
Influence of Estrogen Treatment on ESR1+ and ESR1− Cells in ER+ Breast Cancer: Insights from Single-Cell Analysis of Patient-Derived Xenograft Models

 , ,
, , 
Abstract
Simple Summary
Abstract
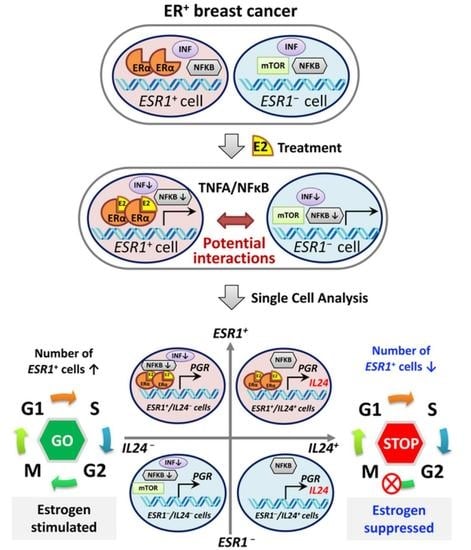
1. Introduction
2. Materials and Methods
2.1. PDX
2.2. In Vivo Animal Study
2.3. Histological Analysis
2.4. Western Blotting
2.5. RPPA Analysis
2.6. Real-Time PCR Analysis
2.7. Bulk RNA-Seq Analysis
2.8. Single-Cell Preparation
2.9. ScRNA-Seq Analysis
2.10. Organoids and In Vitro Treatment Study
2.11. Statistics
3. Results
3.1. SC31 and GS3 Behaved Oppositely Regarding Tumor Growth with Estrogen
3.2. E2 Downregulated the Expression of ERα and Cell Cycle Proliferation Genes in GS3
3.3. Impact of E2 on Gene Expression at the Single-Cell Level in SC31 and GS3
3.4. Characteristics of Single-Cell Clusters in SC31 and GS3
3.5. Comparison of E2-Treated Cells vs. Placebo-Treated Cells in SC31 and GS3
3.6. Effects of E2 Treatment on ESR1+ Cells and ESR1– Cells in SC31 and GS3
3.7. E2-Induced IL-24+ Cells through ERα Only in GS3
3.8. Intermittent E2 Treatment in GS3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chlebowski, R.; Cuzick, J.; Amakye, D.; Bauerfeind, I.; Buzdar, A.; Chiaf, S.; Cutulig, B.; Linforthh, R.; Maassi, N.; Noguchij, S.; et al. Clinical perspectives on the utility of aromatase inhibitors for the adjuvant treatment of breast cancer. Breast 2009, 18, S1–S11. [Google Scholar] [CrossRef]
- Chumsri, S.; Howes, T.; Bao, T.; Sabnis, G.; Brodie, A. Aromatase, aromatase inhibitors, and breast cancer. J. Steroid Biochem. Mol. Biol. 2011, 125, 13–22. [Google Scholar] [CrossRef]
- Lao Romera, J.; Puertolas Hernandez, T.J.; Pelaez Fernandez, I.; Sampedro Gimeno, T.; Fernandez Martinez, R.; Fernandez Perez, I.; Iranzo Gonzalez Cruz, V.; Illarramendi Manas, J.J.; Garcera Juan, S.; Ciruelos Gil, E.M. Update on adjuvant hormonal treatment of early breast cancer. Adv. Ther. 2011, 28, 1–18. [Google Scholar] [CrossRef]
- The BIG 1-98 Collaborative Group. Letrozole Therapy Alone or in Sequence with Tamoxifen in Women with Breast Cancer. N. Engl. J. Med. 2009, 361, 766–776. [Google Scholar] [CrossRef]
- Cuzick, J.; Sestak, I.; Baum, M.; Buzdar, A.; Howell, A.; Dowsett, M.; Forbes, J.F. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol. 2010, 11, 1135–1141. [Google Scholar] [CrossRef]
- van de Velde, C.J.; Rea, D.; Seynaeve, C.; Putter, H.; Hasenburg, A.; Vannetzel, J.M.; Paridaens, R.; Markopoulos, C.; Hozumi, Y.; Hille, E.T.M.; et al. Adjuvant tamoxifen and exemestane in early breast cancer (TEAM): A randomised phase 3 trial. Lancet 2011, 377, 321–331. [Google Scholar] [CrossRef]
- Lønning, P.; Taylor, P.; Anker, G.; Iddon, J.; Wie, L.; Jørgensen, L.-M.; Mella, O.; Howell, A. High-dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res. Treat. 2001, 67, 111–116. [Google Scholar] [CrossRef]
- Agrawal, A.; Robertson, J.F.; Cheung, K.L. Efficacy and tolerability of high dose “ethinylestradiol” in post-menopausal advanced breast cancer patients heavily pre-treated with endocrine agents. World J. Surg. Oncol. 2006, 4, 1–5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ellis, M.J.; Gao, F.; Dehdashti, F.; Jeffe, D.B.; Marcom, P.K.; Carey, L.A.; Dickler, M.N.; Silverman, P.; Fleming, G.F.; Kommareddy, A.; et al. Lower-dose vs. high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: A phase 2 randomized study. JAMA 2009, 302, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Iwase, H.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Murakami, K.I.; Okumura, Y.; Tomita, S.; Inao, T.; Honda, Y.; Omoto, Y.; Iyama, K.I. Ethinylestradiol is beneficial for postmenopausal patients with heavily pre-treated metastatic breast cancer after prior aromatase inhibitor treatment: A prospective study. Br. J. Cancer 2013, 109, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, P.; Stopeck, A.; Clarke, K.; Livingston, R. A pilot study of estradiol followed by exemestane for reversing endocrine resistance in postmenopausal women with hormone receptor-positive metastatic breast cancer. Oncologist 2014, 19, 1127–1128. [Google Scholar] [CrossRef][Green Version]
- Zucchini, G.; Armstrong, A.C.; Wardley, A.M.; Wilson, G.; Misra, V.; Seif, M.; Ryder, W.D.; Cope, J.; Blowers, E.; Howell, A.; et al. A phase II trial of low-dose estradiol in postmenopausal women with advanced breast cancer and acquired resistance to aromatase inhibition. Eur. J. Cancer 2015, 51, 2725–2731. [Google Scholar] [CrossRef]
- Hsu, P.Y.; Wu, V.S.; Kanaya, N.; Petrossian, K.; Hsu, H.K.; Nguyen, D.; Schmolze, D.; Kai, M.; Liu, C.Y.; Lu, H.; et al. Dual mTOR Kinase Inhibitor MLN0128 Sensitizes HR(+)/HER2(+) Breast Cancer Patient-Derived Xenografts to Trastuzumab or Fulvestrant. Clin. Cancer Res. 2018, 24, 395–406. [Google Scholar] [CrossRef]
- Kanaya, N.; Somlo, G.; Wu, J.; Frankel, P.; Kai, M.; Liu, X.; Wu, S.V.; Nguyen, D.; Chan, N.; Hsieh, M.Y.; et al. Characterization of patient-derived tumor xenografts (PDXs) as models for estrogen receptor positive (ER+HER2− and ER+HER2+) breast cancers. J. Steroid Biochem. Mol. Biol. 2017, 170, 65–74. [Google Scholar] [CrossRef]
- Sabnis, G.J.; Goloubeva, O.G.; Kazi, A.A.; Shah, P.; Brodie, A.H. HDAC inhibitor entinostat restores responsiveness of letrozole-resistant MCF-7Ca xenografts to aromatase inhibitors through modulation of Her-2. Mol. Cancer Ther. 2013, 12, 2804–2816. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbons, P.L.; Bartley, A.N.; Connolly, J.L. Template for Reporting Results of Biomarker Testing of Specimens From Patients With Carcinoma of the Breast. In 2018 CAP Accreditation Program Checklist and the CAP/ASCO HER2 Guidelines 2018; Breast Biomarkers 1.2.0.1; College of American Pathologist (CAP): Northfied, MA, USA, 2018; pp. 1–15. [Google Scholar]
- Hennessy, B.T.; Lu, Y.; Gonzalez-Angulo, A.M.; Carey, M.S.; Myhre, S.; Ju, Z.; Davies, M.A.; Liu, W.; Coombes, K.; Meric-Bernstam, F.; et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin. Proteom. 2010, 6, 129–151. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- DeRose, Y.S.; Gligorich, K.M.; Wang, G.; Georgelas, A.; Bowman, P.; Courdy, S.J.; Welm, A.L.; Welm, B.E. Patient-derived models of human breast cancer: Protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr. Protoc. Pharmacol. 2013, 60, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Hosford, S.R.; Shee, K.; Wells, J.D.; Traphagen, N.A.; Fields, J.L.; Hampsch, R.A.; Kettenbach, A.N.; Demidenko, E.; Miller, T.W. Estrogen therapy induces an unfolded protein response to drive cell death in ER+ breast cancer. Mol. Oncol. 2019, 13, 1778–1794. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, I.V.; Su, Z.Z.; Chang, Y.; Kitada, S.; Reed, J.C.; Fisher, P.B. The cancer growth suppressing gene mda-7 induces apoptosis selectively in human melanoma cells. Oncogene 2002, 21, 708–718. [Google Scholar] [CrossRef]
- Sauane, M.; Gopalkrishnan, R.V.; Sarkar, D.; Su, Z.Z.; Lebedeva, I.V.; Dent, P.; Pestka, S.; Fisher, P.B. MDA-7/IL-24: Novel cancer growth suppressing and apoptosis inducing cytokine. Cytokine Growth Factor Rev. 2003, 14, 35–51. [Google Scholar] [CrossRef]
- Saito, Y.; Miyahara, R.; Gopalan, B.; Litvak, A.; Inoue, S.; Shanker, M.; Branch, C.D.; Mhashilkar, A.M.; Roth, J.A.; Chada, S.; et al. Selective induction of cell cycle arrest and apoptosis in human prostate cancer cells through adenoviral transfer of the melanoma differentiation-associated−7 (mda-7)/interleukin-24 (IL-24) gene. Cancer Gene Ther. 2005, 12, 238–247. [Google Scholar] [CrossRef][Green Version]
- Zheng, M.; Bocangel, D.; Doneske, B.; Mhashilkar, A.; Ramesh, R.; Hunt, K.K.; Ekmekcioglu, S.; Sutton, R.B.; Poindexter, N.; Grimm, E.A.; et al. Human interleukin 24 (MDA-7/IL-24) protein kills breast cancer cells via the IL-20 receptor and is antagonized by IL-10. Cancer Immunol. Immunother. 2007, 56, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhao, L.L.; Zhao, H.J.; Cui, J.W.; Li, W.; Wang, N.Y. Lentivirusmediated MDA7/IL24 expression inhibits the proliferation of hepatocellular carcinoma cells. Mol. Med. Rep. 2018, 17, 5764–5773. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: ASCO/CAP Guideline Update. Arch. Pathol. Lab. Med. 2020, 144, 545–563. [Google Scholar] [CrossRef] [PubMed]
- Khongthong, P.; Roseweir, A.K.; Edwards, J. The NF-KB pathway and endocrine therapy resistance in breast cancer. Endocr. -Relat. Cancer 2019, 26, R369–R380. [Google Scholar] [CrossRef]
- Smart, E.; Semina, S.E.; Frasor, J. Update on the Role of NFκB in Promoting Aggressive Phenotypes of Estrogen Receptor– Positive Breast Cancer. Endocrinology 2020, 161, bqaa152. [Google Scholar] [CrossRef]
- Clarke, R.; Leonessa, F.; Welch, J.; Skaar, T. Cellular and Molecular Pharmacology of Antiestrogen Action and Resistance. Pharmacol. Rev. 2001, 53, 25–71. [Google Scholar]
- Hayashi, S.; Niwa, T.; Yamaguchi, Y. Estrogen signaling pathway and its imaging in human breast cancer. Cancer Sci. 2009, 100, 1773–1778. [Google Scholar] [CrossRef]
- Wu, V.S.; Kanaya, N.; Lo, C.; Mortimer, J.; Chen, S. From bench to bedside: What do we know about hormone receptor-positive and human epidermal growth factor receptor 2-positive breast cancer? J. Steroid Biochem. Mol. Biol. 2015, 153, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J.; Song, R.X.; Masamura, S.; Yue, W.; Fan, P.; Sogon, T.; Hayashi, S.; Nakachi, K.; Eguchi, H. Adaptation to estradiol deprivation causes up-regulation of growth factor pathways and hypersensitivity to estradiol in breast cancer cells. Adv. Exp. Med. Biol. 2008, 630, 19–34. [Google Scholar] [CrossRef]
- Martin, L.A.; Farmer, I.; Johnston, S.R.; Ali, S.; Dowsett, M. Elevated ERK1/ERK2/estrogen receptor cross-talk enhances estrogen-mediated signaling during long-term estrogen deprivation. Endocr. Relat. Cancer 2005, 12, S75. [Google Scholar] [CrossRef]
- Sabnis, G.J.; Jelovac, D.; Long, B.; Brodie, A. The role of growth factor receptor pathways in human breast cancer cells adapted to long-term estrogen deprivation. Cancer Res. 2005, 65, 3903–3910. [Google Scholar] [CrossRef]
- Masri, S.; Phung, S.; Wang, X.; Wu, X.; Yuan, Y.C.; Wagman, L.; Chen, S. Genome-wide analysis of aromatase inhibitor-resistant, tamoxifen-resistant, and long-term estrogen-deprived cells reveals a role for estrogen receptor. Cancer Res. 2008, 68, 4910–4918. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, S. TXNIP Links Anticipatory Unfolded Protein Response to Estrogen Reprogramming Glucose Metabolism in Breast Cancer Cells. Endocrinology 2021, 163, bqab212. [Google Scholar] [CrossRef] [PubMed]
- Escher, T.E.; Dandawate, P.; Sayed, A.; Hagan, C.R.; Anant, S.; Lewis-Wambi, J. Enhanced IFNalpha Signaling Promotes Ligand-Independent Activation of ERalpha to Promote Aromatase Inhibitor Resistance in Breast Cancer. Cancers 2021, 13, 5130. [Google Scholar] [CrossRef]
- Hanamura, T.; Hayashi, S.I. Overcoming aromatase inhibitor resistance in breast cancer: Possible mechanisms and clinical applications. Breast Cancer 2018, 25, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S.; Meeke, K.; Osipo, C.; Ross, E.A.; Kidawi, N.; Li, T.; Bell, E.; Chandel, N.S.; Jordan, V.C. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J. Natl. Cancer Inst. 2005, 97, 1746–1759. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Cunliffe, H.E.; Lewis-Wambi, J.S.; Slifker, M.J.; Willis, A.L.; Ramos, P.; Tapia, C.; Kim, H.R.; Yerrum, S.; Sharma, C.G.; et al. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc. Natl. Acad. Sci. USA 2011, 108, 18879. [Google Scholar] [CrossRef]
- Obiorah, I.E.; Fan, P.; Jordan, V.C. Breast cancer cell apoptosis with phytoestrogens is dependent on an estrogen-deprived state. Cancer Prev. Res. 2014, 7, 939–949. [Google Scholar] [CrossRef]
- Fan, P.; Tyagi, A.K.; Agboke, F.A.; Mathur, R.; Pokharel, N.; Jordan, V.C. Modulation of nuclear factor-kappa B activation by the endoplasmic reticulum stress sensor PERK to mediate estrogen-induced apoptosis in breast cancer cells. Cell Death Discov. 2018, 4, 1–14. [Google Scholar] [CrossRef]
- Song, R.X.-D.; Mor, G.; Naftolin, F.; McPherson, R.A.; Song, J.; Zhang, Z.; Yue, W.; Wang, J.; Santen, R.J. Effect of Long-Term Estrogen Deprivation on Apoptotic Responses of Breast Cancer Cells to 17β-Estradiol. J. Natl. Cancer Inst. 2001, 93, 1714–1723. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Raj, G.V.; Sareddy, G.R.; Ma, S.; Lee, T.K.; Viswanadhapalli, S.; Li, R.; Liu, X.; Murakami, S.; Chen, C.C.; Lee, W.R.; et al. Estrogen receptor coregulator binding modulators (ERXs) effectively target estrogen receptor positive human breast cancers. Elife 2017, 6, e26857. [Google Scholar] [CrossRef]
- De Angelis, C.; Fu, X.; Cataldo, M.L.; Nardone, A.; Pereira, R.; Veeraraghavan, J.; Nanda, S.; Qin, L.; Sethunath, V.; Wang, T.; et al. Activation of the IFN Signaling Pathway is Associated with Resistance to CDK4/6 Inhibitors and Immune Checkpoint Activation in ER-Positive Breast Cancer. Clin. Cancer Res. 2021, 27, 4870–4882. [Google Scholar] [CrossRef]
- Stossi, F.; Barnett, D.H.; Frasor, J.; Komm, B.; Lyttle, C.R.; Katzenellenbogen, B.S. Transcriptional profiling of estrogen-regulated gene expression via estrogen receptor (ER) alpha or ERbeta in human osteosarcoma cells: Distinct and common target genes for these receptors. Endocrinology 2004, 145, 3473–3486. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.P.; Mattapallil, M.J.; Raychaudhuri, K.; Bing, S.J.; Wu, S.; Zhong, Y.; Wang, W.; Chen, Z.; Silver, P.B.; Jittayasothorn, Y.; et al. The Cytokine IL-17A Limits Th17 Pathogenicity via a Negative Feedback Loop Driven by Autocrine Induction of IL-24. Immunity 2020, 53, 384–397. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ER+ BC Model | Upregulated in E2 | p-Value 1 | Downregulated in E2 | p-Value 1 |
|---|---|---|---|---|
| SC31 | E2F_TARGETS | 5.94 × 10−84 | INTERFERON_GAMMA_RESPONSE | 5.43 × 10−64 |
| MYC_TARGETS_V1 | 2.88 × 10−76 | INTERFERON_ALPHA_RESPONSE | 6.33 × 10−64 | |
| G2M_CHECKPOINT | 8.99 × 10−72 | TNFA_SIGNALING_VIA_NFKB | 1.70 × 10−33 | |
| ESTROGEN_RESPONSE_EARLY | 5.43 × 10−52 | APOPTOSIS | 6.64 × 10−25 | |
| ESTROGEN_RESPONSE_LATE | 5.43 × 10−52 | P53_PATHWAY | 2.15 × 10−18 | |
| GS3 | ESTROGEN_RESPONSE_LATE | 1.63 × 10−17 | TNFA_SIGNALING_VIA_NFKB | 6.06 × 10−21 |
| ESTROGEN_RESPONSE_EARLY | 4.55 × 10−16 | G2M_CHECKPOINT | 3.95 × 10−13 | |
| P53_PATHWAY | 3.95 × 10−13 | |||
| APOPTOSIS | 5.90 × 10−12 | |||
| E2F_TARGETS | 6.23 × 10−12 |
| Upregulated in ESR1+ Cells | p-Value 1 | Upregulated in ESR1− Cells | p-Value 1 | |
|---|---|---|---|---|
| SC31 | ||||
| Placebo | G2M_CHECKPOINT | 1.99 × 10−08 | HYPOXIA | 2.20 × 10−09 |
| E2F_TARGETS | 8.22 × 10−07 | GLYCOLYSIS | 1.56 × 10−03 | |
| TNFA_SIGNALING_VIA_NFKB | 7.00 × 10−04 | MTORC1_SIGNALING | 1.56 × 10−03 | |
| E2 | ESTROGEN_RESPONSE_EARLY | 1.03 × 10−30 | HYPOXIA | 3.64 × 10−13 |
| ESTROGEN_RESPONSE_LATE | 3.42 × 10−21 | GLYCOLYSIS | 1.08 × 10−09 | |
| G2M_CHECKPOINT | 9.81 × 10−07 | MTORC1_SIGNALING | 4.61 × 10−08 | |
| GS3 | ||||
| Placebo | TNFA_SIGNALING_VIA_NFKB | 4.50 × 10−34 | MYC_TARGETS_V1 | 1.44 × 10−34 |
| APOPTOSIS | 1.45 × 10−12 | E2F_TARGETS | 1.20 × 10−29 | |
| ESTROGEN_RESPONSE_EARLY | 1.54 × 10−11 | G2M_CHECKPOINT | 7.91 × 10−15 | |
| E2 | TNFA_SIGNALING_VIA_NFKB | 7.22 × 10−11 | MYC_TARGETS_V1 | 4.89 × 10−12 |
| UV_RESPONSE_UP | 1.80 × 10−04 | E2F_TARGETS | 4.17 × 10−08 | |
| G2M_CHECKPOINT | 2.68 × 10−06 |
| Upregulated in E2 | p-Value 1 | Downregulated in E2 | p-Value 1 | |
|---|---|---|---|---|
| SC31 | ||||
| ESR1+ cells | E2F_TARGETS | 2.91 × 10−83 | INTERFERON_GAMMA_RESPONSE | 6.11 × 10−64 |
| G2M_CHECKPOINT | 1.23 × 10−72 | INTERFERON_ALPHA_RESPONSE | 6.81 × 10−63 | |
| MYC_TARGETS_V1 | 1.10 × 10−69 | TNFA_SIGNALING_VIA_NFKB | 1.06 × 10−34 | |
| ESTROGEN_RESPONSE_EARLY | 6.52 × 10−57 | APOPTOSIS | 2.82 × 10−23 | |
| ESTROGEN_RESPONSE_LATE | 1.67 × 10−51 | P53_PATHWAY | 3.16 × 10−21 | |
| ESR1– cells | E2F_TARGETS | 1.94 × 10−62 | INTERFERON_ALPHA_RESPONSE | 4.80 × 10−66 |
| MYC_TARGETS_V1 | 1.94 × 10−62 | INTERFERON_GAMMA_RESPONSE | 7.63 × 10−66 | |
| G2M_CHECKPOINT | 1.44 × 10−48 | TNFA_SIGNALING_VIA_NFKB | 7.61 × 10−34 | |
| ESTROGEN_RESPONSE_EARLY | 2.49 × 10−43 | APOPTOSIS | 3.63 × 10−25 | |
| ESTROGEN_RESPONSE_LATE | 4.75 × 10−42 | HYPOXIA | 3.83 × 10−16 | |
| GS3 | ||||
| ESR1+ cells | ESTROGEN_RESPONSE_EARLY | 1.11 × 10−19 | TNFA_SIGNALING_VIA_NFKB | 2.20 × 10−22 |
| ESTROGEN_RESPONSE_LATE | 3.20 × 10−18 | P53_PATHWAY | 3.69 × 10−13 | |
| APOPTOSIS | 5.57 × 10−12 | |||
| ESR1– cells | ESTROGEN_RESPONSE_LATE | 5.61 × 10−17 | G2M_CHECKPOINT | 3.05 × 10−31 |
| ESTROGEN_RESPONSE_EARLY | 1.44 × 10−15 | E2F_TARGETS | 8.25 × 10−23 | |
| TNFA_SIGNALING_VIA_NFKB | 5.48 × 10−14 |
| Upregulated in IL24+ Cells | p-Value 1 | Downregulated in IL24+ Cells | p-Value 1 |
|---|---|---|---|
| TNFA_SIGNALING_VIA_NFKB | 1.33 × 10−17 | G2M_CHECKPOINT | 8.25 × 10−9 |
| ESTROGEN_RESPONSE_ EARLY | 1.73 × 10−9 | E2F_TARGETS | 8.25 × 10−9 |
| ESTROGEN_RESPONSE_ LATE | 1.73 × 10−9 | MYC_TARGETS_V1 | 5.24 × 10−5 |
| APOPTOSIS | 2.45 × 10−6 | P53_PATHWAY | 2.46 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, H.; Saeki, K.; Chang, G.; Wang, J.; Wu, X.; Hsu, P.-Y.; Kanaya, N.; Wang, X.; Somlo, G.; Nakamura, M.; et al. Influence of Estrogen Treatment on ESR1+ and ESR1− Cells in ER+ Breast Cancer: Insights from Single-Cell Analysis of Patient-Derived Xenograft Models. Cancers 2021, 13, 6375. https://doi.org/10.3390/cancers13246375
Mori H, Saeki K, Chang G, Wang J, Wu X, Hsu P-Y, Kanaya N, Wang X, Somlo G, Nakamura M, et al. Influence of Estrogen Treatment on ESR1+ and ESR1− Cells in ER+ Breast Cancer: Insights from Single-Cell Analysis of Patient-Derived Xenograft Models. Cancers. 2021; 13(24):6375. https://doi.org/10.3390/cancers13246375
Chicago/Turabian StyleMori, Hitomi, Kohei Saeki, Gregory Chang, Jinhui Wang, Xiwei Wu, Pei-Yin Hsu, Noriko Kanaya, Xiaoqiang Wang, George Somlo, Masafumi Nakamura, and et al. 2021. "Influence of Estrogen Treatment on ESR1+ and ESR1− Cells in ER+ Breast Cancer: Insights from Single-Cell Analysis of Patient-Derived Xenograft Models" Cancers 13, no. 24: 6375. https://doi.org/10.3390/cancers13246375
APA StyleMori, H., Saeki, K., Chang, G., Wang, J., Wu, X., Hsu, P.-Y., Kanaya, N., Wang, X., Somlo, G., Nakamura, M., Bild, A., & Chen, S. (2021). Influence of Estrogen Treatment on ESR1+ and ESR1− Cells in ER+ Breast Cancer: Insights from Single-Cell Analysis of Patient-Derived Xenograft Models. Cancers, 13(24), 6375. https://doi.org/10.3390/cancers13246375