kroening, P.R.; Barnes, T.W.; Pease, L.; Limper, A.; Kita, H.; Vassallo, R. Cigarette smoke-induced oxidative stress suppresses generation of dendritic cell IL-12 and IL-23 through ERK-dependent pathways. J. Immunol. 2008, 181, 1536–1547. [Google Scholar] [CrossRef]
- Liu, X.; Togo, S.; Al-Mugotir, M.; Kim, H.; Fang, Q.; Kobayashi, T.; Wang, X.; Mao, L.; Bitterman, P.; Rennard, S. NF-kappaB mediates the survival of human bronchial epithelial cells exposed to cigarette smoke extract. Respir. Res. 2008, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Herr, C.; Beisswenger, C.; Hess, C.; Kandler, K.; Suttorp, N.; Welte, T.; Schroeder, J.M.; Vogelmeier, C. Suppression of pulmonary innate host defence in smokers. Thorax 2009, 64, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, J.; Wu, P.; Zhou, L.; Lu, B.; Ying, K.; Chen, E.; Lu, Y.; Liu, P. Smoker and non-smoker lung adenocarcinoma is characterized by distinct tumor immune microenvironments. Oncoimmunology 2018, 7, e1494677. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, S.A.; Gordon, L.I. Inflammation and cancer: Role of phagocyte-generated oxidants in carcinogenesis. Blood 1990, 76, 655–663. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Allavena, P.; Germano, G.; Marchesi, F.; Mantovani, A. Chemokines in cancer related inflammation. Exp. Cell Res. 2011, 317, 664–673. [Google Scholar] [CrossRef]
- Bronte, V.; Cingarlini, S.; Marigo, I.; De Santo, C.; Gallina, G.; Dolcetti, L.; Ugel, S.; Peranzoni, E.; Mandruzzato, S.; Zanovello, P. Leukocyte infiltration in cancer creates an unfavorable environment for antitumor immune responses: A novel target for therapeutic intervention. Immunol. Investig. 2006, 35, 327–357. [Google Scholar] [CrossRef]
- Zaynagetdinov, R.; Sherrill, T.P.; Polosukhin, V.V.; Han, W.; Ausborn, J.A.; McLoed, A.G.; McMahon, F.B.; Gleaves, L.A.; Degryse, A.L.; Stathopoulos, G.T.; et al. A critical role for macrophages in promotion of urethane-induced lung carcinogenesis. J. Immunol. 2011, 187, 5703–5711. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chintala, N.K.; Vadrevu, S.K.; Patel, J.; Karbowniczek, M.; Markiewski, M.M. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J. Immunol. 2015, 194, 5529–5538. [Google Scholar] [CrossRef] [PubMed]
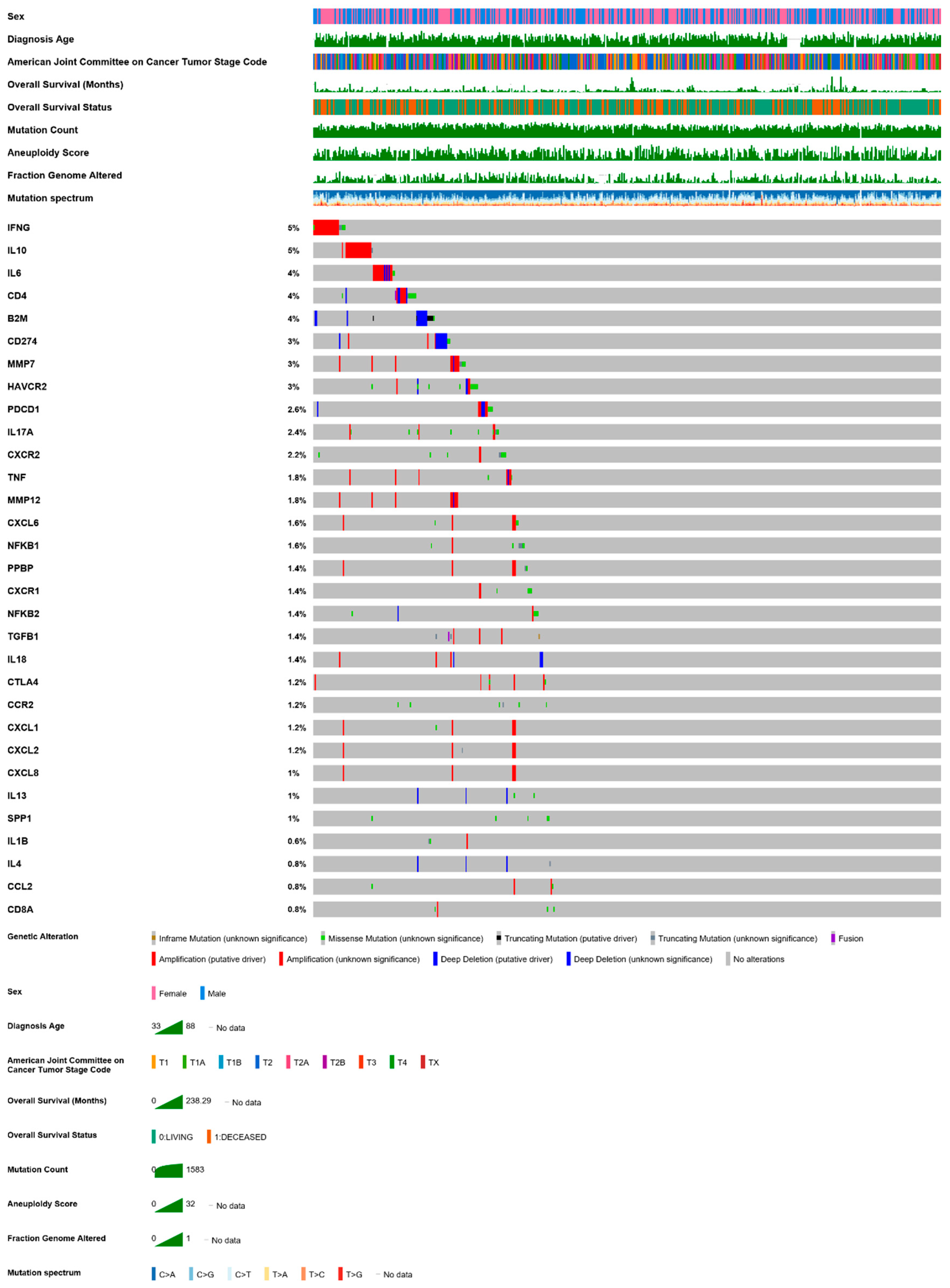
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Zaynagetdinov, R.; Stathopoulos, G.T.; Sherrill, T.P.; Cheng, D.S.; McLoed, A.G.; Ausborn, J.A.; Polosukhin, V.V.; Connelly, L.; Zhou, W.; Fingleton, B.; et al. Epithelial nuclear factor-kappaB signaling promotes lung carcinogenesis via recruitment of regulatory T lymphocytes. Oncogene 2012, 31, 3164–3176. [Google Scholar] [CrossRef]
- Shimizu, K.; Nakata, M.; Hirami, Y.; Yukawa, T.; Maeda, A.; Tanemoto, K. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J Thorac. Oncol. 2010, 5, 585–590. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Andersson, A.; Zhu, L.; Harris-White, M.; Lee, J.M.; Dubinett, S.; Sharma, S. Myeloid suppressor cells and immune modulation in lung cancer. Immunotherapy 2012, 4, 291–304. [Google Scholar] [CrossRef]
- Busch, S.E.; Hanke, M.L.; Kargl, J.; Metz, H.E.; MacPherson, D.; Houghton, A.M. Lung Cancer Subtypes Generate Unique Immune Responses. J. Immunol. 2016, 197, 4493–4503. [Google Scholar] [CrossRef]
- Ji, H.; Houghton, A.M.; Mariani, T.J.; Perera, S.; Kim, C.B.; Padera, R.; Tonon, G.; McNamara, K.; Marconcini, L.A.; Hezel, A.; et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene 2006, 25, 2105–2112. [Google Scholar] [CrossRef]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef]
- Giannou, A.D.; Marazioti, A.; Spella, M.; Kanellakis, N.I.; Apostolopoulou, H.; Psallidas, I.; Prijovich, Z.M.; Vreka, M.; Zazara, D.E.; Lilis, I.; et al. Mast cells mediate malignant pleural effusion formation. J. Clin. Investig. 2015, 125, 2317–2334. [Google Scholar] [CrossRef]
- Kim, J.W.; Koh, Y.; Kim, D.W.; Ahn, Y.O.; Kim, T.M.; Han, S.W.; Oh, D.Y.; Lee, S.H.; Im, S.A.; Kim, T.Y.; et al. Clinical Implications of VEGF, TGF-beta1, and IL-1beta in Patients with Advanced Non-small Cell Lung Cancer. Cancer Res. Treat. 2013, 45, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Gagné-Ouellet, V.J.É.; Boucher-Lafleur, A.M.; Plante, S.; Bouchard, L.; Chakir, J.; Laprise, C. Tobacco smoke induces changes in IL-1 family in bronchial epithelial cells obtained from asthmatic individuals. All Asth. Clin. Immun. 2014, 10, A55. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- McLoed, A.G.; Sherrill, T.P.; Cheng, D.S.; Han, W.; Saxon, J.A.; Gleaves, L.A.; Wu, P.; Polosukhin, V.V.; Karin, M.; Yull, F.E.; et al. Neutrophil-Derived IL-1beta Impairs the Efficacy of NF-kappaB Inhibitors against Lung Cancer. Cell Rep. 2016, 16, 120–132. [Google Scholar] [CrossRef]
- Testorelli, C.; Bussini, S.; De Filippi, R.; Marelli, O.; Orlando, L.; Greiner, J.W.; Grohmann, U.; Tentori, L.; Giuliani, A.; Bonmassar, E.; et al. Dacarbazine-induced immunogenicity of a murine leukemia is attenuated in cells transfected with mutated K-ras gene. J. Exp. Clin. Cancer Res. 1997, 16, 15–22. [Google Scholar]
- Weijzen, S.; Velders, M.P.; Kast, W.M. Modulation of the immune response and tumor growth by activated Ras. Leukemia 1999, 13, 502–513. [Google Scholar] [CrossRef]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef]
- Granville, C.A.; Memmott, R.M.; Balogh, A.; Mariotti, J.; Kawabata, S.; Han, W.; Lopiccolo, J.; Foley, J.; Liewehr, D.J.; Steinberg, S.M.; et al. A central role for Foxp3+ regulatory T cells in K-Ras-driven lung tumorigenesis. PLoS ONE 2009, 4, e5061. [Google Scholar] [CrossRef]
- Chang, S.H. Tumorigenic Th17 cells in oncogenic Kras-driven and inflammation-accelerated lung cancer. Oncoimmunology 2015, 4, e955704. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Mirabolfathinejad, S.G.; Katta, H.; Cumpian, A.M.; Gong, L.; Caetano, M.S.; Moghaddam, S.J.; Dong, C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 5664–5669. [Google Scholar] [CrossRef] [PubMed]
- Smakman, N.; Veenendaal, L.M.; van Diest, P.; Bos, R.; Offringa, R.; Borel Rinkes, I.H.; Kranenburg, O. Dual effect of Kras(D12) knockdown on tumorigenesis: Increased immune-mediated tumor clearance and abrogation of tumor malignancy. Oncogene 2005, 24, 8338–8342. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.Y.; Chen, H.J.; Sun, Y.L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef]
- Huynh, T.G.; Morales-Oyarvide, V.; Campo, M.J.; Gainor, J.F.; Bozkurtlar, E.; Uruga, H.; Zhao, L.; Gomez-Caraballo, M.; Hata, A.N.; Mark, E.J.; et al. Programmed Cell Death Ligand 1 Expression in Resected Lung Adenocarcinomas: Association with Immune Microenvironment. J. Thorac. Oncol. 2016, 11, 1869–1878. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Herter-Sprie, G.S.; Koyama, S.; Korideck, H.; Hai, J.; Deng, J.; Li, Y.Y.; Buczkowski, K.A.; Grant, A.K.; Ullas, S.; Rhee, K.; et al. Synergy of radiotherapy and PD-1 blockade in Kras-mutant lung cancer. JCI Insight 2016, 1, e87415. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Ribas, A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra137. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Oh, S.A.; Wu, D.C.; Cheung JNavarro, A.; Xiong, H.; Cubas, R.; Totpal, K.; Chiu, H.; Wu, Y.; Comps-Agrar, L.; Leader, A.M.; et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer 2020, 681–691. [Google Scholar] [CrossRef]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wolfel, T.; Holzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Knutson, K.L.; Lu, H.; Stone, B.; Reiman, J.M.; Behrens, M.D.; Prosperi, C.M.; Gad, E.A.; Smorlesi, A.; Disis, M.L. Immunoediting of cancers may lead to epithelial to mesenchymal transition. J. Immunol. 2006, 177, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Santisteban, M.; Reiman, J.M.; Asiedu, M.K.; Behrens, M.D.; Nassar, A.; Kalli, K.R.; Haluska, P.; Ingle, J.N.; Hartmann, L.C.; Manjili, M.H.; et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009, 69, 2887–2895. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.K.; Wu, Y.L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Horn, L.; Gandhi, L.; Spigel, D.R.; Antonia, S.J.; Rizvi, N.A.; Powderly, J.D.; Heist, R.S.; Carvajal, R.D.; Jackman, D.M.; et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients with Previously Treated Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2015, 33, 2004–2012. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Kinoshita, T.; Kudo-Saito, C.; Muramatsu, R.; Fujita, T.; Saito, M.; Nagumo, H.; Sakurai, T.; Noji, S.; Takahata, E.; Yaguchi, T.; et al. Determination of poor prognostic immune features of tumour microenvironment in non-smoking patients with lung adenocarcinoma. Eur. J. Cancer. 2017, 86, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Maeurer, M.J.; Gollin, S.M.; Storkus, W.J.; Swaney, W.; Karbach, J.; Martin, D.; Castelli, C.; Salter, R.; Knuth, A.; Lotze, M.T. Tumor escape from immune recognition: Loss of HLA-A2 melanoma cell surface expression is associated with a complex rearrangement of the short arm of chromosome 6. Clin. Cancer Res. 1996, 2, 641–652. [Google Scholar] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e1211. [Google Scholar] [CrossRef] [PubMed]
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic HLA class I loss is a widespread mechanism of immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef]
- Bar, J.; Ofek, E.; Barshack, I.; Gottfried, T.; Zadok, O.; Kamer, I.; Urban, D.; Perelman, M.; Onn, A. Transformation to small cell lung cancer as a mechanism of resistance to immunotherapy in non-small cell lung cancer. Lung Cancer. 2019, 138, 109–115. [Google Scholar] [CrossRef]
- Imakita, T.; Fujita, K.; Kanai, O.; Terashima, T.; Mio, T. Small cell lung cancer transformation during immunotherapy with nivolumab: A case report. Respir. Med. Case Rep. 2017, 21, 52–55. [Google Scholar] [CrossRef]
- Weinstein, I.B. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Barlesi, F.; Mazieres, J.; Merlio, J.P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Costa, C.; Molina, M.A.; Drozdowskyj, A.; Gimenez-Capitan, A.; Bertran-Alamillo, J.; Karachaliou, N.; Gervais, R.; Massuti, B.; Wei, J.; Moran, T.; et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin. Cancer Res. 2014, 20, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef]
- Arcila, M.E.; Oxnard, G.R.; Nafa, K.; Riely, G.J.; Solomon, S.B.; Zakowski, M.F.; Kris, M.G.; Pao, W.; Miller, V.A.; Ladanyi, M. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin. Cancer Res. 2011, 17, 1169–1180. [Google Scholar] [CrossRef]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Tricker, E.M.; Xu, C.; Uddin, S.; Capelletti, M.; Ercan, D.; Ogino, A.; Pratilas, C.A.; Rosen, N.; Gray, N.S.; Wong, K.K.; et al. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer Discov. 2015, 5, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Ikushima, S.; Ono, R.; Awano, N.; Kondo, K.; Furuhata, Y.; Fukumoto, K.; Kumasaka, T. Transformation to small-cell lung cancer as a mechanism of acquired resistance to crizotinib and alectinib. Jpn. J. Clin. Oncol. 2016, 46, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Tartour, E.; Zitvogel, L. Lung cancer: Potential targets for immunotherapy. Lancet Respir. Med. 2013, 1, 551–563. [Google Scholar] [CrossRef]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef]
- Arce Vargas, F.; Furness, A.J.S.; Solomon, I.; Joshi, K.; Mekkaoui, L.; Lesko, M.H.; Miranda Rota, E.; Dahan, R.; Georgiou, A.; Sledzinska, A.; et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 2017, 46, 577–586. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| A | B | Neither | A Not B | B Not A | Both | Log2 Odds Ratio | p-Value | q-Value | Tendency |
|---|---|---|---|---|---|---|---|---|---|
| CDKN2A | CDKN2B | 402 | 22 | 0 | 83 | >3 | <0.001 | <0.001 | COOC |
| CDKN2B | MTAP | 422 | 21 | 2 | 62 | >3 | <0.001 | <0.001 | COOC |
| CDKN2A | MTAP | 400 | 43 | 2 | 62 | >3 | <0.001 | <0.001 | COOC |
| NKX2-1 | FOXA1 | 433 | 20 | 5 | 49 | >3 | <0.001 | <0.001 | COOC |
| PCLO | RELN | 361 | 58 | 45 | 43 | 2.572 | <0.001 | <0.001 | COOC |
| KRAS | EGFR | 265 | 163 | 73 | 6 | −2.904 | <0.001 | <0.001 | MUEX |
| LRP1B | RELN | 288 | 131 | 33 | 55 | 1.873 | <0.001 | <0.001 | COOC |
| LRP1B | PCLO | 278 | 128 | 43 | 58 | 1.551 | <0.001 | <0.001 | COOC |
| TP53 | LRP1B | 179 | 142 | 64 | 122 | 1.265 | <0.001 | <0.001 | COOC |
| TP53 | PCLO | 215 | 191 | 28 | 73 | 1.553 | <0.001 | <0.001 | COOC |
| ALK | EML4 | 462 | 28 | 9 | 8 | >3 | <0.001 | <0.001 | COOC |
| KEAP1 | STK11 | 359 | 68 | 49 | 31 | 1.74 | <0.001 | <0.001 | COOC |
| TP53 | KRAS | 139 | 199 | 104 | 65 | −1.196 | <0.001 | <0.001 | MUEX |
| KRAS | STK11 | 301 | 126 | 37 | 43 | 1.473 | <0.001 | <0.001 | COOC |
| TP53 | TERT | 225 | 212 | 18 | 52 | 1.616 | <0.001 | <0.001 | COOC |
| TP53 | NF1 | 226 | 216 | 17 | 48 | 1.563 | <0.001 | 0.001 | COOC |
| LRP1B | PTPRD | 261 | 124 | 60 | 62 | 1.121 | <0.001 | 0.002 | COOC |
| CDKN2A | ROS1 | 384 | 89 | 18 | 16 | 1.939 | <0.001 | 0.003 | COOC |
| TP53 | EGFR | 219 | 209 | 24 | 55 | 1.264 | <0.001 | 0.005 | COOC |
| KRAS | NF1 | 283 | 159 | 55 | 10 | −1.628 | <0.001 | 0.005 | MUEX |
| LRP1B | EGFR | 258 | 170 | 63 | 16 | −1.375 | <0.001 | 0.005 | MUEX |
| TP53 | RELN | 215 | 204 | 28 | 60 | 1.175 | <0.001 | 0.005 | COOC |
| PTPRD | PCLO | 321 | 85 | 64 | 37 | 1.126 | 0.001 | 0.009 | COOC |
| STK11 | TERT | 360 | 77 | 67 | 3 | −2.256 | 0.002 | 0.013 | MUEX |
| PTPRD | RELN | 329 | 90 | 56 | 32 | 1.063 | 0.003 | 0.022 | COOC |
| KRAS | ROS1 | 308 | 165 | 30 | 4 | −2.006 | 0.003 | 0.022 | MUEX |
| KEAP1 | EGFR | 336 | 92 | 72 | 7 | −1.494 | 0.005 | 0.033 | MUEX |
| STK11 | EGFR | 353 | 75 | 74 | 5 | −1.653 | 0.006 | 0.041 | MUEX |
| TP53 | PTPRD | 197 | 188 | 46 | 76 | 0.792 | 0.006 | 0.041 | COOC |
| TP53 | STK11 | 194 | 233 | 49 | 31 | −0.925 | 0.007 | 0.042 | MUEX |
| LRP1B | KRAS | 227 | 111 | 94 | 75 | 0.706 | 0.007 | 0.046 | COOC |
| A | B | Neither | A Not B | B Not A | Both | Log2 Odds Ratio | p-Value | q-Value | Tendency |
|---|---|---|---|---|---|---|---|---|---|
| MMP7 | MMP12 | 492 | 6 | 0 | 9 | >3 | <0.001 | <0.001 | Co-occurrence |
| CXCL1 | CXCL8 | 501 | 1 | 0 | 5 | >3 | <0.001 | <0.001 | Co-occurrence |
| CXCL2 | CXCL8 | 501 | 1 | 0 | 5 | >3 | <0.001 | <0.001 | Co-occurrence |
| PPBP | CXCL8 | 500 | 2 | 0 | 5 | >3 | <0.001 | <0.001 | Co-occurrence |
| CXCL1 | CXCL2 | 500 | 1 | 1 | 5 | >3 | <0.001 | <0.001 | Co-occurrence |
| CXCL6 | CXCL8 | 499 | 3 | 0 | 5 | >3 | <0.001 | <0.001 | Co-occurrence |
| PPBP | CXCL1 | 499 | 2 | 1 | 5 | >3 | <0.001 | <0.001 | Co-occurrence |
| PPBP | CXCL2 | 499 | 2 | 1 | 5 | >3 | <0.001 | <0.001 | Co-occurrence |
| CXCL6 | CXCL1 | 498 | 3 | 1 | 5 | >3 | <0.001 | <0.001 | Co-occurrence |
| CXCL6 | CXCL2 | 498 | 3 | 1 | 5 | >3 | <0.001 | <0.001 | Co-occurrence |
| CXCL6 | PPBP | 497 | 3 | 2 | 5 | >3 | <0.001 | <0.001 | Co-occurrence |
| IL13 | IL4 | 501 | 2 | 1 | 3 | >3 | <0.001 | <0.001 | Co-occurrence |
| TGFB1 | IL18 | 496 | 4 | 4 | 3 | >3 | <0.001 | 0.002 | Co-occurrence |
| MMP12 | IL18 | 494 | 6 | 4 | 3 | >3 | <0.001 | 0.004 | Co-occurrence |
| MMP7 | IL18 | 488 | 12 | 4 | 3 | >3 | <0.001 | 0.021 | Co-occurrence |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spella, M.; Stathopoulos, G.T. Immune Resistance in Lung Adenocarcinoma. Cancers 2021, 13, 384. https://doi.org/10.3390/cancers13030384
Spella M, Stathopoulos GT. Immune Resistance in Lung Adenocarcinoma. Cancers. 2021; 13(3):384. https://doi.org/10.3390/cancers13030384
Chicago/Turabian StyleSpella, Magda, and Georgios T. Stathopoulos. 2021. "Immune Resistance in Lung Adenocarcinoma" Cancers 13, no. 3: 384. https://doi.org/10.3390/cancers13030384
APA StyleSpella, M., & Stathopoulos, G. T. (2021). Immune Resistance in Lung Adenocarcinoma. Cancers, 13(3), 384. https://doi.org/10.3390/cancers13030384