
MiR-195 and Its Target SEMA6D Regulate Chemoresponse in Breast Cancer

 , , ,
, , ,  , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics, Patients and Tissue Samples
2.2. Laser Capture Microdissection, RNA Extraction and microRNA (miRNA) Profiling
2.3. Cell Culture and Transfections
2.4. Chemoresponse Assays
2.5. Expression Analyses (qPCR, Western Blots)
2.6. Biotinylated miRNA Mimic Pulldowns, RNA-seq, and Data Analysis
2.7. Data Mining
2.8. Statistical Analyses
3. Results
3.1. MiR-195 and miR-26b Expressions Were Consistently Changed Post-NAC
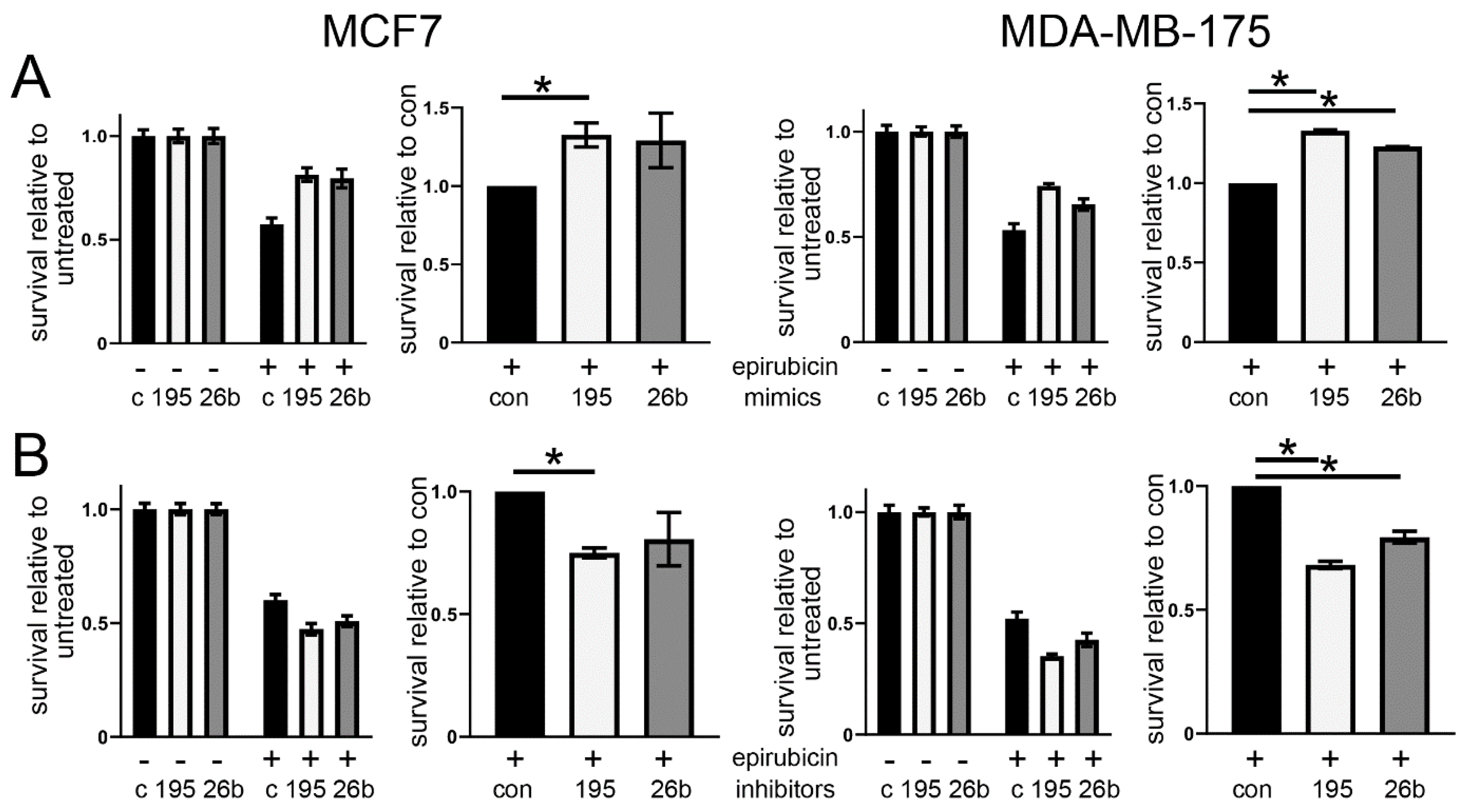
3.2. Altered Expression of miR-195 or miR-26b Controls Chemoresponse in ER Positive Breast Cancer Cell Lines
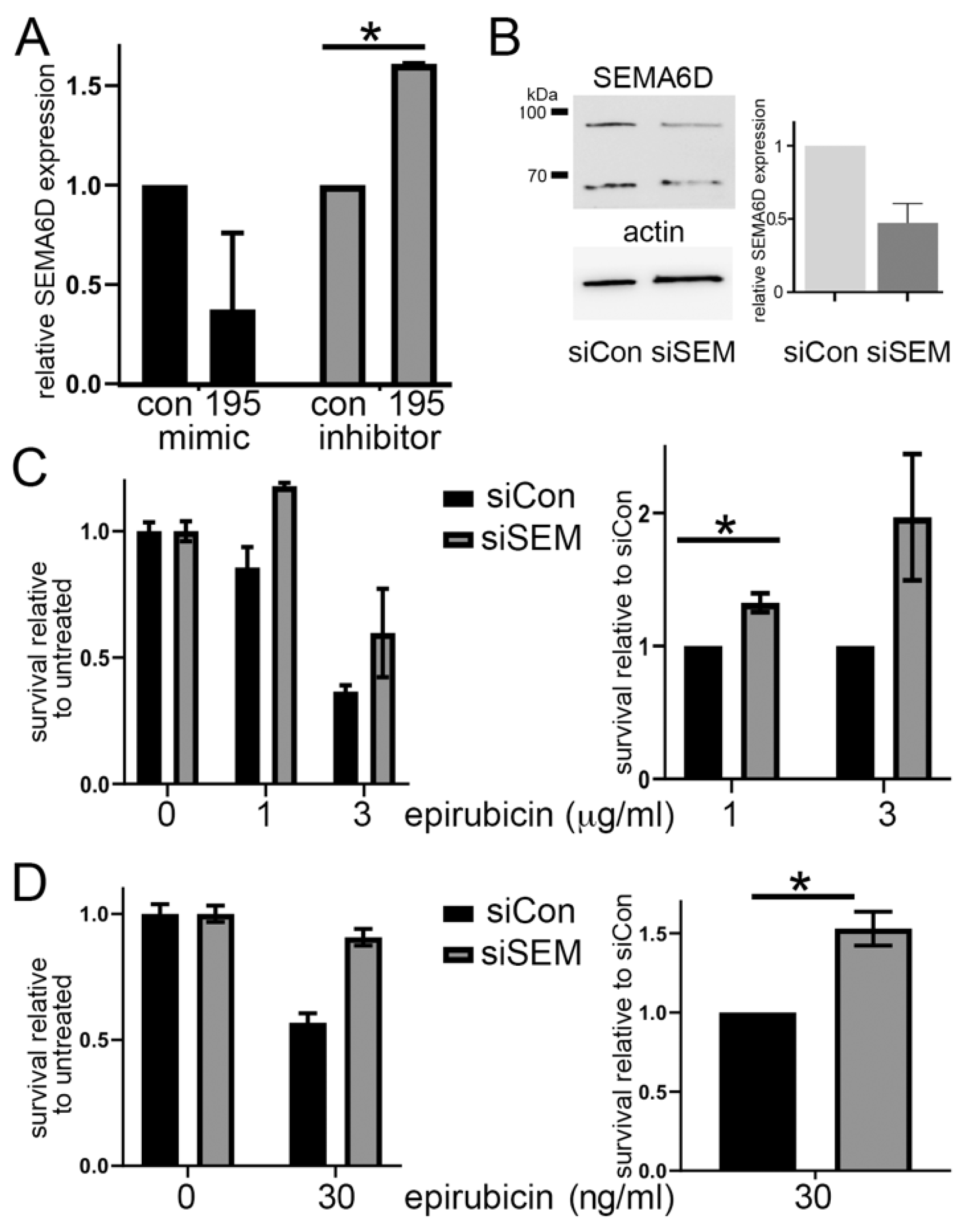
3.3. SEMA6D Is a Candidate Target of miR-195 and miR-26b
3.4. SEMA6D Is Targeted by miR-195 and Mediates Chemoresistance In Vitro
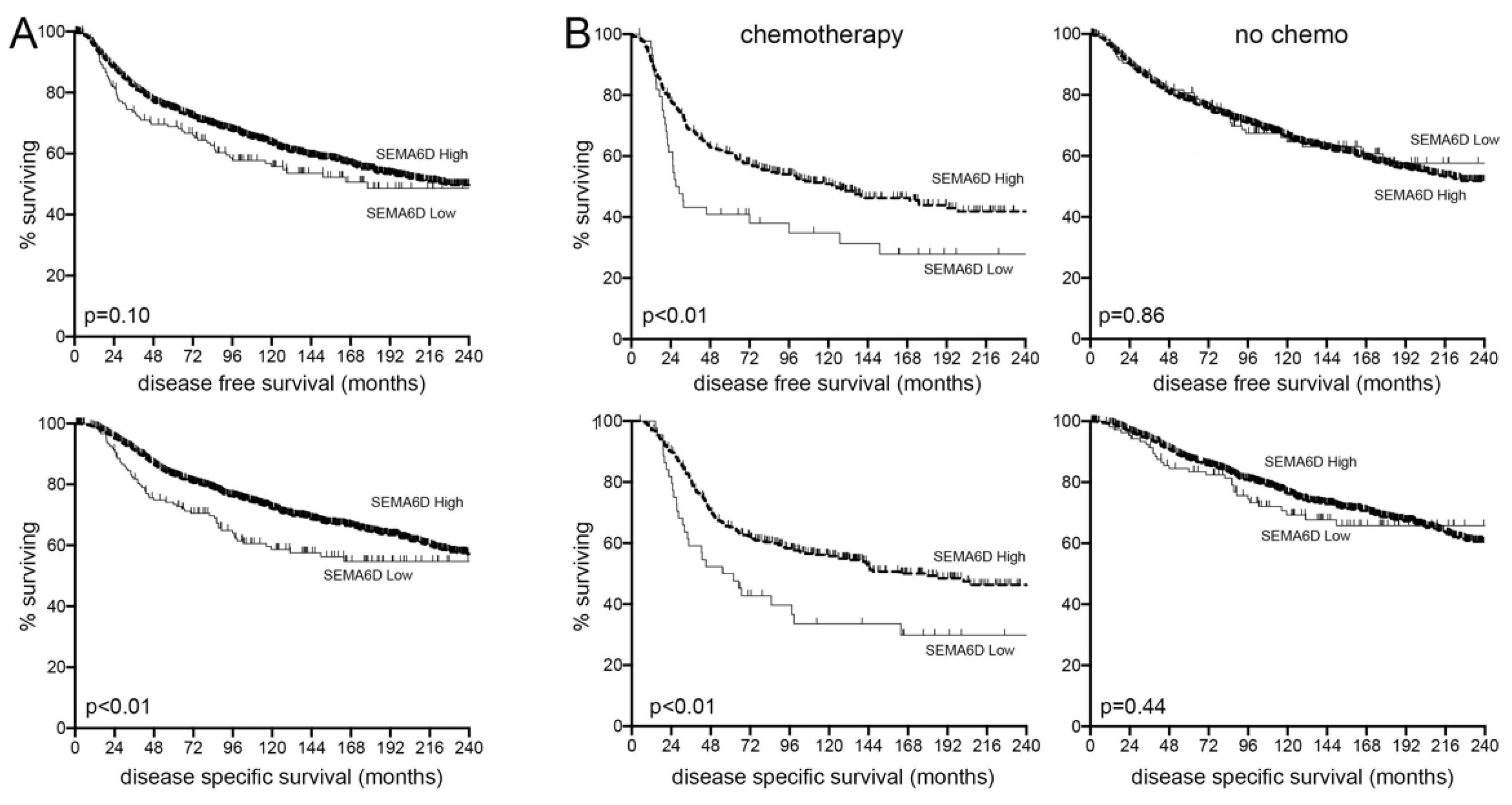
3.5. SEMA6D Is a Predictive Marker of Chemotherapy Response in Breast Cancer Patients
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- NCRAS; CRUK. Chemotherapy, Radiotherapy and Tumour Resections in England: 2013–2014; NCRAS: London, UK, 2017. [Google Scholar]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Lainetti, P.d.F.; Leis-Filho, A.F.; Laufer-Amorim, R.; Battazza, A.; Fonseca-Alves, C.E. Mechanisms of Resistance to Chemotherapy in Breast Cancer and Possible Targets in Drug Delivery Systems. Pharmaceutics 2020, 12, 1193. [Google Scholar] [CrossRef]
- Karakatsanis, A.; Tasoulis, M.K.; Warnberg, F.; Nilsson, G.; MacNeill, F. Meta-analysis of neoadjuvant therapy and its impact in facilitating breast conservation in operable breast cancer. Br. J. Surg. 2018, 105, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Cain, H.; Macpherson, I.R.; Beresford, M.; Pinder, S.E.; Pong, J.; Dixon, J.M. Neoadjuvant Therapy in Early Breast Cancer: Treatment Considerations and Common Debates in Practice. Clin. Oncol. 2017, 29, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Al Amri, W.S.; Allinson, L.M.; Baxter, D.E.; Bell, S.M.; Hanby, A.M.; Jones, S.J.; Shaaban, A.M.; Stead, L.F.; Verghese, E.T.; Hughes, T.A. Genomic and Expression Analyses Define MUC17 and PCNX1 as Predictors of Chemotherapy Response in Breast Cancer. Mol. Cancer Ther. 2020, 19, 945–955. [Google Scholar] [CrossRef]
- Al Amri, W.S.; Baxter, D.E.; Hanby, A.M.; Stead, L.F.; Verghese, E.T.; Thorne, J.L.; Hughes, T.A. Identification of candidate mediators of chemoresponse in breast cancer through therapy-driven selection of somatic variants. Breast Cancer Res. Treat. 2020, 183, 607–616. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Payne, S.; Wang, F.; Claus, P.; Su, Z.; Groth, J.; Geradts, J.; de Ridder, G.; Alvarez, R.; Marcom, P.K.; et al. Nuclear basic fibroblast growth factor regulates triple-negative breast cancer chemo-resistance. Breast Cancer Res. 2015, 17, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Fatayer, H.; Hanby, A.M.; Horgan, K.; Perry, S.L.; Valleley, E.M.; Verghese, E.T.; Williams, B.J.; Thorne, J.L.; Hughes, T.A. Neoadjuvant chemotherapy induces expression levels of breast cancer resistance protein that predict disease-free survival in breast cancer. PLoS ONE 2013, 8, e62766. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Angulo, A.M.; Iwamoto, T.; Liu, S.Y.; Chen, H.Q.; Do, K.A.; Hortobagyi, G.N.; Mills, G.B.; Meric-Bernstam, F.; Symmans, W.F.; Pusztai, L. Gene Expression, Molecular Class Changes, and Pathway Analysis after Neoadjuvant Systemic Therapy for Breast Cancer. Clin. Cancer Res. 2012, 18, 1109–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, E.M.; Aure, M.R.; Haugen, M.H.; Sahlberg, K.K.; Kristensen, V.N.; Nebdal, D.; Børresen-Dale, A.-L.; Lingjaerde, O.C.; Engebraaten, O. miRNA expression changes during the course of neoadjuvant bevacizumab and chemotherapy treatment in breast cancer. Mol. Oncol. 2019, 13, 2278–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luengo-Gil, G.; García-Martínez, E.; Chaves-Benito, A.; Conesa-Zamora, P.; Navarro-Manzano, E.; González-Billalabeitia, E.; García-Garre, E.; Martínez-Carrasco, A.; Vicente, V.; de la Peña, F.A. Clinical and biological impact of miR-18a expression in breast cancer after neoadjuvant chemotherapy. Cell. Oncol. 2019, 42, 627–644. [Google Scholar] [CrossRef]
- Chen, X.; Lu, P.; Wang, D.D.; Yang, S.J.; Wu, Y.; Shen, H.Y.; Zhong, S.L.; Zhao, J.H.; Tang, J.H. The role of miRNAs in drug resistance and prognosis of breast cancer formalin-fixed paraffin-embedded tissues. Gene 2016, 595, 221–226. [Google Scholar] [CrossRef]
- Al-Khanbashi, M.; Caramuta, S.; Alajmi, A.M.; Al-Haddabi, I.; Al-Riyami, M.; Lui, W.-O.; Al-Moundhri, M.S. Tissue and Serum miRNA Profile in Locally Advanced Breast Cancer (LABC) in Response to Neo-Adjuvant Chemotherapy (NAC) Treatment. PLoS ONE 2016, 11, e0152032. [Google Scholar] [CrossRef] [PubMed]
- Frères, P.; Josse, C.; Bovy, N.; Boukerroucha, M.; Struman, I.; Bours, V.; Jerusalem, G. Neoadjuvant Chemotherapy in Breast Cancer Patients Induces miR-34a and miR-122 Expression. J. Cell. Physiol. 2015, 230, 473–481. [Google Scholar] [CrossRef]
- Rajan, R.; Poniecka, A.; Smith, T.L.; Yang, Y.; Frye, D.; Pusztai, L.; Fiterman, D.J.; Gal-Gombos, E.; Whitman, G.; Rouzier, R.; et al. Change in tumor cellularity of breast carcinoma after neoadjuvant chemotherapy as a variable in the pathologic assessment of response. Cancer 2004, 100, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, K.; Pinder, S.E. The importance of histological assessment after neoadjuvant therapy and the need for standardisation. Clin. Radiol. 2018, 73, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.H.; Lal, S.; Lee, J.E.; Choi, Y.-L.; Wen, J.; Ram, S.; Ding, Y.; Lee, S.-H.; Powell, E.; Lee, S.K.; et al. Chemotherapy induces dynamic immune responses in breast cancers that impact treatment outcome. Nat. Commun. 2020, 11, 6175. [Google Scholar] [CrossRef] [PubMed]
- Hamy, A.S.; Bonsang-Kitzis, H.; De Croze, D.; Laas, E.; Darrigues, L.; Topciu, L.; Menet, E.; Vincent-Salomon, A.; Lerebours, F.; Pierga, J.Y.; et al. Interaction between Molecular Subtypes and Stromal Immune Infiltration before and after Treatment in Breast Cancer Patients Treated with Neoadjuvant Chemotherapy. Clin. Cancer Res. 2019, 25, 6731–6741. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.C.; Wooten, E.C.; Tsimelzon, A.; Hilsenbeck, S.G.; Gutierrez, M.C.; Tham, Y.L.; Kalidas, M.; Elledge, R.; Mohsin, S.; Osborne, C.K.; et al. Patterns of resistance and incomplete response to docetaxel by gene expression profiling in breast cancer patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Gentile, L.F.; Plitas, G.; Zabor, E.C.; Stempel, M.; Morrow, M.; Barrio, A.V. Tumor Biology Predicts Pathologic Complete Response to Neoadjuvant Chemotherapy in Patients Presenting with Locally Advanced Breast Cancer. Ann. Surg. Oncol. 2017, 24, 3896–3902. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Broad, R.V.; Jones, S.J.; Teske, M.C.; Wastall, L.M.; Hanby, A.M.; Thorne, J.L.; Hughes, T.A. Inhibition of interferon-signalling halts cancer-associated fibroblast-dependent protection of breast cancer cells from chemotherapy. Br. J. Cancer 2021, 124, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Veeneman, B.A.; Shukla, S.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Nesvizhskii, A.I. Two-pass alignment improves novel splice junction quantification. Bioinformatics 2016, 32, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-H.; Li, J.-H.; Shao, P.; Zhou, H.; Chen, Y.-Q.; Qu, L.-H. starBase: A database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011, 39, D202–D209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, S.A.; Lianto, P.; Roberg-Larsen, H.; Battaglia, S.; Hughes, T.A.; Thorne, J.L. ER-Negative Breast Cancer Is Highly Responsive to Cholesterol Metabolite Signalling. Nutrients 2019, 11, 2618. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, S.; Lester, S.C. Pathology of breast carcinomas after neoadjuvant chemotherapy: An overview with recommendations on specimen processing and reporting. Arch. Pathol. Lab. Med. 2009, 133, 633–642. [Google Scholar] [CrossRef]
- Thomson, D.W.; Bracken, C.P.; Goodall, G.J. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011, 39, 6845–6853. [Google Scholar] [CrossRef] [Green Version]
- Amirkhah, R.; Meshkin, H.N.; Farazmand, A.; Rasko, J.E.J.; Schmitz, U. Computational and Experimental Identification of Tissue-Specific MicroRNA Targets. Methods Mol. Biol. 2017, 1580, 127–147. [Google Scholar] [CrossRef]
- Ørom, U.A.; Lund, A.H. Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods 2007, 43, 162–165. [Google Scholar] [CrossRef]
- Tan, S.M.; Kirchner, R.; Jin, J.; Hofmann, O.; McReynolds, L.; Hide, W.; Lieberman, J. Sequencing of captive target transcripts identifies the network of regulated genes and functions of primate-specific miR-522. Cell Rep. 2014, 8, 1225–1239. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- St Laurent, G.; Shtokalo, D.; Tackett, M.R.; Yang, Z.; Vyatkin, Y.; Milos, P.M.; Seilheimer, B.; McCaffrey, T.A.; Kapranov, P. On the importance of small changes in RNA expression. Methods 2013, 63, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, D.; Zheng, S.; Su, R.; Tang, Y. Serum microRNA-195 as a potential diagnostic biomarker for breast cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Pathol. 2019, 12, 3982–3991. [Google Scholar] [PubMed]
- McGuire, A.; Casey, M.-C.; Waldron, R.M.; Heneghan, H.; Kalinina, O.; Holian, E.; McDermott, A.; Lowery, A.J.; Newell, J.; Dwyer, R.M.; et al. Prospective Assessment of Systemic MicroRNAs as Markers of Response to Neoadjuvant Chemotherapy in Breast Cancer. Cancers 2020, 12, 1820. [Google Scholar] [CrossRef]
- Shen, M.; Dong, C.; Ruan, X.; Yan, W.; Cao, M.; Pizzo, D.; Wu, X.; Yang, L.; Liu, L.; Ren, X.; et al. Chemotherapy-Induced Extracellular Vesicle miRNAs Promote Breast Cancer Stemness by Targeting ONECUT2. Cancer Res. 2019, 79, 3608–3621. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhao, Y.; Liu, C.; Chen, X.; Qi, Y.; Jiang, Y.; Zou, C.; Zhang, X.; Liu, S.; Wang, X.; et al. Analysis of MiR-195 and MiR-497 expression, regulation and role in breast cancer. Clin. Cancer Res. 2011, 17, 1722–1730. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.X.; Li, X.J.; Zhang, B.; Liang, Y.J.; Zhou, C.X.; Cao, D.X.; He, M.; Chen, G.Q.; He, J.R.; Zhao, Q. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett. 2011, 585, 1363–1367. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Kong, X.; Zhang, J.; Luo, Q.; Li, X.; Fang, L. MiRNA-26b inhibits proliferation by targeting PTGS2 in breast cancer. Cancer Cell. Int. 2013, 13, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, J.-M.; Ang, D.A.; Sim, N.; Budiman, A.; Li, Y. Approaches to Identify and Characterise the Post-Transcriptional Roles of lncRNAs in Cancer. Noncoding RNA 2021, 7, 19. [Google Scholar] [CrossRef]
- Mastrantonio, R.; You, H.; Tamagnone, L. Semaphorins as emerging clinical biomarkers and therapeutic targets in cancer. Theranostics 2021, 11, 3262–3277. [Google Scholar] [CrossRef]
- Toyofuku, T.; Zhang, H.; Kumanogoh, A.; Takegahara, N.; Yabuki, M.; Harada, K.; Hori, M.; Kikutani, H. Guidance of myocardial patterning in cardiac development by Sema6D reverse signalling. Nat. Cell Biol. 2004, 6, 1204–1211. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Eun, S.Y.; Ye, Z.; Zozulya, A.L.; Lich, J.D.; Moore, C.B.; Iocca, H.A.; Roney, K.E.; Holl, E.K.; Wu, Q.P.; et al. Semaphorin 6D regulates the late phase of CD4+ T cell primary immune responses. Proc. Natl. Acad. Sci. USA 2008, 105, 13015–13020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Kalambogias, J.; Yoshioka, S.; Han, W.; Li, Z.; Kawasawa, Y.I.; Pochareddy, S.; Li, Z.; Liu, F.; Xu, X.; et al. Control of species-dependent cortico-motoneuronal connections underlying manual dexterity. Science 2017, 357, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Li, Y.; Wang, L.; Jiao, K. SEMA6D Expression and Patient Survival in Breast Invasive Carcinoma. Int. J. Breast Cancer 2015, 2015, 539721. [Google Scholar] [CrossRef] [Green Version]
- Encinas, G.; Sabelnykova, V.Y.; de Lyra, E.C.; Katayama, M.L.H.; Maistro, S.; Valle, P.W.M.d.V.; de Lima Pereira, G.F.; Rodrigues, L.M.; Serio, P.A.d.M.P.; de Gouvêa, A.C.R.C.; et al. Somatic mutations in early onset luminal breast cancer. Oncotarget 2018, 9, 22460–22479. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, L.; Chen, Y.; Li, M.; Ha, M.; Li, S. Screening and identification of biomarkers associated with the diagnosis and prognosis of lung adenocarcinoma. J. Clin. Lab. Anal. 2020, 34, e23450. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Li, W.; Dong, L.; Qu, F. CircUBAP2 promotes SEMA6D expression to enhance the cisplatin resistance in osteosarcoma through sponging miR-506-3p by activating Wnt/β-catenin signaling pathway. J. Mol. Histol. 2020, 51, 329–340. [Google Scholar] [CrossRef]
- Beretta, G.L.; Zunino, F. Molecular mechanisms of anthracycline activity. Top. Curr. Chem. 2008, 283, 1–19. [Google Scholar] [CrossRef]
- Jung, Y.; Lippard, S.J. Direct cellular responses to platinum-induced DNA damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Fu, X.; Johansson, I.; Capparuccia, L.; Andersson, F.; Giustacchini, A.; Squadrito, M.L.; Venneri, M.A.; Mazzone, M.; Larsson, E.; et al. Systemic and targeted delivery of semaphorin 3A inhibits tumor angiogenesis and progression in mouse tumor models. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 741–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, L.A.T.; Fritz, J.; Pierdant-Mancera, M.; Bagnard, D. Current drug design to target the Semaphorin/Neuropilin/Plexin complexes. Cell Adhes. Migr. 2016, 10, 700–708. [Google Scholar] [CrossRef] [Green Version]
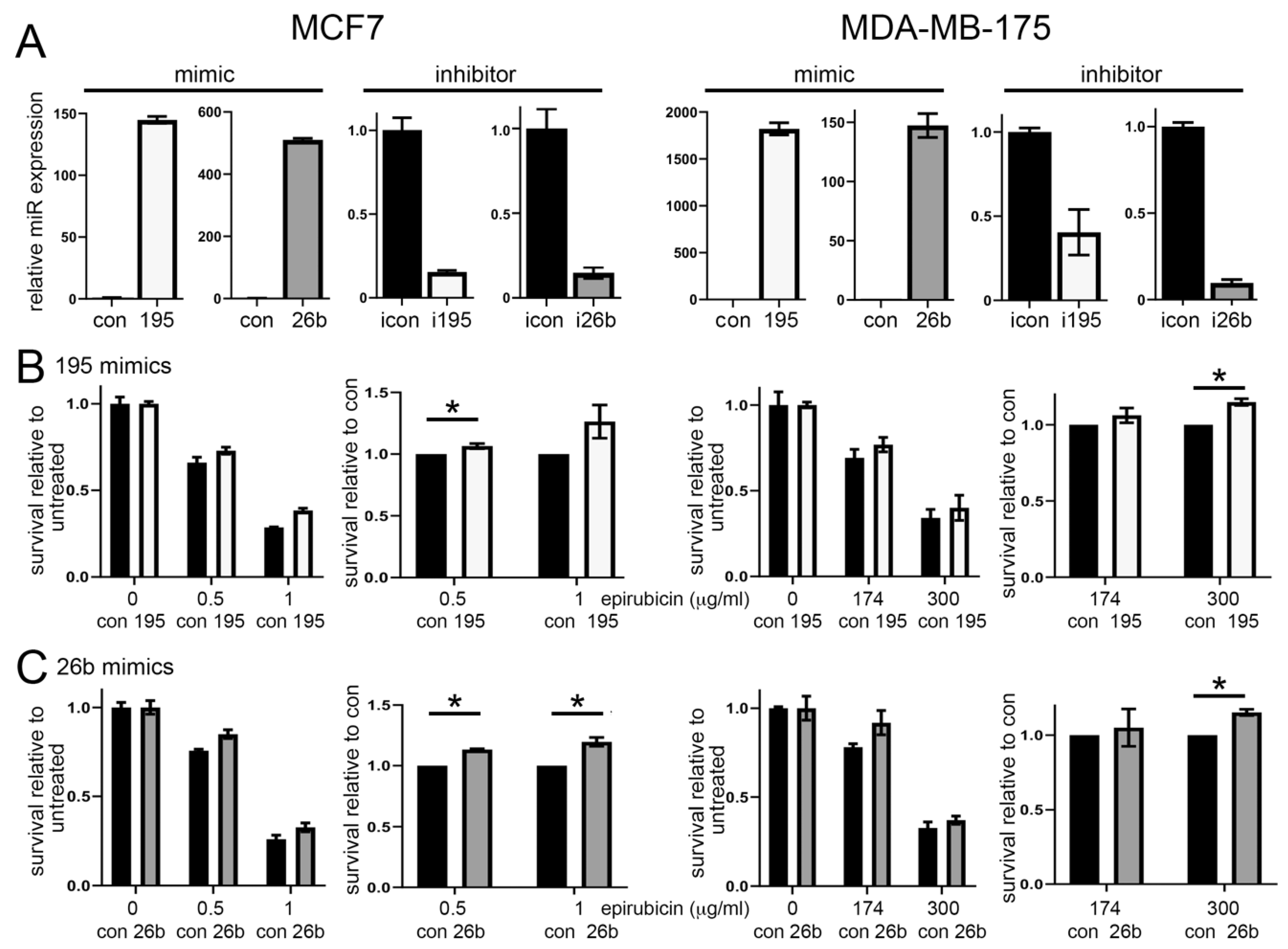

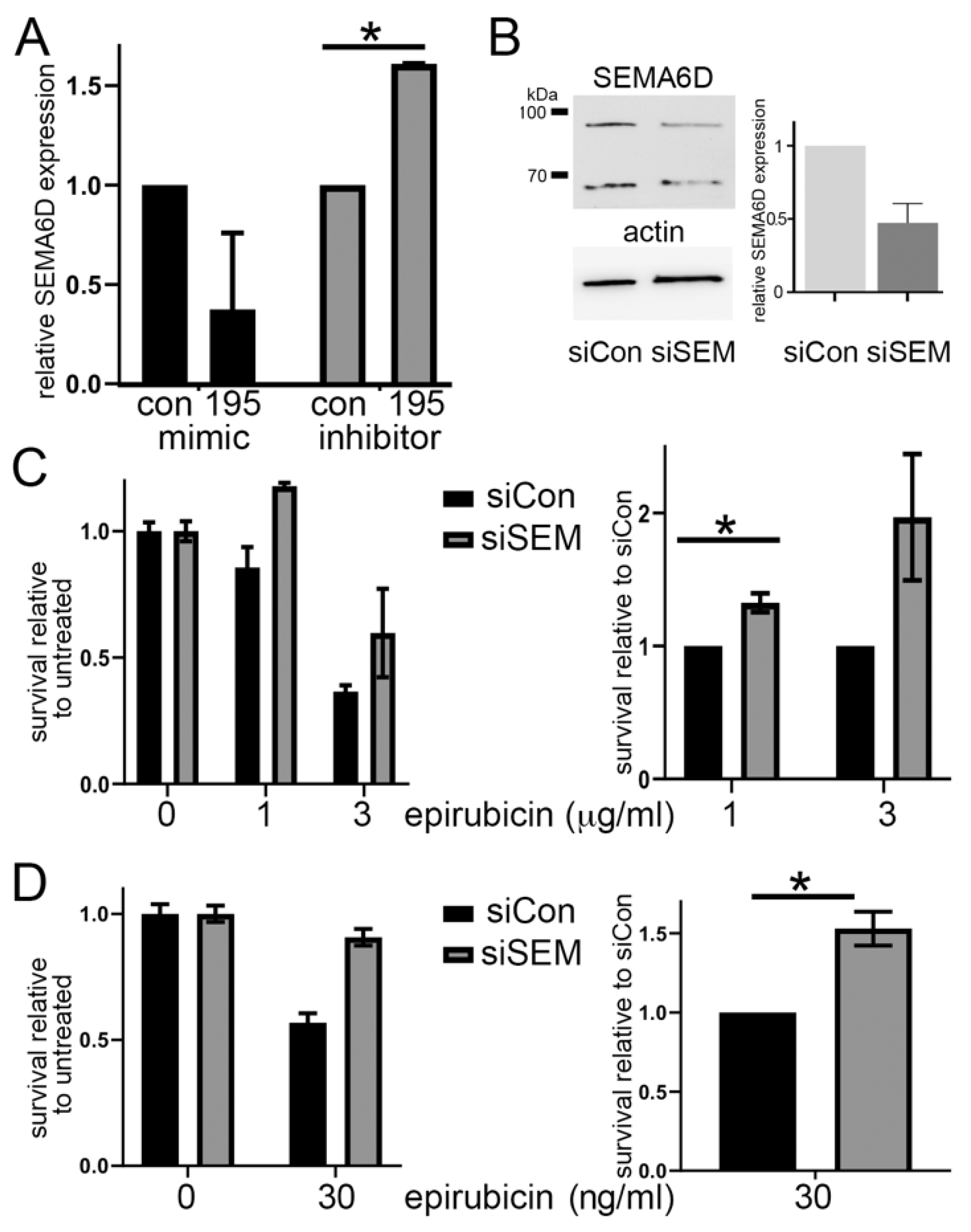
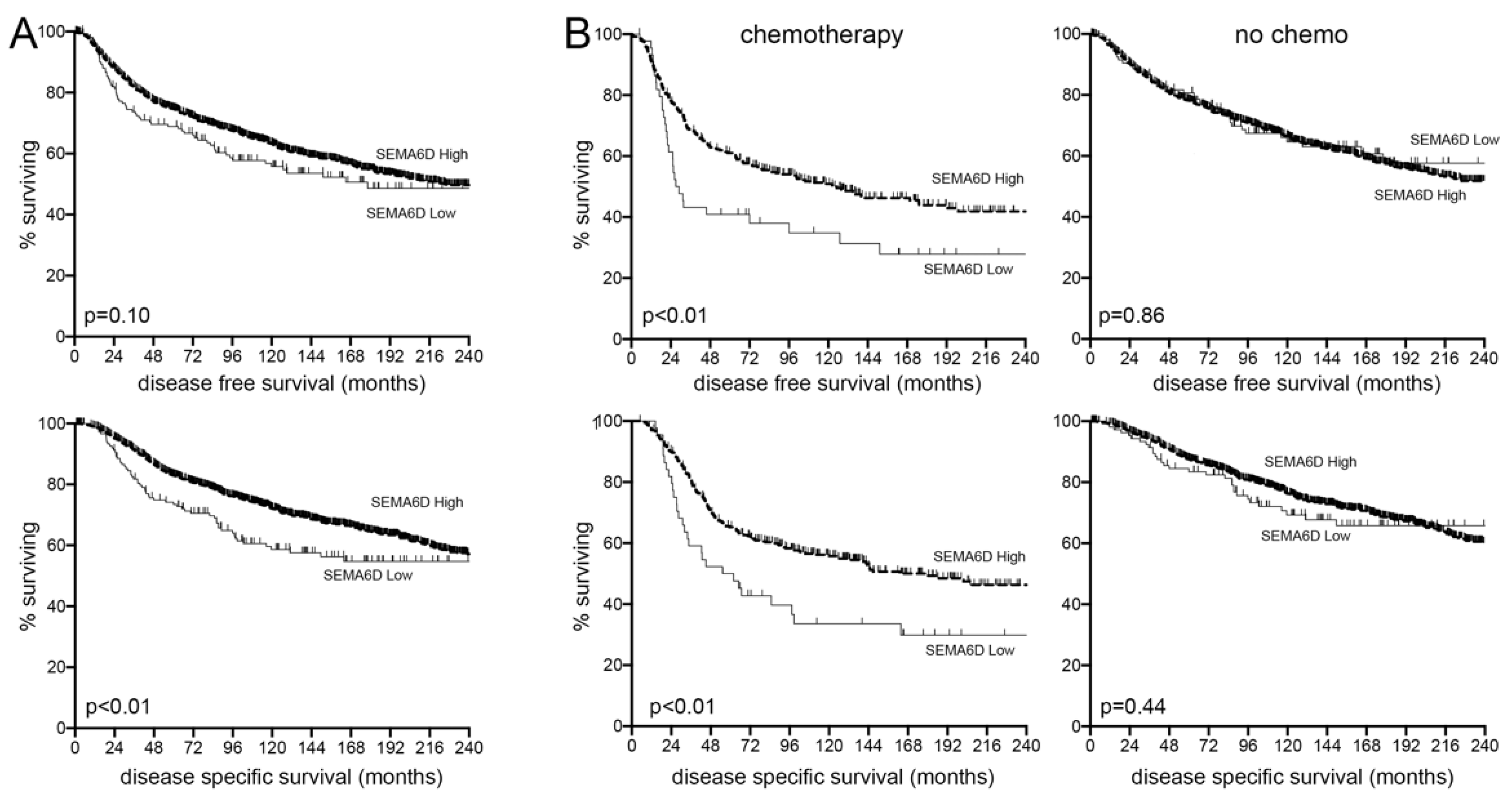

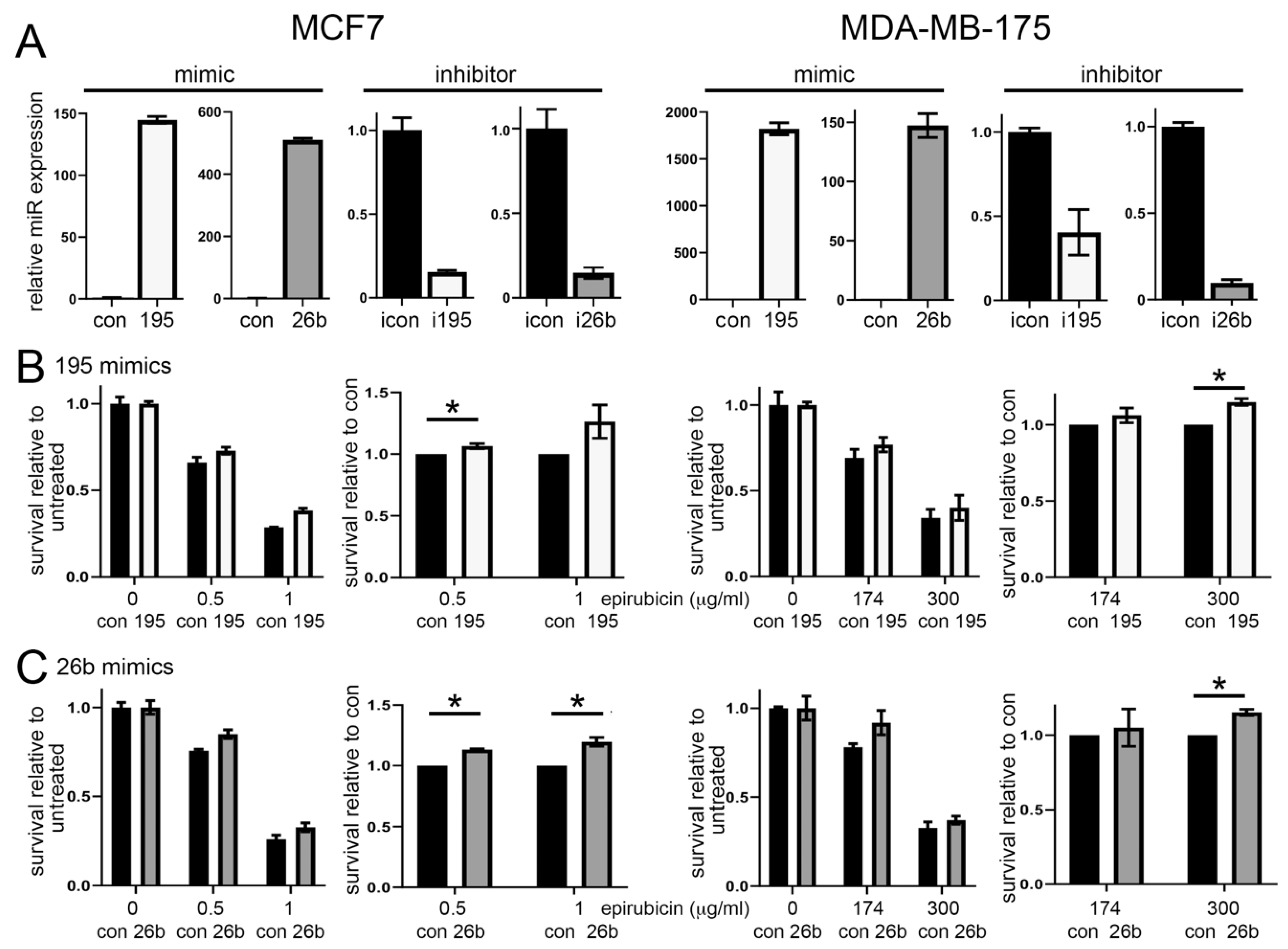{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MiRNA | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Mean |
|---|---|---|---|---|---|---|
| miR-195 | 3.46 | 1.84 | 1.70 | 1.33 | 4.50 | 2.57 |
| miR-26b | 3.39 | 1.54 | 1.92 | 1.79 | 15.7 | 4.87 |
| let-7c | 1.71 | 2.76 | 1.99 | 1.15 | 1.94 | 1.91 |
| miR-10a | −2.33 | −1.42 | −14.4 | −1.27 | −1.36 | −1.83 |
| miR-26a | 1.72 | 1.18 | 2.83 | 1.54 | 2.69 | 1.99 |
| miR-330 | 1.70 | 1.32 | 1.11 | 1.82 | >30 * | 1.49 |
| miR-335 | 3.15 | 4.94 | 1.16 | 1.55 | >120 * | 2.70 |
| miR-362 | 1.68 | 1.03 | 1.27 | 2.85 | >100 * | 1.71 |
| miR-483-5p | 7020 | 3.34 | 1.13 | 2.27 | 19.9 | 1410 |
| miR-885-5p | >870 * | 1.14 | 42.0 | 2.69 | >20 * | 15.29 |
| miR-625 | 27.8 | 1.24 | 1.10 | 4.04 | >10 * | 8.53 |
| miR-365 | −1.18 | −1.47 | −4.03 | −1.19 | −1.66 | −1.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baxter, D.E.; Allinson, L.M.; Al Amri, W.S.; Poulter, J.A.; Pramanik, A.; Thorne, J.L.; Verghese, E.T.; Hughes, T.A. MiR-195 and Its Target SEMA6D Regulate Chemoresponse in Breast Cancer. Cancers 2021, 13, 5979. https://doi.org/10.3390/cancers13235979
Baxter DE, Allinson LM, Al Amri WS, Poulter JA, Pramanik A, Thorne JL, Verghese ET, Hughes TA. MiR-195 and Its Target SEMA6D Regulate Chemoresponse in Breast Cancer. Cancers. 2021; 13(23):5979. https://doi.org/10.3390/cancers13235979
Chicago/Turabian StyleBaxter, Diana E., Lisa M. Allinson, Waleed S. Al Amri, James A. Poulter, Arindam Pramanik, James L. Thorne, Eldo T. Verghese, and Thomas A. Hughes. 2021. "MiR-195 and Its Target SEMA6D Regulate Chemoresponse in Breast Cancer" Cancers 13, no. 23: 5979. https://doi.org/10.3390/cancers13235979
APA StyleBaxter, D. E., Allinson, L. M., Al Amri, W. S., Poulter, J. A., Pramanik, A., Thorne, J. L., Verghese, E. T., & Hughes, T. A. (2021). MiR-195 and Its Target SEMA6D Regulate Chemoresponse in Breast Cancer. Cancers, 13(23), 5979. https://doi.org/10.3390/cancers13235979