
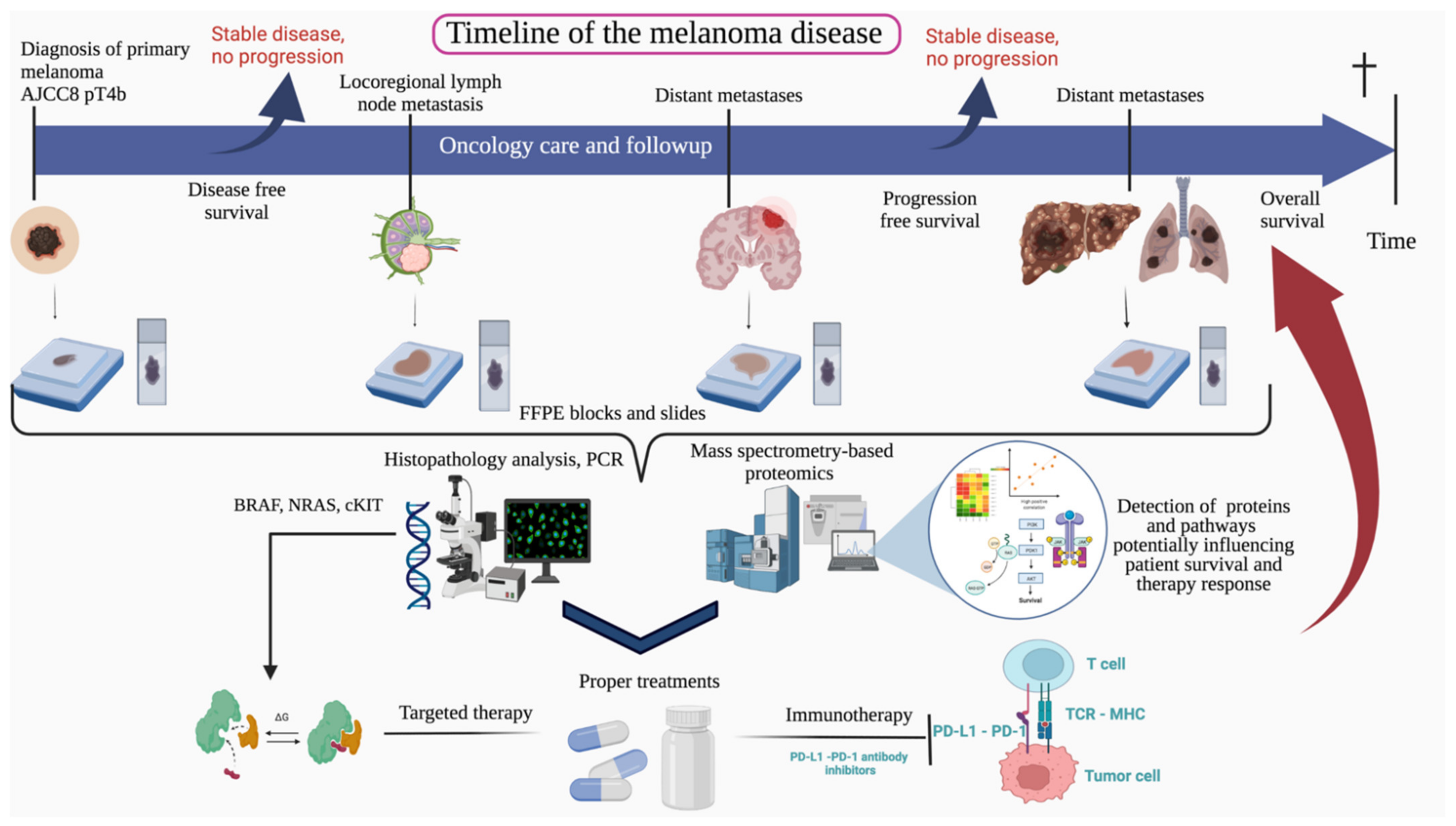
Deep Proteomic Analysis on Biobanked Paraffine-Archived Melanoma with Prognostic/Predictive Biomarker Read-Out

 , ,
, ,  ,
,  , , , ,
, , , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
Survival Analysis of Patient Cohort
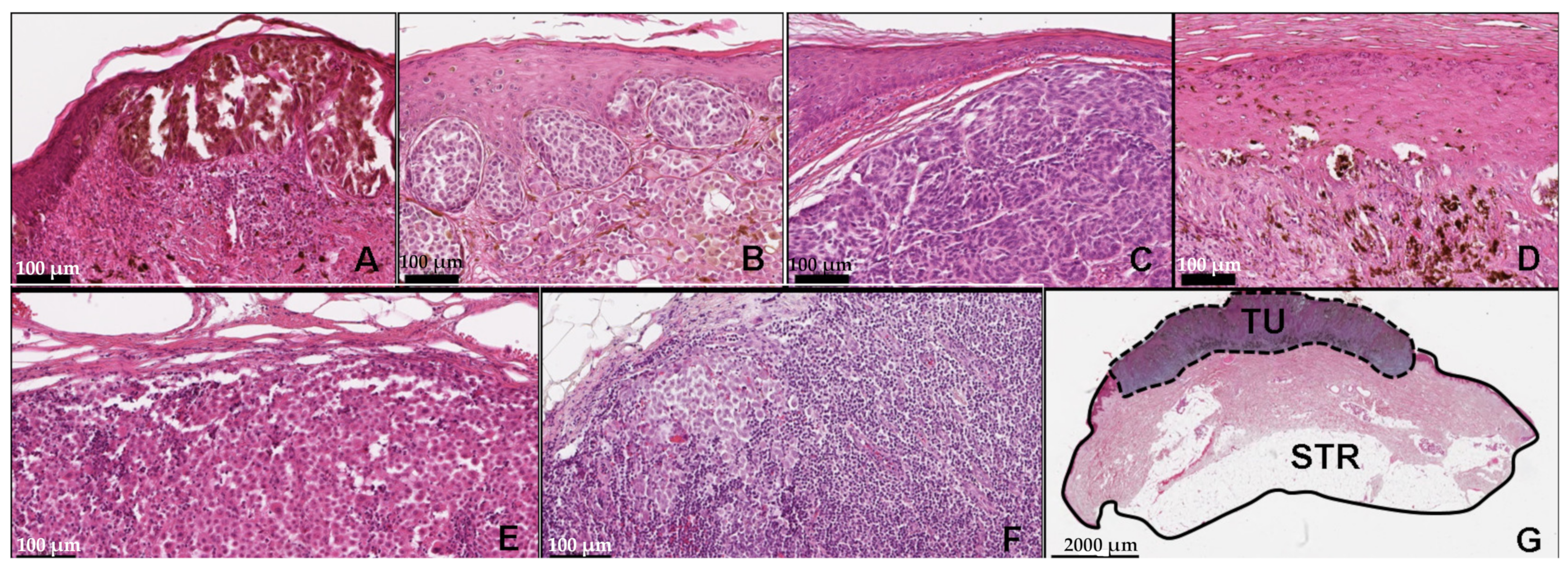
2.2. FFPE Sample Preparation and Histology Validation
FFPE Sample Processing
2.3. Protein Digestion
2.4. LC-MS/MS Analysis
2.5. Database Searching
2.6. Data Normalization and Batch Effect Correction
2.7. Proteomics Data Analysis
2.8. Data Availability
2.9. Figure Illustration
3. Results and Discussion
3.1. The Clinical Significance of the Study
Clinicopathologic Characteristics of the Patient Cohort
3.2. Proteomic Analysis Reveals Sample Clustering According to Relevant Histopathological and Clinical Parameters Associated with Survival and Tumor Progression
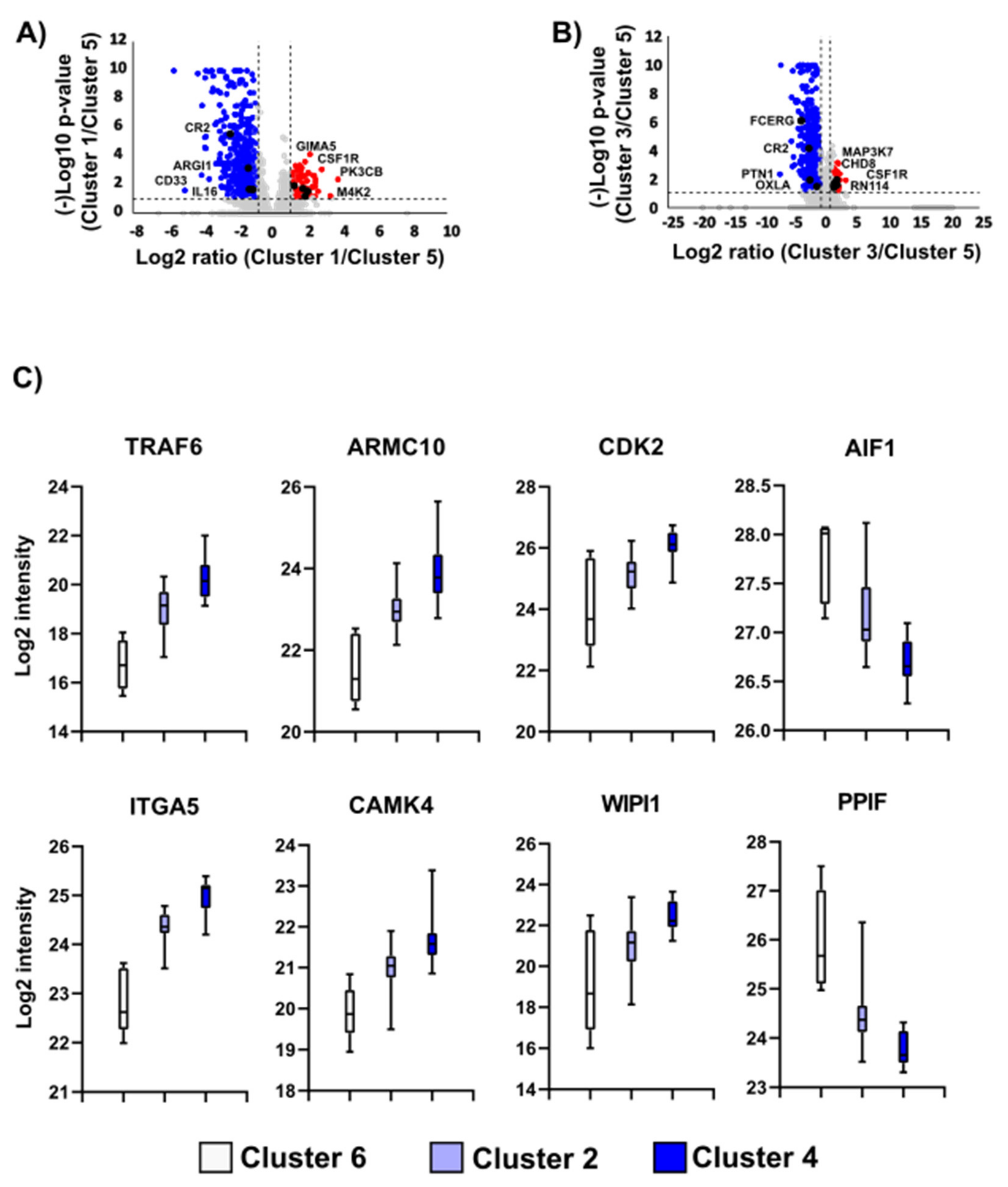
3.3. Imbalance of Immune-Related Proteins as Molecular Fingerprint of the Sample Clusters with Varying Survival Outcomes
3.4. Protein Translation and Immune System Stand out in the Proteome Profile of the Sample Clusters with Different Progression-Free Survival
3.5. Proteome Mapping Detects Gradual Proteomic Changes in Sample Clusters Exhibiting Different Disease-Free Survival
3.6. Proteomic Differences with Regard to Tumor Type and Clinical Stage
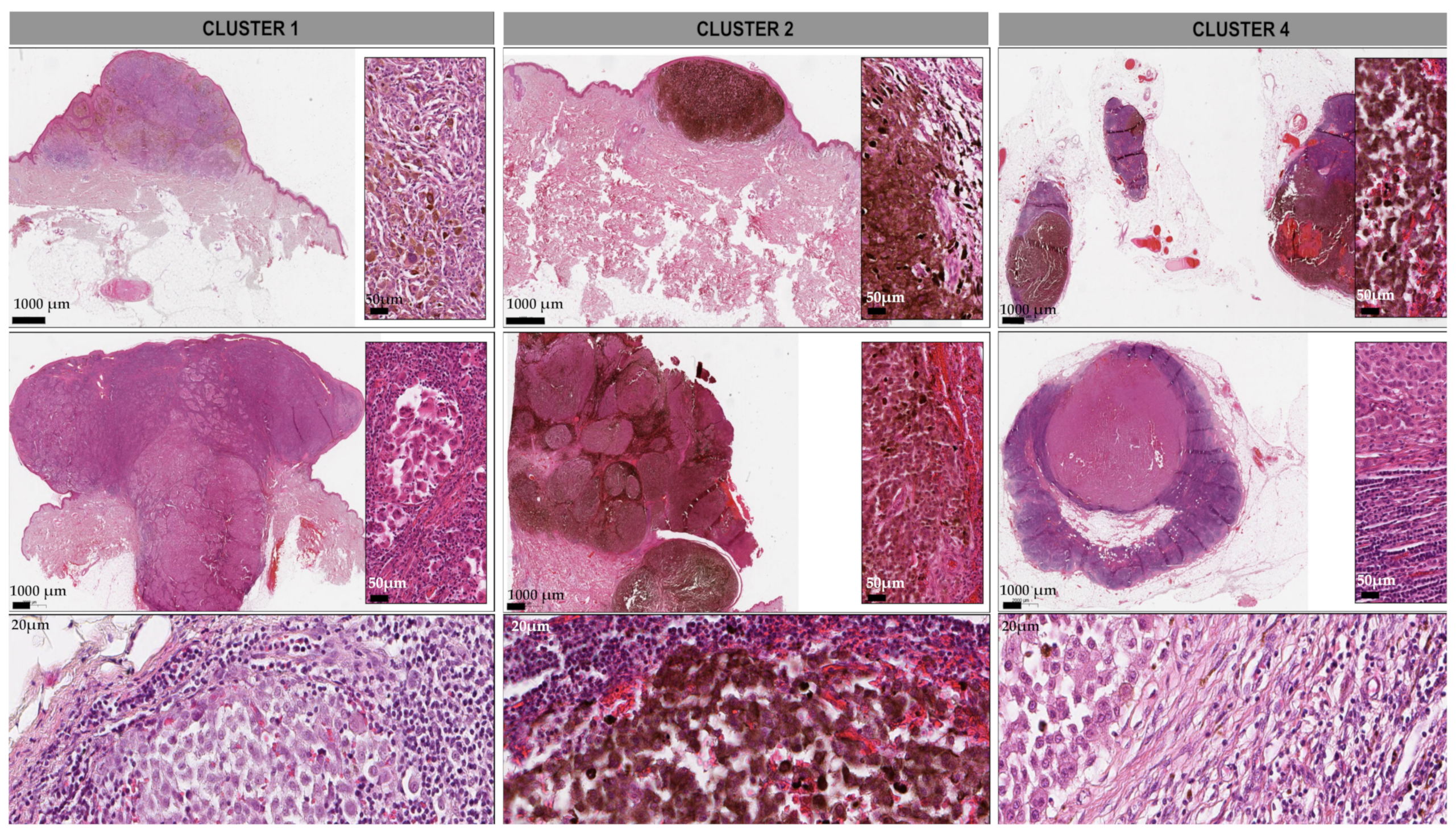
3.7. Histopathologic Relations to the Clustered Sample Cohort
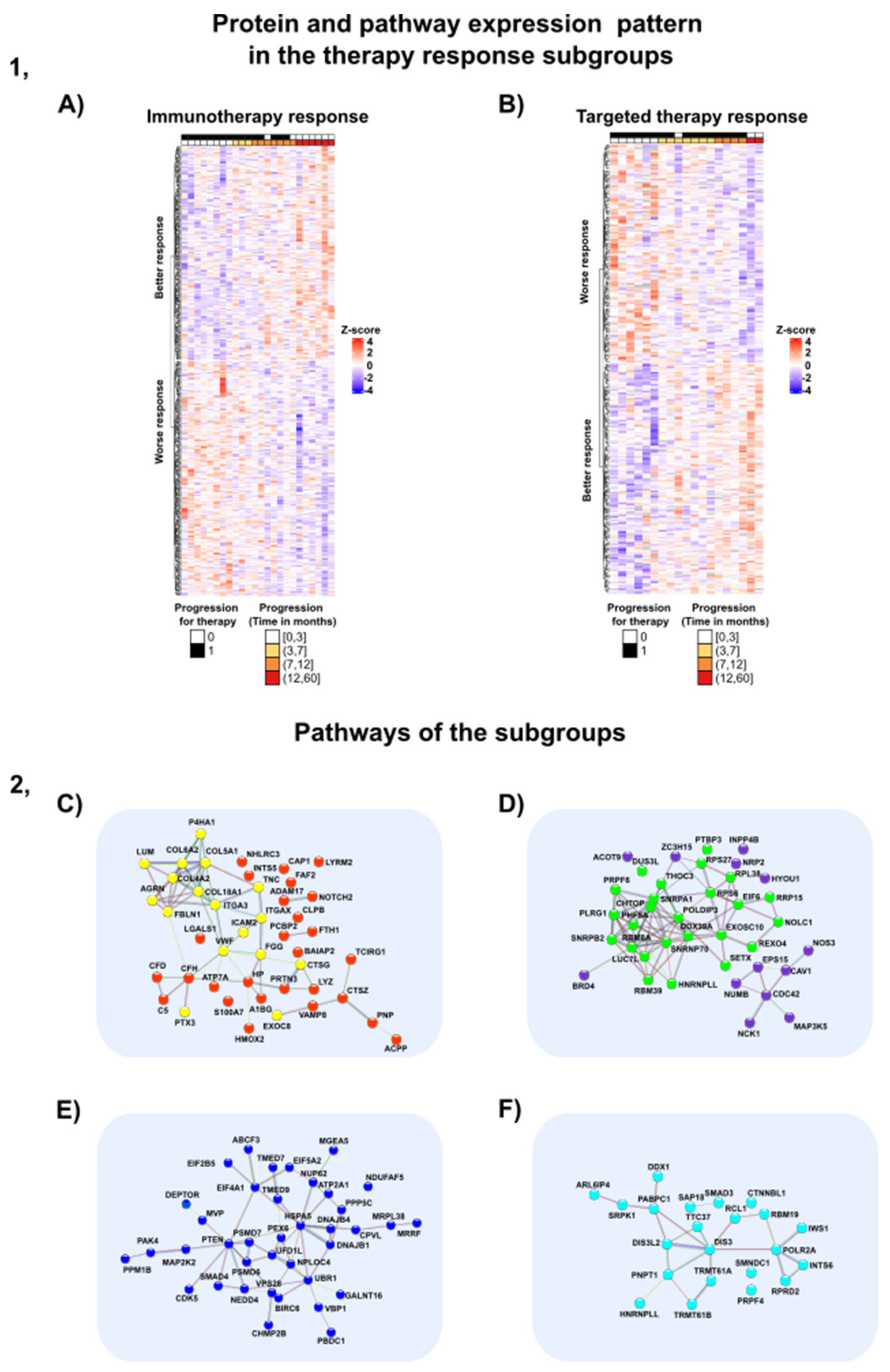
3.8. Proteins and Pathways Predicting Therapy Response
3.8.1. Proteins and Pathways Indicating Worse Outcome in the Immunotherapy-Treated Subgroup
3.8.2. Proteins and Pathways Indicating Better Outcome in the Immunotherapy-Treated Subgroup
3.8.3. Proteins and Pathways Altering the Patient Outcome in the Targeted Therapy Subgroup
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIF1 | apoptosis-inducing factor 1 |
| Akt | protein kinase B (collective name of the serine/threonine-specific protein kinases (Akt1,2,3) |
| ARGI1 | arginase-1 |
| ARGN | isoform 6 of Agrin |
| ARMC10 | armadillo repeat-containing protein 10 |
| BRAF | B-raf proto-oncogene, serine/threonine kinase |
| CAMK4 | calcium/calmodulin dependent protein kinase IV |
| CD33 | myeloid cell surface antigen CD33 (cluster of differentiation 33) |
| CD46 | cluster of differentiation 46 |
| CDK2 | cyclin-dependent kinase 2 |
| CHD8 | Chromodomain-helicase-DNA-binding protein 8 |
| CKIT | KIT proto-oncogene, receptor tyrosine kinase |
| c-Myc | MYC proto-oncogene, bHLH transcription factor |
| COL4A2 | collagen type IV alpha 2 chain |
| COL6A2 | collagen type VI alpha 2 chain |
| CR2 | complement C3d receptor 2 |
| CSF1R | Colony stimulating factor 1 receptor |
| CXCR2 | C-X-C motif chemokine receptor 2 |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| DEPTOR | DEP domain containing mTOR interacting protein |
| ERK | extracellular signal regulated kinases, classical MAP kinases |
| FCERG | fusion protein erythroblast transformation specific related gene |
| FDR | False Discovery Rate |
| GIMA5 | GTPase IMAP family member 5 |
| HCT-116 | human colorectal carcinoma cell line |
| HT-29 | human colon cancer cell line |
| ICAM2 | intercellular adhesion molecule 2 |
| IFNγ | interferon-gamma |
| IL-16 | interleukin-16 |
| IRE genes | iron-responsive element genes |
| ITGA5 | Integrin Subunit Alpha 5 |
| MAPK | mitogen-activated protein kinase pathway |
| MAP2K2 | mitogen-activated protein kinase kinase 2 |
| MAP3K7 | mitogen-activated protein kinase kinase kinase 7 |
| M4K2 | mitogen-activated protein kinase kinase kinase kinase 2 |
| MCM9 | minichromosome maintenance 9 homologous recombination repair factor |
| MDM2 | mouse double minute 2 homolog, E3 ubiquitin-protein ligase, proto-oncogene |
| MITF | melanocyte inducing transcription factor |
| mTOR | mammalian target of rapamycin |
| NF1 | neurofibrin 1 |
| NKAP | NFkB activating protein |
| OXLA | l-amino-acid oxidase |
| PAK4 | P21 (RAC1) activated kinase 4 |
| PELO | pelota mRNA surveillance and ribosome rescue factor |
| PI3K | phosphoinositide 3-kinase |
| PIK3CA | phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha |
| PIK3CB | phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta |
| PPIF | peptidyl-prolyl cis-trans isomerase F |
| PRTN3 | Proteinase 3 |
| PSMD3 | proteasome 26S subunit, non-ATPase 3 |
| PTEN | phosphatase and tensin homolog |
| PTN1 | Protein Tyrosine Phosphatase Non-Receptor Type 1 |
| RNF114 | E3 ubiquitin-protein ligase |
| RPS12 | ribosomal protein S12 |
| SNRNP70 | small nuclear ribonucleoprotein U1 subunit 70 |
| SNRPA1 | small nuclear ribonucleoprotein polypeptide A’ |
| SNRPB2 | small nuclear ribonucleoprotein polypeptide B2 |
| TP53 | tumor protein p53 |
| TRAF6 | tumor necrosis factor receptor-associated factor 6 |
| VEGFA | VEGFR pathway–vascular endothelial growth factor A—vascular endothelial growth factor R pathway |
| WIPI1 | WD repeat domain, phosphoinositide interacting 1 |
| WNT signaling pathway | wingless-related integration site signal pathway |
| WT | wild type |
References
- Tímár, J.; Vizkeleti, L.; Doma, V.; Barbai, T.; Rásó, E. Genetic Progression of Malignant Melanoma. Cancer Metastasis Rev. 2016, 35, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Rassy, E.; Samartzis, E.; Moschetta, M.; Sheriff, M.; Pérez-Fidalgo, J.A.; Pavlidis, N. Melanoma of Unknown Primary: New Perspectives for an Old Story. Crit. Rev. Oncol. Hematol. 2021, 158, 103208. [Google Scholar] [CrossRef] [PubMed]
- Cancer Today. Available online: http://gco.iarc.fr/today/home (accessed on 24 November 2021).
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Thomas, D.; Bello, D.M. Adjuvant Immunotherapy for Melanoma. J. Surg. Oncol. 2021, 123, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Bastholt, L.; Bataille, V.; del Marmol, V.; Dréno, B.; Fargnoli, M.C.; et al. European Consensus-Based Interdisciplinary Guideline for Melanoma. Part 2: Treatment—Update 2019. Eur. J. Cancer 2020, 126, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Betancourt, L.H.; Szasz, A.M.; Kuras, M.; Rodriguez Murillo, J.; Sugihara, Y.; Pla, I.; Horvath, Z.; Pawłowski, K.; Rezeli, M.; Miharada, K.; et al. The Hidden Story of Heterogeneous B-Raf V600E Mutation Quantitative Protein Expression in Metastatic Melanoma-Association with Clinical Outcome and Tumor Phenotypes. Cancers 2019, 11, 1981. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.-N.J.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E Inhibition Enhances T-Cell Recognition of Melanoma without Affecting Lymphocyte Function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF Inhibition Increases Tumor Infiltration by T Cells and Enhances the Antitumor Activity of Adoptive Immunotherapy in Mice. Clin. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-Tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [CrossRef]
- Velasquez, E.; Szadai, L.; Zhou, Q.; Kim, Y.; Pla, I.; Sanchez, A.; Appelqvist, R.; Oskolas, H.; Marko-Varga, M.; Lee, B.; et al. A Biobanking Turning-Point in the Use of Formalin-Fixed, Paraffin Tumor Blocks to Unveil Kinase Signaling in Melanoma. Clin. Transl. Med. 2021, 11, e466. [Google Scholar] [CrossRef]
- Čuklina, J. Computational Challenges in Biomarker Discovery from High-Throughput Proteomic Data. Ph.D. Thesis, ETH Zurich, Zurich, Switzerland, 2018. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- Vu, V. Ggbiplot. Available online: https://github.com/vqv/ggbiplot (accessed on 24 November 2021).
- Wilke, C.O. Cowplot: Streamlined Plot Theme and Plot Annotations for “Ggplot2”. Available online: https://CRAN.R-project.org/package=cowplot (accessed on 24 November 2021).
- Auguie, B.; Antonov, A. GridExtra: Miscellaneous Functions for “Grid” Graphics. Available online: https://CRAN.R-project.org/package=gridExtra (accessed on 24 November 2021).
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining Clusters from a Hierarchical Cluster Tree: The Dynamic Tree Cut Package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S2666675821000667 (accessed on 24 November 2021).
- Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles | PNAS. Available online: https://www.pnas.org/content/102/43/15545 (accessed on 24 November 2021).
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting Communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Gene Ontology Consortium the Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Keung, E.Z.; Gershenwald, J.E. The Eighth Edition American Joint Committee on Cancer (AJCC) Melanoma Staging System: Implications for Melanoma Treatment and Care. Expert Rev. Anticancer Ther. 2018, 18, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.E.; Massi, D.; Scolyer, R.A.; Willemze, R. WHO Classification of Skin Tumours; IARC: Lyon, France, 2018; ISBN 92-832-2440-X. [Google Scholar]
- Dusek, R.L.; Attardi, L.D. Desmosomes: New Perpetrators in Tumour Suppression. Nat. Rev. Cancer 2011, 11, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Yang, L.; Gray, A.; Srivastava, A.K.; Li, C.; Zhang, G.; Cui, T. The Role of Desmosomes in Carcinogenesis. OncoTargets Ther. 2017, 10, 4059–4063. [Google Scholar] [CrossRef]
- Valenzuela-Iglesias, A.; Burks, H.E.; Arnette, C.R.; Yalamanchili, A.; Nekrasova, O.; Godsel, L.M.; Green, K.J. Desmoglein 1 Regulates Invadopodia by Suppressing EGFR/Erk Signaling in an Erbin-Dependent Manner. Mol. Cancer Res. 2019, 17, 1195–1206. [Google Scholar] [CrossRef]
- Peitsch, W.K.; Doerflinger, Y.; Fischer-Colbrie, R.; Huck, V.; Bauer, A.T.; Utikal, J.; Goerdt, S.; Schneider, S.W. Desmoglein 2 Depletion Leads to Increased Migration and Upregulation of the Chemoattractant Secretoneurin in Melanoma Cells. PLoS ONE 2014, 9, e89491. [Google Scholar] [CrossRef]
- Hütz, K.; Zeiler, J.; Sachs, L.; Ormanns, S.; Spindler, V. Loss of Desmoglein 2 Promotes Tumorigenic Behavior in Pancreatic Cancer Cells. Mol. Carcinog. 2017, 56, 1884–1895. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kim, J.-M.; Lee, D.G.; Lee, J.; Park, J.-G.; Han, T.-S.; Cho, H.-S.; Cho, Y.-L.; Bae, K.-H.; Park, Y.-J.; et al. Loss of Desmoglein-2 Promotes Gallbladder Carcinoma Progression and Resistance to EGFR-Targeted Therapy through Src Kinase Activation. Cell Death Differ. 2021, 28, 968–984. [Google Scholar] [CrossRef]
- Tan, L.Y.; Mintoff, C.; Johan, M.Z.; Ebert, B.W.; Fedele, C.; Zhang, Y.F.; Szeto, P.; Sheppard, K.E.; McArthur, G.A.; Foster-Smith, E.; et al. Desmoglein 2 Promotes Vasculogenic Mimicry in Melanoma and Is Associated with Poor Clinical Outcome. Oncotarget 2016, 7, 46492–46508. [Google Scholar] [CrossRef]
- Lo, A.K.; Yuen, P.W.; Liu, Y.; Wang, X.H.; Cheung, A.L.; Wong, Y.C.; Tsao, S.W. Downregulation of Hemidesmosomal Proteins in Nasopharyngeal Carcinoma Cells. Cancer Lett. 2001, 163, 117–123. [Google Scholar] [CrossRef]
- Kim, H.J.; Maiti, P.; Barrientos, A. Mitochondrial Ribosomes in Cancer. Semin. Cancer Biol. 2017, 47, 67–81. [Google Scholar] [CrossRef]
- Huang, G.; Li, H.; Zhang, H. Abnormal Expression of Mitochondrial Ribosomal Proteins and Their Encoding Genes with Cell Apoptosis and Diseases. Int. J. Mol. Sci. 2020, 21, 8879. [Google Scholar] [CrossRef]
- Liu, J.; Wu, S.; Xie, X.; Wang, Z.; Lei, Q. Identification of Potential Crucial Genes and Key Pathways in Osteosarcoma. Hereditas 2020, 157, 29. [Google Scholar] [CrossRef]
- Tang, N.-Y.; Chueh, F.-S.; Yu, C.-C.; Liao, C.-L.; Lin, J.-J.; Hsia, T.-C.; Wu, K.-C.; Liu, H.-C.; Lu, K.-W.; Chung, J.-G. Benzyl Isothiocyanate Alters the Gene Expression with Cell Cycle Regulation and Cell Death in Human Brain Glioblastoma GBM 8401 Cells. Oncol. Rep. 2016, 35, 2089–2096. [Google Scholar] [CrossRef]
- Sotgia, F.; Fiorillo, M.; Lisanti, M.P. Mitochondrial Markers Predict Recurrence, Metastasis and Tamoxifen-Resistance in Breast Cancer Patients: Early Detection of Treatment Failure with Companion Diagnostics. Oncotarget 2017, 8, 68730–68745. [Google Scholar] [CrossRef]
- Federico, A.; Rienzo, M.; Abbondanza, C.; Costa, V.; Ciccodicola, A.; Casamassimi, A. Pan-Cancer Mutational and Transcriptional Analysis of the Integrator Complex. Int. J. Mol. Sci. 2017, 18, 936. [Google Scholar] [CrossRef]
- Yadavilli, S.; Mayo, L.D.; Higgins, M.; Lain, S.; Hegde, V.; Deutsch, W.A. Ribosomal Protein S3: A Multi-Functional Protein That Interacts with Both P53 and MDM2 through Its KH Domain. DNA Repair 2009, 8, 1215–1224. [Google Scholar] [CrossRef]
- Murray, L.B.; Lau, Y.-K.I.; Yu, Q. Merlin Is a Negative Regulator of Human Melanoma Growth. PLoS ONE 2012, 7, e43295. [Google Scholar] [CrossRef]
- Pio, R.; Corrales, L.; Lambris, J.D. The Role of Complement in Tumor Growth. Adv. Exp. Med. Biol. 2014, 772, 229–262. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Q.; Li, T.; Liao, Q.; Zhao, Y. Role of the Complement System in the Tumor Microenvironment. Cancer Cell Int. 2019, 19, 300. [Google Scholar] [CrossRef]
- Simpson-Haidaris, P.J.; Rybarczyk, B. Tumors and Fibrinogen. The Role of Fibrinogen as an Extracellular Matrix Protein. Ann. N. Y. Acad. Sci. 2001, 936, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Kanterman, J.; Klieger, Y.; Ish-Shalom, E.; Olga, M.; Saragovi, A.; Shtainberg, H.; Lotem, M.; Baniyash, M. Clinical Significance of Circulating CD33+CD11b+HLA-DR- Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 5661–5672. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Kim, Y.J.; Yun, K.A.; Won, C.H.; Lee, M.W.; Choi, J.H.; Chang, S.E.; Lee, W.J. The Prognostic Significance of VISTA and CD33-Positive Myeloid Cells in Cutaneous Melanoma and Their Relationship with PD-1 Expression. Sci. Rep. 2020, 10, 14372. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef]
- Pajerowski, A.G.; Nguyen, C.; Aghajanian, H.; Shapiro, M.J.; Shapiro, V.S. NKAP Is a Transcriptional Repressor of Notch Signaling and Is Required for T Cell Development. Immunity 2009, 30, 696–707. [Google Scholar] [CrossRef]
- Shu, W.; Liu, G.; Dai, Y.; Feng, A.; Chen, Z.; Han, J.; Li, X. The Oncogenic Role of NKAP in the Growth and Invasion of Colon Cancer Cells. Oncol. Rep. 2019, 42, 2130–2138. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, M.; Kan, Y.; Wang, W.; Liu, J.; Gong, J.; Yang, J. Nuclear Factor-ΚB Activating Protein Plays an Oncogenic Role in Neuroblastoma Tumorigenesis and Recurrence Through the Phosphatidylinositol 3-Kinase/Protein Kinase B Signaling Pathway. Front. Cell Dev. Biol. 2020, 8, 622793. [Google Scholar] [CrossRef]
- Patterson, A.R.; Endale, M.; Lampe, K.; Aksoylar, H.I.; Flagg, A.; Woodgett, J.R.; Hildeman, D.; Jordan, M.B.; Singh, H.; Kucuk, Z.; et al. Gimap5-Dependent Inactivation of GSK3β Is Required for CD4 + T Cell Homeostasis and Prevention of Immune Pathology. Nat. Commun. 2018, 9, 430. [Google Scholar] [CrossRef]
- Patterson, A.R.; Bolcas, P.; Lampe, K.; Cantrell, R.; Ruff, B.; Lewkowich, I.; Hogan, S.P.; Janssen, E.M.; Bleesing, J.; Khurana Hershey, G.K.; et al. Loss of GTPase of Immunity-Associated Protein 5 (Gimap5) Promotes Pathogenic CD4+ T-Cell Development and Allergic Airway Disease. J. Allergy Clin. Immunol. 2019, 143, 245–257.e6. [Google Scholar] [CrossRef]
- Dai, P.; Tang, Z.; Ruan, P.; Bajinka, O.; Liu, D.; Tan, Y. Gimap5 Inhibits Lung Cancer Growth by Interacting With M6PR. Front. Oncol. 2021, 11, 699847. [Google Scholar] [CrossRef]
- Chuang, H.-C.; Wang, X.; Tan, T.-H. MAP4K Family Kinases in Immunity and Inflammation. Adv. Immunol. 2016, 129, 277–314. [Google Scholar] [CrossRef]
- Jauliac, S.; Mazerolles, F.; Jabado, N.; Pallier, A.; Bernard, F.; Peake, J.; Fischer, A.; Hivroz, C. Ligands of CD4 Inhibit the Association of Phospholipase Cgamma1 with Phosphoinositide 3 Kinase in T Cells: Regulation of This Association by the Phosphoinositide 3 Kinase Activity. Eur. J. Immunol. 1998, 28, 3183–3191. [Google Scholar] [CrossRef]
- Whale, A.D.; Colman, L.; Lensun, L.; Rogers, H.L.; Shuttleworth, S.J. Functional Characterization of a Novel Somatic Oncogenic Mutation of PIK3CB. Signal Transduct. Target. Ther. 2017, 2, 17063. [Google Scholar] [CrossRef]
- Lin, C.-W.; Lai, G.-M.; Chen, K.-C.; Lin, T.-H.; Fan, J.-J.; Hsu, R.-L.; Chou, C.-M.; Lin, C.-M.; Kandaswami, C.C.; Lee, M.-T.; et al. RPS12 Increases the Invasiveness in Cervical Cancer Activated by C-Myc and Inhibited by the Dietary Flavonoids Luteolin and Quercetin. J. Funct. Foods 2015, 19, 236–247. [Google Scholar] [CrossRef]
- Sadik, A.; Somarribas Patterson, L.F.; Öztürk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F.; Pfänder, P.; Loth, S.; Salem, H.; Prentzell, M.T.; et al. IL4I1 Is a Metabolic Immune Checkpoint That Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270.e34. [Google Scholar] [CrossRef]
- Wright, K.D.; Miller, B.S.; El-Meanawy, S.; Tsaih, S.-W.; Banerjee, A.; Geurts, A.M.; Sheinin, Y.; Sun, Y.; Kalyanaraman, B.; Rui, H.; et al. The P52 Isoform of SHC1 Is a Key Driver of Breast Cancer Initiation. Breast Cancer Res. 2019, 21, 74. [Google Scholar] [CrossRef]
- Luo, Z.; Zhang, X.; Zeng, W.; Su, J.; Yang, K.; Lu, L.; Lim, C.B.; Tang, W.; Wu, L.; Zhao, S.; et al. TRAF6 Regulates Melanoma Invasion and Metastasis through Ubiquitination of Basigin. Oncotarget 2016, 7, 7179–7192. [Google Scholar] [CrossRef]
- Lin, B.; Ke, Q.; Leaman, D.W.; Goel, V.; Agarwal, A. Regulation of RANKL-Induced Osteoclastogenesis by RING Finger Protein RNF114. J. Orthop. Res. 2018, 36, 159–166. [Google Scholar] [CrossRef]
- Landström, M. The TAK1-TRAF6 Signalling Pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef]
- Du, J.; Widlund, H.R.; Horstmann, M.A.; Ramaswamy, S.; Ross, K.; Huber, W.E.; Nishimura, E.K.; Golub, T.R.; Fisher, D.E. Critical Role of CDK2 for Melanoma Growth Linked to Its Melanocyte-Specific Transcriptional Regulation by MITF. Cancer Cell 2004, 6, 565–576. [Google Scholar] [CrossRef]
- Desai, B.M.; Villanueva, J.; Nguyen, T.-T.K.; Lioni, M.; Xiao, M.; Kong, J.; Krepler, C.; Vultur, A.; Flaherty, K.T.; Nathanson, K.L.; et al. The Anti-Melanoma Activity of Dinaciclib, a Cyclin-Dependent Kinase Inhibitor, Is Dependent on P53 Signaling. PLoS ONE 2013, 8, e59588. [Google Scholar] [CrossRef]
- Huang, R.; Xing, Z.; Luan, Z.; Wu, T.; Wu, X.; Hu, G. A Specific Splicing Variant of SVH, a Novel Human Armadillo Repeat Protein, Is up-Regulated in Hepatocellular Carcinomas. Cancer Res. 2003, 63, 3775–3782. [Google Scholar] [PubMed]
- Zhou, X.; Yang, G.; Huang, R.; Chen, X.; Hu, G. SVH-B Interacts Directly with P53 and Suppresses the Transcriptional Activity of P53. FEBS Lett. 2007, 581, 4943–4948. [Google Scholar] [CrossRef] [PubMed]
- Selimovic, D.; Sprenger, A.; Hannig, M.; Haïkel, Y.; Hassan, M. Apoptosis Related Protein-1 Triggers Melanoma Cell Death via Interaction with the Juxtamembrane Region of P75 Neurotrophin Receptor. J. Cell. Mol. Med. 2012, 16, 349–361. [Google Scholar] [CrossRef]
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Higgins, A.M.; Tomar, D.; Nemani, N.; Hines, K.J.; Smith, D.J.; Eguchi, A.; Vallem, S.; et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell 2015, 60, 47–62. [Google Scholar] [CrossRef]
- CD46 Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000117335-CD46 (accessed on 24 November 2021).
- Geller, A.; Yan, J. The Role of Membrane Bound Complement Regulatory Proteins in Tumor Development and Cancer Immunotherapy. Front. Immunol. 2019, 10, 1074. [Google Scholar] [CrossRef]
- Cardone, J.; Le Friec, G.; Vantourout, P.; Roberts, A.; Fuchs, A.; Jackson, I.; Suddason, T.; Lord, G.; Atkinson, J.P.; Cope, A.; et al. Complement Regulator CD46 Temporally Regulates Cytokine Production by Conventional and Unconventional T Cells. Nat. Immunol. 2010, 11, 862–871. [Google Scholar] [CrossRef]
- C, K.; Ac, C.; Jm, G.; Ka, B.; Km, M.; Jp, A. Activation of Human CD4+ Cells with CD3 and CD46 Induces a T-Regulatory Cell 1 Phenotype. Nature 2003, 421. [Google Scholar] [CrossRef]
- Truscott, S.M.; Abate, G.; Price, J.D.; Kemper, C.; Atkinson, J.P.; Hoft, D.F. CD46 Engagement on Human CD4+ T Cells Produces T Regulatory Type 1-like Regulation of Antimycobacterial T Cell Responses. Infect. Immun. 2010, 78, 5295–5306. [Google Scholar] [CrossRef][Green Version]
- The Cancer Genome Atlas Program—National Cancer Institute. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 24 November 2021).
- Han, W.; Wu, Y.-Z.; Zhao, X.-Y.; Gong, Z.-H.; Shen, G.-L. Integrative Analysis of Minichromosome Maintenance Proteins and Their Prognostic Significance in Melanoma. Front. Oncol. 2021, 11, 3203. [Google Scholar] [CrossRef]
- PELO Pelota MRNA Surveillance and Ribosome Rescue Factor [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/53918 (accessed on 24 November 2021).
- Patel, V.N.; Gokulrangan, G.; Chowdhury, S.A.; Chen, Y.; Sloan, A.E.; Koyutürk, M.; Barnholtz-Sloan, J.; Chance, M.R. Network Signatures of Survival in Glioblastoma Multiforme. PLoS Comput. Biol. 2013, 9, e1003237. [Google Scholar] [CrossRef]
- Chen, J.; Wu, F.; Shi, Y.; Yang, D.; Xu, M.; Lai, Y.; Liu, Y. Identification of Key Candidate Genes Involved in Melanoma Metastasis. Mol. Med. Rep. 2019, 20, 903–914. [Google Scholar] [CrossRef]
- Alessi, C.; Scapulatempo Neto, C.; Viana, C.R.; Vazquez, V. de L. PD-1/PD-L1 and VEGF-A/VEGF-C Expression in Lymph Node Microenvironment and Association with Melanoma Metastasis and Survival. Melanoma Res. 2017, 27, 565–572. [Google Scholar] [CrossRef]
- Frezzetti, D.; Gallo, M.; Maiello, M.R.; D’Alessio, A.; Esposito, C.; Chicchinelli, N.; Normanno, N.; De Luca, A. VEGF as a Potential Target in Lung Cancer. Expert Opin. Ther. Targets 2017, 21, 959–966. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Novotny, W. Bevacizumab (Avastin), a Humanized Anti-VEGF Monoclonal Antibody for Cancer Therapy. Biochem. Biophys. Res. Commun. 2005, 333, 328–335. [Google Scholar] [CrossRef]
- VEGFA Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000112715-VEGFA (accessed on 24 November 2021).
- De Melo, F.H.M.; Gonçalves, D.A.; de Sousa, R.X.; Icimoto, M.Y.; de Fernandes, D.C.; Laurindo, F.R.M.; Jasiulionis, M.G. Metastatic Melanoma Progression Is Associated with Endothelial Nitric Oxide Synthase Uncoupling Induced by Loss of ENOS:BH4 Stoichiometry. Int. J. Mol. Sci. 2021, 22, 9556. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.; Xu, Y.-M.; Li, J.; Huang, L.-F.; Lin, J.; Zhang, J.; Min, Q.-H.; Yang, W.-M.; et al. Mechanism of Alternative Splicing and Its Regulation (Review). Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef]
- Deng, K.; Yao, J.; Huang, J.; Ding, Y.; Zuo, J. Abnormal Alternative Splicing Promotes Tumor Resistance in Targeted Therapy and Immunotherapy. Transl. Oncol. 2021, 14, 101077. [Google Scholar] [CrossRef]
- Eymin, B. Targeting the Spliceosome Machinery: A New Therapeutic Axis in Cancer? Biochem. Pharmacol. 2021, 189, 114039. [Google Scholar] [CrossRef]
- Effenberger, K.A.; Urabe, V.K.; Jurica, M.S. Modulating Splicing with Small Molecular Inhibitors of the Spliceosome. Wiley Interdiscip. Rev. 2017, 8. [Google Scholar] [CrossRef]
- Kuckleburg, C.J.; Newman, P.J. Neutrophil Proteinase 3 Acts on Protease-Activated Receptor-2 to Enhance Vascular Endothelial Cell Barrier Function. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 275–284. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alatrash, G.; Vence, L.M.; Woodward, W.; Ueno, N.T.; Molldrem, J.J. Leukemia-Associated Primary Granule Proteins (PGPs) Elastase-2 and Proteinase-3 Are Aberrantly Expressed in Solid Tumors: A Potential Therapeutic Target for PR1-Directed Immunotherapy. Blood 2008, 112, 5440. [Google Scholar] [CrossRef]
- Babačić, H.; Lehtiö, J.; Pico de Coaña, Y.; Pernemalm, M.; Eriksson, H. In-Depth Plasma Proteomics Reveals Increase in Circulating PD-1 during Anti-PD-1 Immunotherapy in Patients with Metastatic Cutaneous Melanoma. J. Immunother. Cancer 2020, 8, e000204. [Google Scholar] [CrossRef] [PubMed]
- Fejza, A.; Polano, M.; Camicia, L.; Poletto, E.; Carobolante, G.; Toffoli, G.; Mongiat, M.; Andreuzzi, E. The Efficacy of Anti-PD-L1 Treatment in Melanoma Is Associated with the Expression of the ECM Molecule EMILIN2. Int. J. Mol. Sci. 2021, 22, 7511. [Google Scholar] [CrossRef]
- Wang, H.; Pan, J.; Barsky, L.; Jacob, J.C.; Zheng, Y.; Gao, C.; Wang, S.; Zhu, W.; Sun, H.; Lu, L.; et al. Characteristics of Pre-Metastatic Niche: The Landscape of Molecular and Cellular Pathways. Mol. Biomed. 2021, 2, 3. [Google Scholar] [CrossRef]
- Ostrowski, S.M.; Fisher, D.E. Biology of Melanoma. Hematol. Oncol. Clin. N. Am. 2021, 35, 29–56. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The Genetic Landscape of Clinical Resistance to RAF Inhibition in Metastatic Melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF Inhibitor Resistance: A Multicenter Meta-Analysis of the Spectrum and Frequencies, Clinical Behaviour, and Phenotypic Associations of Resistance Mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF Inhibitor Resistance Mechanisms in Metastatic Melanoma: Spectrum and Clinical Impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef]
- Teh, J.L.F.; Cheng, P.F.; Purwin, T.J.; Nikbakht, N.; Patel, P.; Chervoneva, I.; Ertel, A.; Fortina, P.M.; Kleiber, I.; HooKim, K.; et al. In Vivo E2F Reporting Reveals Efficacious Schedules of MEK1/2-CDK4/6 Targeting and MTOR-S6 Resistance Mechanisms. Cancer Discov. 2018, 8, 568–581. [Google Scholar] [CrossRef]
- Apostoli, P.; Minoia, C.; Hamilton, E.I. Significance and Utility of Reference Values in Occupational Medicine. Sci. Total Environ. 1998, 209, 69–77. [Google Scholar] [CrossRef]
- Goel, V.K.; Lazar, A.J.F.; Warneke, C.L.; Redston, M.S.; Haluska, F.G. Examination of Mutations in BRAF, NRAS, and PTEN in Primary Cutaneous Melanoma. J. Investig. Dermatol. 2006, 126, 154–160. [Google Scholar] [CrossRef]
- Tsao, H.; Goel, V.; Wu, H.; Yang, G.; Haluska, F.G. Genetic Interaction between NRAS and BRAF Mutations and PTEN/MMAC1 Inactivation in Melanoma. J. Investig. Dermatol. 2004, 122, 337–341. [Google Scholar] [CrossRef]
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic Silencing of the PTEN Gene in Melanoma. Cancer Res. 2006, 66, 6546–6552. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Properties | ||||
|---|---|---|---|---|
| Patients | Variable | n | Median | Range |
| Age | Age at primary 1 | 77 | 64 yrs | 54–74 yrs |
| Survival | DFS (m) 1 | 74 | 17 months | 0–45 months |
| PFS (m) 1 | 74 | 42 months | 3–81 months | |
| OS (m) 1 | 74 | 51 months | 6–96 months | |
| Treatments | Variable | n | % of total (n = 77) | |
| Immunotherapy | 22 | 28.57 | ||
| CTL4i | 2 | 2.59 | ||
| PD-1i, PD-L1i | 22 | 28.57 | ||
| Targeted therapy | 15 | 19.48 | ||
| BRAFi | 15 | 19.48 | ||
| MEKi | 14 | 18.18 | ||
| Targeted-plus Immunotherapy | 3 | 3.89 | ||
| Other therapies | 59 | 76.62 | ||
| irradiation | 35 | 45.45 | ||
| chemotherapy | 9 | 11.68 | ||
| ECT | 4 | 5.19 | ||
| IFN-therapy | 13 | 16.88 | ||
| No treatments | 18 | 23.37 | ||
| Clinical stage (AJCC8) | St. I | 7 | 9.1 | |
| St. II | 24 | 31.16 | ||
| St. III | 31 | 40.26 | ||
| St. IV | 14 | 18.18 | ||
| NA | 1 | 1.3 | ||
| Tumor samples | Variable | n | % of total (n = 90) | |
| Primary tumors | 53 | 58.9 | ||
| SSM with vertical growth | 15/53 | 28.3 | ||
| SSM | 5/53 | 9.43 | ||
| NM | 27/53 | 51 | ||
| ALM | 3/53 | 5.66 | ||
| ALM with vertical growth | 1/53 | 1.88 | ||
| ALM with SSM | 1/53 | 1.88 | ||
| LMM | 1/53 | 1.88 | ||
| Locoregional lymph. met. | 24/90 | 26.66 | ||
| Cutaneous met. | 13/90 | 14.44 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szadai, L.; Velasquez, E.; Szeitz, B.; Almeida, N.P.d.; Domont, G.; Betancourt, L.H.; Gil, J.; Marko-Varga, M.; Oskolas, H.; Jánosi, Á.J.; et al. Deep Proteomic Analysis on Biobanked Paraffine-Archived Melanoma with Prognostic/Predictive Biomarker Read-Out. Cancers 2021, 13, 6105. https://doi.org/10.3390/cancers13236105
Szadai L, Velasquez E, Szeitz B, Almeida NPd, Domont G, Betancourt LH, Gil J, Marko-Varga M, Oskolas H, Jánosi ÁJ, et al. Deep Proteomic Analysis on Biobanked Paraffine-Archived Melanoma with Prognostic/Predictive Biomarker Read-Out. Cancers. 2021; 13(23):6105. https://doi.org/10.3390/cancers13236105
Chicago/Turabian StyleSzadai, Leticia, Erika Velasquez, Beáta Szeitz, Natália Pinto de Almeida, Gilberto Domont, Lazaro Hiram Betancourt, Jeovanis Gil, Matilda Marko-Varga, Henriett Oskolas, Ágnes Judit Jánosi, and et al. 2021. "Deep Proteomic Analysis on Biobanked Paraffine-Archived Melanoma with Prognostic/Predictive Biomarker Read-Out" Cancers 13, no. 23: 6105. https://doi.org/10.3390/cancers13236105
APA StyleSzadai, L., Velasquez, E., Szeitz, B., Almeida, N. P. d., Domont, G., Betancourt, L. H., Gil, J., Marko-Varga, M., Oskolas, H., Jánosi, Á. J., Boyano-Adánez, M. d. C., Kemény, L., Baldetorp, B., Malm, J., Horvatovich, P., Szász, A. M., Németh, I. B., & Marko-Varga, G. (2021). Deep Proteomic Analysis on Biobanked Paraffine-Archived Melanoma with Prognostic/Predictive Biomarker Read-Out. Cancers, 13(23), 6105. https://doi.org/10.3390/cancers13236105