
HDAC6 Inhibition Extinguishes Autophagy in Cancer: Recent Insights

 , ,
, ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Autophagy and Cancer
2.1. Autophagy and Tumorigenesis
2.2. Autophagy and Cancer Treatment Resistance
2.3. Therapeutic Agents Targeting Autophagy
3. HDACi Trigger the Autophagy Process
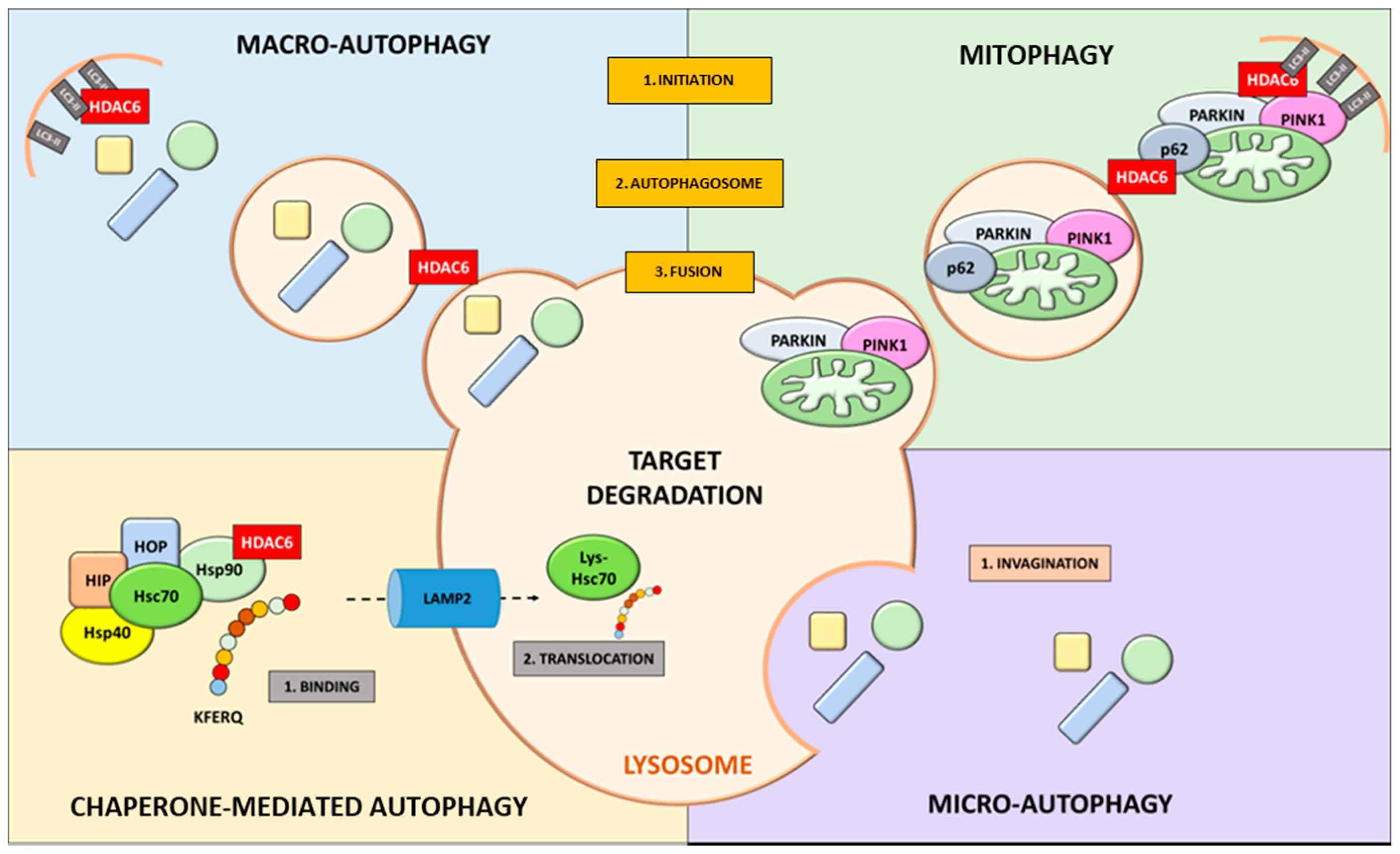
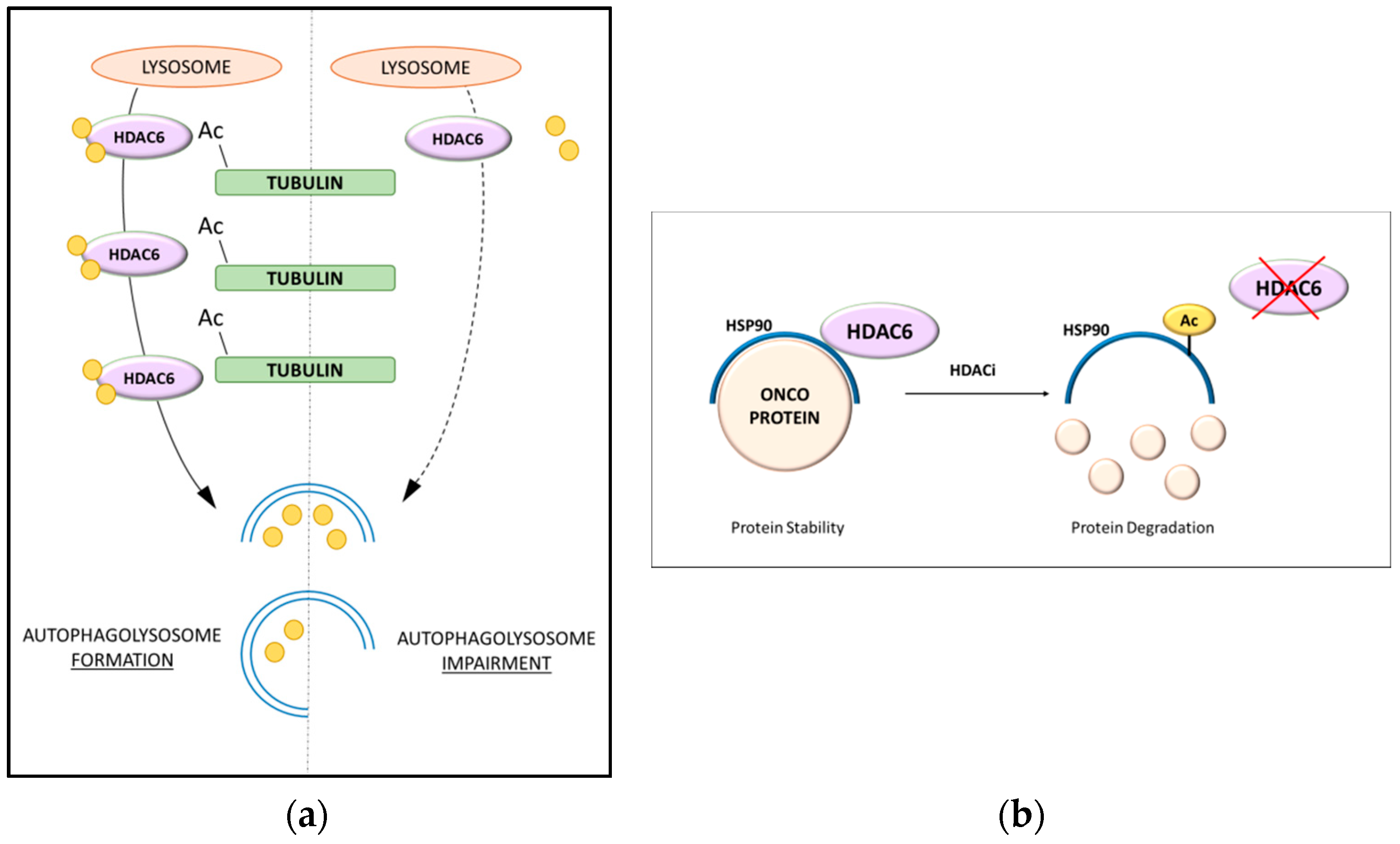
4. Role of HDAC6 in Autophagic Processes
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condello, M.; Pellegrini, E.; Caraglia, M.; Meschini, S. Targeting Autophagy to Overcome Human Diseases. Int. J. Mol. Sci. 2019, 20, 725. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; White, E. Autophagy, Metabolism, and Cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 73–78. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Raj, S.; Chandel, V.; Kumar, A.; Kesari, K.K.; Asthana, S.; Ruokolainen, J.; Kamal, M.A.; Kumar, D. Molecular mechanisms of interplay between autophagy and metabolism in cancer. Life Sci. 2020, 259, 118184. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Arias, E.; Cuervo, A.M. Chaperone-mediated autophagy in protein quality control. Curr. Opin. Cell Biol. 2011, 23, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.S. Autophagy and cancer. Exp. Mol. Med. 2012, 44, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Meythaler, J.G.; Garcia-Mayea, Y.; Mir, C.; Kondoh, H.; ME, L.L. Autophagy Takes Center Stage as a Possible Cancer Hallmark. Front. Oncol. 2020, 10, 586069. [Google Scholar] [CrossRef]
- Djajadikerta, A.; Keshri, S.; Pavel, M.; Prestil, R.; Ryan, L.; Rubinsztein, D.C. Autophagy Induction as a Therapeutic Strategy for Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2799–2821. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Lin, S.R.; Fu, Y.S.; Tsai, M.J.; Cheng, H.; Weng, C.F. Natural Compounds from Herbs that can Potentially Execute as Autophagy Inducers for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1412. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Rucker, E.B., 3rd; Zhou, B.P. Autophagy regulation in the development and treatment of breast cancer. Acta Biochim Biophys. Sin. (Shanghai) 2016, 48, 60–74. [Google Scholar] [CrossRef] [Green Version]
- Colhado Rodrigues, B.L.; Lallo, M.A.; Perez, E.C. The Controversial Role of Autophagy in Tumor Development: A Systematic Review. Immunol. Investig. 2020, 49, 386–396. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.; Qin, H.; Zhang, K.; Li, B.; Zhang, X. Targeting autophagy in chemotherapy-resistant of hepatocellular carcinoma. Am. J. Cancer Res. 2018, 8, 354–365. [Google Scholar] [PubMed]
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: Further developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef]
- Antunes, F.; Erustes, A.G.; Costa, A.J.; Nascimento, A.C.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. Autophagy and intermittent fasting: The connection for cancer therapy? Clinics 2018, 73, e814s. [Google Scholar] [CrossRef]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging Mechanisms in Initiating and Terminating Autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Sharma, V.; Verma, S.; Seranova, E.; Sarkar, S.; Kumar, D. Selective Autophagy and Xenophagy in Infection and Disease. Front. Cell Dev. Biol. 2018, 6, 147. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Perez-Hernandez, M.; Arias, A.; Martinez-Garcia, D.; Perez-Tomas, R.; Quesada, R.; Soto-Cerrato, V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manic, G.; Obrist, F.; Kroemer, G.; Vitale, I.; Galluzzi, L. Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell Oncol. 2014, 1, e29911. [Google Scholar] [CrossRef]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H.; Kim, K.I. Epigenetic Control of Autophagy: Nuclear Events Gain More Attention. Mol. Cell 2017, 65, 781–785. [Google Scholar] [CrossRef] [Green Version]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [Green Version]
- Cosenza, M.; Pozzi, S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. Int. J. Mol. Sci. 2018, 19, 2337. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Rikiishi, H. Autophagic and apoptotic effects of HDAC inhibitors on cancer cells. J. Biomed. Biotechnol. 2011, 2011, 830260. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Rosato, R.R.; Almenara, J.A.; Dai, Y.; Grant, S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol. Cancer Ther. 2003, 2, 1273–1284. [Google Scholar]
- Carew, J.S.; Nawrocki, S.T.; Cleveland, J.L. Modulating autophagy for therapeutic benefit. Autophagy 2007, 3, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Mahapatra, K.K.; Patra, S.; Mishra, S.R.; Behera, B.P.; Bhutia, S.K. Epigenetic modifications of autophagy in cancer and cancer therapeutics. Semin. Cancer Biol. 2020, 66, 22–33. [Google Scholar] [CrossRef]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammoh, N.; Marks, P.A.; Jiang, X. Curbing autophagy and histone deacetylases to kill cancer cells. Autophagy 2012, 8, 1521–1522. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef] [Green Version]
- Mrakovcic, M.; Frohlich, L.F. Molecular Determinants of Cancer Therapy Resistance to HDAC Inhibitor-Induced Autophagy. Cancers 2019, 12, 109. [Google Scholar] [CrossRef] [Green Version]
- Mrakovcic, M.; Kleinheinz, J.; Frohlich, L.F. Histone Deacetylase Inhibitor-Induced Autophagy in Tumor Cells: Implications for p53. Int. J. Mol. Sci. 2017, 18, 1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, S.B. A matter of balance between life and death: Targeting reactive oxygen species (ROS)-induced autophagy for cancer therapy. Autophagy 2010, 6, 835–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci. 2007, 120, 4155–4166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrakovcic, M.; Bohner, L.; Hanisch, M.; Frohlich, L.F. Epigenetic Targeting of Autophagy via HDAC Inhibition in Tumor Cells: Role of p53. Int. J. Mol. Sci. 2018, 19, 3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, K.F.; Yeung, P.L.; Chiang, A.K. Induction of MAPK- and ROS-dependent autophagy and apoptosis in gastric carcinoma by combination of romidepsin and bortezomib. Oncotarget 2016, 7, 4454–4467. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Lei, Y.; An, G.L.; He, L. Down-regulation of deacetylase HDAC6 inhibits the melanoma cell line A375.S2 growth through ROS-dependent mitochondrial pathway. PLoS ONE 2015, 10, e0121247. [Google Scholar] [CrossRef] [Green Version]
- Banreti, A.; Sass, M.; Graba, Y. The emerging role of acetylation in the regulation of autophagy. Autophagy 2013, 9, 819–829. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Bhat, Z.R.; Jena, G. Role of autophagy and histone deacetylases in diabetic nephropathy: Current status and future perspectives. Genes Dis. 2016, 3, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Finkel, T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 2009, 284, 6322–6328. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.Y.; Li, T.Y.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.M.; Lian, G.; Liu, Q.; Ruan, K.; et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012, 336, 477–481. [Google Scholar] [CrossRef]
- Stankov, M.V.; El Khatib, M.; Kumar Thakur, B.; Heitmann, K.; Panayotova-Dimitrova, D.; Schoening, J.; Bourquin, J.P.; Schweitzer, N.; Leverkus, M.; Welte, K.; et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 2014, 28, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Talebian, S.; Daghagh, H.; Yousefi, B.; Ozkul, Y.; Ilkhani, K.; Seif, F.; Alivand, M.R. The role of epigenetics and non-coding RNAs in autophagy: A new perspective for thorough understanding. Mech. Ageing Dev. 2020, 190, 111309. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Wang, H.; Zhang, L.; Pei, F.; Chen, Z. HDAC6 Regulates the Fusion of Autophagosome and Lysosome to Involve in Odontoblast Differentiation. Front. Cell Dev. Biol. 2020, 8, 605609. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Kekatpure, V.D.; Dannenberg, A.J.; Subbaramaiah, K. Withdrawal: HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J. Biol. Chem. 2020, 295, 297. [Google Scholar] [CrossRef] [Green Version]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12 (Suppl. 2), 1542–1552. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. The molecular machinery of autophagy: Unanswered questions. J. Cell Sci. 2005, 118, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. MicroBiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy—unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hook, S.S.; Orian, A.; Cowley, S.M.; Eisenman, R.N. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 13425–13430. [Google Scholar] [CrossRef] [Green Version]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.S.; Hubbert, C.C.; Lu, J.; Lee, Y.S.; Lee, J.Y.; Yao, T.P. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol. Cell Biol. 2007, 27, 8637–8647. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE 2013, 8, e76016. [Google Scholar] [CrossRef] [Green Version]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Mun, S.; Harada, A.; Ohkawa, Y.; Inagaki, A.; Sano, S.; Takahashi, K.; Izumi, Y.; Osada-Oka, M.; Wanibuchi, H.; et al. Hsc70 contributes to cancer cell survival by preventing Rab1A degradation under stress conditions. PLoS ONE 2014, 9, e96785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, S.; Liu, N.; Zhang, Y.; Liu, M.; Li, D.; Seto, E.; Yao, T.P.; Shui, W.; Zhou, J. Proteomic identification and functional characterization of MYH9, Hsc70, and DNAJA1 as novel substrates of HDAC6 deacetylase activity. Protein Cell 2015, 6, 42–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef]
- Li, Z.Y.; Li, Q.Z.; Chen, L.; Chen, B.D.; Zhang, C.; Wang, X.; Li, W.P. HPOB, an HDAC6 inhibitor, attenuates corticosterone-induced injury in rat adrenal pheochromocytoma PC12 cells by inhibiting mitochondrial GR translocation and the intrinsic apoptosis pathway. Neurochem. Int. 2016, 99, 239–251. [Google Scholar] [CrossRef]
- Su, M.; Guan, H.; Zhang, F.; Gao, Y.; Teng, X.; Yang, W. HDAC6 Regulates the Chaperone-Mediated Autophagy to Prevent Oxidative Damage in Injured Neurons after Experimental Spinal Cord Injury. Oxid. Med. Cell Longev. 2016, 2016, 7263736. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Bloemberg, D.; Quadrilatero, J. Autophagy, apoptosis, and mitochondria: Molecular integration and physiological relevance in skeletal muscle. Am. J. Physiol. Cell Physiol. 2019, 317, C111–C130. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Bertos, N.R.; Gilquin, B.; Chan, G.K.; Yen, T.J.; Khochbin, S.; Yang, X.J. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J. Biol. Chem. 2004, 279, 48246–48254. [Google Scholar] [CrossRef] [Green Version]
- Osko, J.D.; Christianson, D.W. Structural Basis of Catalysis and Inhibition of HDAC6 CD1, the Enigmatic Catalytic Domain of Histone Deacetylase 6. Biochemistry 2019, 58, 4912–4924. [Google Scholar] [CrossRef]
- Yan, J. Interplay between HDAC6 and its interacting partners: Essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol. 2014, 33, 567–580. [Google Scholar] [CrossRef]
- Li, Z.Y.; Zhang, C.; Zhang, Y.; Chen, L.; Chen, B.D.; Li, Q.Z.; Zhang, X.J.; Li, W.P. A novel HDAC6 inhibitor Tubastatin A: Controls HDAC6-p97/VCP-mediated ubiquitination-autophagy turnover and reverses Temozolomide-induced ER stress-tolerance in GBM cells. Cancer Lett. 2017, 391, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.; Le, J.; Sausville, E.; Emadi, A. Autophagy modulation: A target for cancer treatment development. Cancer Chemother. Pharmacol. 2015, 75, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, U.K.; Chaudhary, A. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med. Chem. 2015, 7, 1535–1542. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.Y.; Hofman, F.M.; Louie, S.G.; Schonthal, A.H.; Chen, T.C. Quinoline-based antimalarial drugs: A novel class of autophagy inhibitors. Neurosurg. Focus 2015, 38, E12. [Google Scholar] [CrossRef]
- Li, Y.; McGreal, S.; Zhao, J.; Huang, R.; Zhou, Y.; Zhong, H.; Xia, M.; Ding, W.X. A cell-based quantitative high-throughput image screening identified novel autophagy modulators. Pharmacol. Res. 2016, 110, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, J.; Li, K.; Deng, L.; Wang, H. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front. Pharmacol. 2020, 11, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poklepovic, A.; Gewirtz, D.A. Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy 2014, 10, 1478–1480. [Google Scholar] [CrossRef] [Green Version]
- Kaliszczak, M.; Trousil, S.; Aberg, O.; Perumal, M.; Nguyen, Q.D.; Aboagye, E.O. A novel small molecule hydroxamate preferentially inhibits HDAC6 activity and tumour growth. Br. J. Cancer 2013, 108, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Kaliszczak, M.; van Hechanova, E.; Li, Y.; Alsadah, H.; Parzych, K.; Auner, H.W.; Aboagye, E.O. The HDAC6 inhibitor C1A modulates autophagy substrates in diverse cancer cells and induces cell death. Br. J. Cancer 2018, 119, 1278–1287. [Google Scholar] [CrossRef]
- Liu, J.R.; Yu, C.W.; Hung, P.Y.; Hsin, L.W.; Chern, J.W. High-selective HDAC6 inhibitor promotes HDAC6 degradation following autophagy modulation and enhanced antitumor immunity in glioblastoma. Biochem. Pharmacol. 2019, 163, 458–471. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Beer, P.A.; Cartwright, C.A.; Haymaker, C.; Vo, H.H.; Kiany, S.; Cecil, A.R.L.; Dow, J.; Haque, K.; Silva, F.A.; et al. Preclinical Development and First-in-Human Study of KA2507, a Selective and Potent Inhibitor of Histone Deacetylase 6, for Patients with Refractory Solid Tumors. Clin. Cancer Res. 2021, 27, 3584–3594. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Lv, W.; Wang, L.; Xu, J.; Yuan, P.; Huang, S.; He, Z.; Hu, J. Ricolinostat (ACY-1215) suppresses proliferation and promotes apoptosis in esophageal squamous cell carcinoma via miR-30d/PI3K/AKT/mTOR and ERK pathways. Cell Death Dis. 2018, 9, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amengual, J.E.; Lue, J.K.; Ma, H.; Lichtenstein, R.; Shah, B.; Cremers, S.; Jones, S.; Sawas, A. First-in-Class Selective HDAC6 Inhibitor (ACY-1215) Has a Highly Favorable Safety Profile in Patients with Relapsed and Refractory Lymphoma. Oncologist 2021, 26, 184-e366. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Compound | Target | Source | Chemical Class | Isoform Selectivity | Study Phase - Clinical trial |
|---|---|---|---|---|---|
| Vorinostat (SAHA) Belinostat (PXD-101) Panobinostat (LBH-589) Trichostatin A (TSA) Quisinostat (JNJ-16241199) WW437 Dacinostat (LAQ824) | Pan-HDAC | Synthetic Synthetic Synthetic Natural Synthetic Synthetic Synthetic | Hydroxamic acid | Class I, II and IV Class I and II Class I, II and IV Class I and II Class I and II HDAC 2 and 4 Class I and II | FDA approval for Cutaneous T-Cell Lymphoma FDA approval for Peripheral T Cell lymphoma (PTCL) FDA approval (PTCL and multiple myelomas) Toxic–Phase I Phase II Cutaneous T-Cell Lymphoma Pre-clinical Phase I |
| Pivaloyloxmethylbutyrate (AN-9) Sodium Butyrate (NaB) Sodium Phenylbutyrate (4-PB) Valproate (valproic acid) | Pan-HDAC | Synthetic Natural Synthetic Synthetic | Short chain fatty acids | Class I and IIa Class I and IIa Class I and IIa Class I and IIa | Phase II Melanoma and Phase I Chronic lymphocytic leukemia (CLL) Phase I Colorectal cancer FDA approval (urea cycle disorders) Phase I (Brain and Central Nervous System Tumors) |
| Romidepsin (FK228) | Pan-HDAC | Natural | Cyclic tetrapeptides | Class I (HDAC1, 2, 4, 6) | Phase II |
| Entinostat (MS-275) Tacedinaline (CI-994) Mocetinostat (MG-0103) | Pan-HDAC | Synthetic Synthetic Synthetic | Benzamides | Class I Class I Class I and IV | Phase II (Hodgkin’s Lymphoma) Phase II (Myeloma) Phase I (Hodgkin’s Lymphoma) |
| Trapoxins (TPX) α-ketoamides Heterocyclic ketones | Pan-HDAC | Natural Synthetic Synthetic | Ketones | Class I NA NA | NA (Not approved) NA NA |
| Cambinol EX-527 Sirtinol Nicotinamide | Sirtuin inhibitors | Synthetic Synthetic Synthetic Synthetic | SIRT1 and 2 SIRT1 and 2 SIRT1 and 2 class III | Pre-clinical Pre-clinical Pre-clinical Phase III (laryngeal cancer) | |
| Azelaic Bishydroxamic Acid (ABHA) m-carboxycinnamic acid bis-hydroxamide (CBHA) Ricolinosta (ACY-1215) Tubacin | Selective HDAC | Synthetic Synthetic Synthetic Synthetic | Hydroxamate Derivatives Benzamides | HDAC3 HDAC3 HDAC6 HDAC6 | NA Pre-clinical Phase II Pre-clinical Acute lymphocytic leukemia (ALL) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passaro, E.; Papulino, C.; Chianese, U.; Toraldo, A.; Congi, R.; Del Gaudio, N.; Nicoletti, M.M.; Benedetti, R.; Altucci, L. HDAC6 Inhibition Extinguishes Autophagy in Cancer: Recent Insights. Cancers 2021, 13, 6280. https://doi.org/10.3390/cancers13246280
Passaro E, Papulino C, Chianese U, Toraldo A, Congi R, Del Gaudio N, Nicoletti MM, Benedetti R, Altucci L. HDAC6 Inhibition Extinguishes Autophagy in Cancer: Recent Insights. Cancers. 2021; 13(24):6280. https://doi.org/10.3390/cancers13246280
Chicago/Turabian StylePassaro, Eugenia, Chiara Papulino, Ugo Chianese, Antonella Toraldo, Raffaella Congi, Nunzio Del Gaudio, Maria Maddalena Nicoletti, Rosaria Benedetti, and Lucia Altucci. 2021. "HDAC6 Inhibition Extinguishes Autophagy in Cancer: Recent Insights" Cancers 13, no. 24: 6280. https://doi.org/10.3390/cancers13246280
APA StylePassaro, E., Papulino, C., Chianese, U., Toraldo, A., Congi, R., Del Gaudio, N., Nicoletti, M. M., Benedetti, R., & Altucci, L. (2021). HDAC6 Inhibition Extinguishes Autophagy in Cancer: Recent Insights. Cancers, 13(24), 6280. https://doi.org/10.3390/cancers13246280