
Early TP53 Alterations Shape Gastric and Esophageal Cancer Development

 ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Gastric and Esophageal Adenocarcinomas Are Similar Cancers
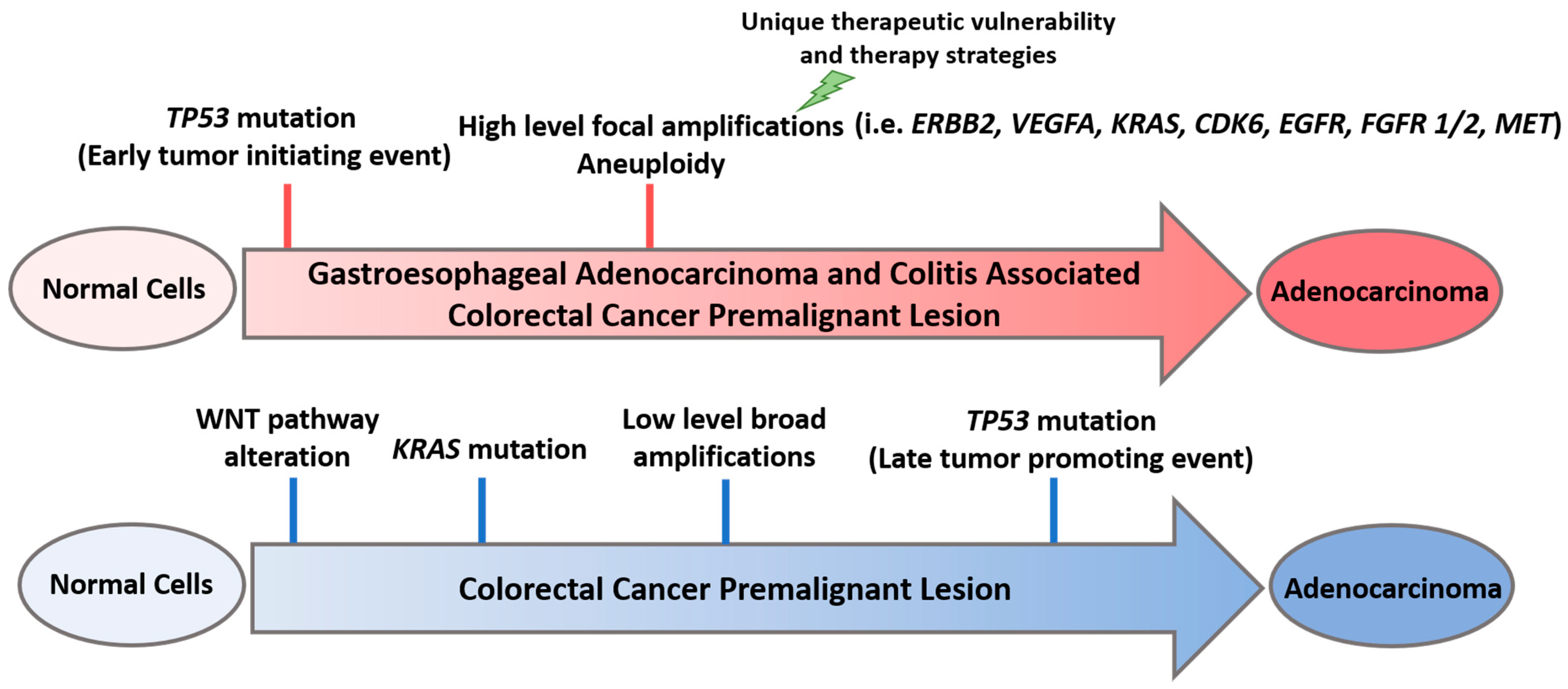
2. TP53 Alteration Is an Early Event in GE Premalignancy
3. Context Matters: Environmental Conditions Contribute to Selection of Early TP53 Alterations
4. Early TP53 Mutations Shape the Method of Genomic Alterations in Cancer-Promoting Pathways
5. Therapeutic Vulnerabilities Imparted by Early TP53 Mutations
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Karlić, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.S.; Reynolds, A.; Rynes, E.; Vlahoviček, K.; Stamatoyannopoulos, J.A.; et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nat. Cell Biol. 2015, 518, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Analysis Working Group: Asan University; BC Cancer Agency; Brigham and Women’s Hospital; Broad Institute; Brown University; Case Western Reserve University; Dana-Farber Cancer Institute; Duke University; Greater Poland Cancer Centre; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef]
- Dulak, A.M.; Schumacher, S.E.; van Lieshout, J.; Imamura, Y.; Fox, C.; Shim, B.; Ramos, A.H.; Saksena, G.; Baca, S.C.; Baselga, J.; et al. Gastrointestinal Adenocarcinomas of the Esophagus, Stomach, and Colon Exhibit Distinct Patterns of Genome Instability and Oncogenesis. Cancer Res. 2012, 72, 4383–4393. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.W.; Karakasheva, T.A.; Lee, D.-J.; Lee, J.-S.; Long, Q.; Bass, A.J.; Wong, K.K.; Rustgi, A.K. Comparative transcriptomes of adenocarcinomas and squamous cell carcinomas reveal molecular similarities that span classical anatomic boundaries. PLoS Genet. 2017, 13, e1006938. [Google Scholar] [CrossRef]
- Sukawa, Y.; Yamamoto, H.; Nosho, K.; Kunimoto, H.; Suzuki, H.; Adachi, Y.; Nakazawa, M.; Nobuoka, T.; Kawayama, M.; Mikami, M.; et al. Alterations in the human epidermal growth factor receptor 2-phosphatidylinositol 3-kinase-v-Akt pathway in gastric cancer. World J. Gastroenterol. 2012, 18, 6577–6586. [Google Scholar] [CrossRef]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735.e728. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nat. Cell Biol. 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Wang, L.; Guo, H.; Lin, C.; Yang, L.; Wang, X. Enrichment and characterization of cancer stem-like cells from a cervical cancer cell line. Mol. Med. Rep. 2014, 9, 2117–2123. [Google Scholar] [CrossRef]
- Van De Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.-S.; Hsu, L.-J.; Lin, Y.-S.; Lai, F.-J.; Sheu, H.-M. WW domain-containing oxidoreductase: A candidate tumor suppressor. Trends Mol. Med. 2007, 13, 12–22. [Google Scholar] [CrossRef]
- Aqeilan, R.I.; Kuroki, T.; Pekarsky, Y.; Albagha, O.; Trapasso, F.; Baffa, R.; Huebner, K.; Edmonds, P.; Croce, C.M. Loss of WWOX Expression in Gastric Carcinoma. Clin. Cancer Res. 2004, 10, 3053–3058. [Google Scholar] [CrossRef] [PubMed]
- Del Mare, S.; Husanie, H.; Iancu, O.; Abu-Odeh, M.; Evangelou, K.; Lovat, F.; Volinia, S.; Gordon, J.; Amir, G.; Stein, J.; et al. WWOX and p53 Dysregulation Synergize to Drive the Development of Osteosarcoma. Cancer Res. 2016, 76, 6107–6117. [Google Scholar] [CrossRef]
- Abdeen, S.K.; Aqeilan, R.I. Decoding the link between WWOX and p53 in aggressive breast cancer. Cell Cycle 2019, 18, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Rustgi, A.K.; El-Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef]
- Giroux, V.; Rustgi, A.K. Metaplasia: Tissue injury adaptation and a precursor to the dysplasia–cancer sequence. Nat. Rev. Cancer 2017, 17, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.K.; Ramnarayanan, K.; Zhu, F.; Srivastava, S.; Xu, C.; Tan, A.L.K.; Lee, M.; Tay, S.; Das, K.; Xing, M.; et al. Genomic and Epigenomic Profiling of High-Risk Intestinal Metaplasia Reveals Molecular Determinants of Progression to Gastric Cancer. Cancer Cell 2018, 33, 137–150.e135. [Google Scholar] [CrossRef]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Becq, J.; Warren, A.; Cheetham, R.K.; Northen, H.; O’Donovan, M.; Malhotra, S.; Di Pietro, M.; Ivakhno, S.; He, M.; et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett’s esophagus and esophageal adenocarcinoma. Nat. Genet. 2015, 47, 1038–1046. [Google Scholar] [CrossRef]
- Stachler, M.D.; Camarda, N.D.; Deitrick, C.; Kim, A.; Agoston, A.T.; Odze, R.D.; Hornick, J.L.; Nag, A.; Thorner, A.R.; Ducar, M.; et al. Detection of Mutations in Barrett’s Esophagus Before Progression to High-Grade Dysplasia or Adenocarcinoma. Gastroenterology 2018, 155, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.-J.; Iwasa, S.; Sugimoto, N.; Ryu, M.-H.; Sakai, D.; Chung, H.-C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N. Engl. J. Med. 2020, 382, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Bang, Y.-J.; Qin, S.K.; Chung, H.C.; Xu, J.M.; Park, J.O.; Jeziorski, K.; Shparyk, Y.; Hoff, P.M.; Sobrero, A.; et al. Lapatinib in Combination With Capecitabine Plus Oxaliplatin in Human Epidermal Growth Factor Receptor 2–Positive Advanced or Metastatic Gastric, Esophageal, or Gastroesophageal Adenocarcinoma: TRIO-013/LOGiC—A Randomized Phase III Trial. J. Clin. Oncol. 2016, 34, 443–451. [Google Scholar] [CrossRef]
- Ito, Y.; Suenaga, M.; Hatake, K.; Takahashi, S.; Yokoyama, M.; Onozawa, Y.; Yamazaki, K.; Hironaka, S.; Hashigami, K.; Hasegawa, H.; et al. Safety, Efficacy and Pharmacokinetics of Neratinib (HKI-272) in Japanese Patients with Advanced Solid Tumors: A Phase 1 Dose-escalation Study. Jpn. J. Clin. Oncol. 2012, 42, 278–286. [Google Scholar] [CrossRef][Green Version]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.-C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.-Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Kim, S.T.; Lee, J.; Lee, S.J.; Park, S.H.; Jung, S.-H.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Park, J.O. Prospective phase II trial of pazopanib plus CapeOX (capecitabine and oxaliplatin) in previously untreated patients with advanced gastric cancer. Oncotarget 2016, 7, 24088–24096. [Google Scholar] [CrossRef]
- Moehler, M.; Gepfner-Tuma, I.; Maderer, A.; Thuss-Patience, P.C.; Ruessel, J.; Hegewisch-Becker, S.; Wilke, H.; Al-Batran, S.-E.; Rafiyan, M.-R.; Weißinger, F.; et al. Sunitinib added to FOLFIRI versus FOLFIRI in patients with chemorefractory advanced adenocarcinoma of the stomach or lower esophagus: A randomized, placebo-controlled phase II AIO trial with serum biomarker program. BMC Cancer 2016, 16, 699. [Google Scholar] [CrossRef]
- Ohtsu, A.; Shah, M.A.; Van Cutsem, E.; Rha, S.Y.; Sawaki, A.; Park, S.R.; Lim, H.Y.; Yamada, Y.; Wu, J.; Langer, B.; et al. Bevacizumab in Combination With Chemotherapy As First-Line Therapy in Advanced Gastric Cancer: A Randomized, Double-Blind, Placebo-Controlled Phase III Study. J. Clin. Oncol. 2011, 29, 3968–3976. [Google Scholar] [CrossRef]
- Sun, W.; Powell, M.; O’Dwyer, P.J.; Catalano, P.; Ansari, R.H.; Benson, A.B., III. Phase II Study of Sorafenib in Combination With Docetaxel and Cisplatin in the Treatment of Metastatic or Advanced Gastric and Gastroesophageal Junction Adenocarcinoma: ECOG. J. Clin. Oncol. 2010, 28, 2947–2951. [Google Scholar] [CrossRef]
- Fukuoka, S.; Hara, H.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y.; et al. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J. Clin. Oncol. 2020, 38, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, N.; Sjoquist, K.M.; Martin, A.J.; Tsobanis, E.; Yip, S.; Kang, Y.-K.; Bang, Y.-J.; Alcindor, T.; O’Callaghan, C.J.; Burnell, M.J.; et al. Regorafenib for the Treatment of Advanced Gastric Cancer (INTEGRATE): A Multinational Placebo-Controlled Phase II Trial. J. Clin. Oncol. 2016, 34, 2728–2735. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-Y.; Doi, T.; Shirao, K.; Lee, K.-W.; Park, S.R.; Chen, Y.; Yang, L.; Valota, O.; Bang, Y.-J. Phase I Study of Axitinib in Combination with Cisplatin and Capecitabine in Patients with Previously Untreated Advanced Gastric Cancer. Cancer Res. Treat. 2015, 47, 687–696. [Google Scholar] [CrossRef]
- Kawazoe, A.; Fukuoka, S.; Nakamura, Y.; Kuboki, Y.; Wakabayashi, M.; Nomura, S.; Mikamoto, Y.; Shima, H.; Fujishiro, N.; Higuchi, T.; et al. Lenvatinib plus pembrolizumab in patients with advanced gastric cancer in the first-line or second-line setting (EPOC1706): An open-label, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1057–1065. [Google Scholar] [CrossRef]
- Karasic, T.B.; O’Hara, M.H.; Teitelbaum, U.R.; Damjanov, N.; Giantonio, B.J.; D’Entremont, T.S.; Gallagher, M.; Zhang, P.J.; O’Dwyer, P.J. Phase II Trial of Palbociclib in Patients with Advanced Esophageal or Gastric Cancer. Oncologist 2020, 25, e1864–e1868. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Blaszkowsky, L.S.; Enzinger, P.C.; Ryan, D.P.; Abrams, T.A.; Zhu, A.X.; Temel, J.S.; Schrag, D.; Bhargava, P.; Meyerhardt, J.A.; et al. A multicenter phase II trial of single-agent cetuximab in advanced esophageal and gastric adenocarcinoma. Ann. Oncol. 2011, 22, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Di Fabio, F.; Siena, S.; Cascinu, S.; Llimpe, F.R.; Ceccarelli, C.; Mutri, V.; Giannetta, L.; Giaquinta, S.; Funaioli, C.; et al. Phase II study of cetuximab in combination with FOLFIRI in patients with untreated advanced gastric or gastroesophageal junction adenocarcinoma (FOLCETUX study). Ann. Oncol. 2007, 18, 510–517. [Google Scholar] [CrossRef]
- Waddell, T.; Chau, I.; Cunningham, D.; Gonzalez, D.; Okines, A.F.C.; Wotherspoon, A.; Saffery, C.; Middleton, G.; Wadsley, J.; Ferry, D.; et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): A randomised, open-label phase 3 trial. Lancet Oncol. 2013, 14, 481–489. [Google Scholar] [CrossRef]
- Dragovich, T.; McCoy, S.; Fenoglio-Preiser, C.M.; Wang, J.; Benedetti, J.K.; Baker, A.F.; Hackett, C.B.; Urba, S.G.; Zaner, K.S.; Blanke, C.D.; et al. Phase II Trial of Erlotinib in Gastroesophageal Junction and Gastric Adenocarcinomas: SWOG. J. Clin. Oncol. 2006, 24, 4922–4927. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Rasco, D.; Lee, J.; Rha, S.Y.; Lee, K.-W.; Bang, Y.J.; Bendell, J.; Enzinger, P.; Marina, N.; Xiang, H.; et al. Phase I Escalation and Expansion Study of Bemarituzumab (FPA144) in Patients With Advanced Solid Tumors and FGFR2b-Selected Gastroesophageal Adenocarcinoma. J. Clin. Oncol. 2020, 38, 2418–2426. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Enzinger, P.C.; Kang, Y.-K.; Yamaguchi, K.; Qin, S.; Lee, K.-W.; Oh, S.C.; Li, J.; Turk, H.M.; Teixeira, A.C.; et al. Randomized double-blind placebo-controlled phase 2 study of bemarituzumab combined with modified FOLFOX6 (mFOLFOX6) in first-line (1L) treatment of advanced gastric/gastroesophageal junction adenocarcinoma (FIGHT). J. Clin. Oncol. 2021, 39, 160. [Google Scholar] [CrossRef]
- Lennerz, J.K.; Kwak, E.L.; Ackerman, A.; Michael, M.; Fox, S.B.; Bergethon, K.; Lauwers, G.Y.; Christensen, J.G.; Wilner, K.D.; Haber, D.A.; et al. MET Amplification Identifies a Small and Aggressive Subgroup of Esophagogastric Adenocarcinoma With Evidence of Responsiveness to Crizotinib. J. Clin. Oncol. 2011, 29, 4803–4810. [Google Scholar] [CrossRef]
- Bang, Y.J.; Su, W.C.; Schuler, M.; Nam, D.H.; Lim, W.T.; Bauer, T.M.; Azaro, A.; Poon, R.T.P.; Hong, D.; Lin, C.C.; et al. Phase 1 study of capmatinib in MET-positive solid tumor patients: Dose escalation and expansion of selected cohorts. Cancer Sci. 2020, 111, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Kurzrock, R.; Amin, H.M.; Xiong, W.; Fu, S.; Piha-Paul, S.A.; Janku, F.; Eskandari, G.; Catenacci, D.V.T.; Klevesath, M.B.; et al. First-in-Man Phase I Trial of the Selective MET Inhibitor Tepotinib in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2019, 26, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, T.A.; Crispin, D.A.; Rabinovitch, P.S.; Haggitt, R.C.; Rubin, C.E.; Stevens, A.C.; Burmer, G.C. Mutations in the p53 gene: An early marker of neoplastic progression in ulcerative colitis. Gastroenterology 1994, 107, 369–378. [Google Scholar] [CrossRef]
- Rabinovitch, P.S.; Dziadon, S.; Brentnall, T.A.; Emond, M.J.; Crispin, D.A.; Haggitt, R.C.; Bronner, M.P. Pancolonic chro-mosomal instability precedes dysplasia and cancer in ulcerative colitis. Cancer Res. 1999, 59, 5148–5153. [Google Scholar] [PubMed]
- Yin, J.; Harpaz, N.; Tong, Y.; Huang, Y.; Laurin, J.; Greenwald, B.D.; Hontanosas, M.; Newkirk, C.; Meltzer, S.J. p53 Point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology 1993, 104, 1633–1639. [Google Scholar] [CrossRef]
- Yaeger, R.; Shah, M.A.; Miller, V.A.; Kelsen, J.R.; Wang, K.; Heins, Z.J.; Ross, J.S.; He, Y.; Sanford, E.; Yantiss, R.K.; et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology 2016, 151, 278–287.e276. [Google Scholar] [CrossRef]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease−Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef]
- Saraggi, D.; Fassan, M.; Mescoli, C.; Scarpa, M.; Valeri, N.; Michielan, A.; D’Incá, R.; Rugge, M. The molecular landscape of colitis-associated carcinogenesis. Dig. Liver Dis. 2017, 49, 326–330. [Google Scholar] [CrossRef]
- Christakis, J.N.A.; Odze, R.; Hamilton, M.; Parrack, P.; Goldblum, J.; Pankaj, A.; Deshpande, V.; Lindeman, N.; Patil, D. Abstracts from USCAP 2021: Gastrointestinal Pathology (311–404). Mod. Pathol. 2021, 34, 397. [Google Scholar] [CrossRef]
- Wanders, L.K.; Cordes, M.; Voorham, Q.; Sie, D.; De Vries, S.D.; D’Haens, G.; de Boer, N.K.H.; Ylstra, B.; Van Grieken, N.C.T.; Meijer, G.A.; et al. IBD-Associated Dysplastic Lesions Show More Chromosomal Instability Than Sporadic Adenomas. Inflamm. Bowel Dis. 2020, 26, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.-M.; Cross, W.; Curtius, K.; Al Bakir, I.; Choi, C.-H.R.; Davis, H.L.; Temko, D.; Biswas, S.; Martinez, P.; Williams, M.J.; et al. Evolutionary history of human colitis-associated colorectal cancer. Gut 2019, 68, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Olafsson, S.; McIntyre, R.E.; Coorens, T.; Butler, T.; Jung, H.; Robinson, P.S.; Lee-Six, H.; Sanders, M.A.; Arestang, K.; Dawson, C.; et al. Somatic Evolution in Non-neoplastic IBD-Affected Colon. Cell 2020, 182, 672–684.e611. [Google Scholar] [CrossRef]
- Chatila, W.K.; (Weill Cornell Medical College, New York, NY, USA); Yaeger, R.; (Memorial Sloan Kettering, New York, NY, USA). Personal communication, 2021.
- Chen, S.-L.; Mo, J.-Z.; Cao, Z.-J.; Chen, X.-Y.; Xiao, S.-D. Effects of bile reflux on gastric mucosal lesions in patients with dyspepsia or chronic gastritis. World J. Gastroenterol. 2005, 11, 2834–2837. [Google Scholar] [CrossRef]
- Sobala, G.M.; O’Connor, H.J.; Dewar, E.P.; King, R.F.; Axon, A.T.; Dixon, M.F. Bile reflux and intestinal metaplasia in gastric mucosa. J. Clin. Pathol. 1993, 46, 235–240. [Google Scholar] [CrossRef]
- Stefaniwsky, A.B.; Tint, G.S.; Speck, J.; Shefer, S.; Salen, G. Ursodeoxycholic acid treatment of bile reflux gastritis. Gastroenterology 1985, 89, 1000–1004. [Google Scholar] [CrossRef]
- Shimizu, T.; Marusawa, H.; Matsumoto, Y.; Inuzuka, T.; Ikeda, A.; Fujii, Y.; Minamiguchi, S.; Miyamoto, S.; Kou, T.; Sakai, Y.; et al. Accumulation of Somatic Mutations in TP53 in Gastric Epithelium With Helicobacter pylori Infection. Gastroenterology 2014, 147, 407–417.e403. [Google Scholar] [CrossRef]
- Li, J.-H.; Shi, X.-Z.; Lv, S.; Liu, M.; Xu, G.-W. Effect ofHelicobacter pyloriinfection onp53expression of gastric mucosa and adenocarcinoma with microsatellite instability. World J. Gastroenterol. 2005, 11, 4363–4366. [Google Scholar] [CrossRef]
- Petersson, F.; Franzén, L.E.; Borch, K. Characterization of the Gastric Cardia in Volunteers from the General Population. Type of mucosa, Helicobacter pylori infection, inflammation, mucosal proliferative activity, p53 and p21 expression, and relations to gastritis. Dig. Dis. Sci. 2010, 55, 46–53. [Google Scholar] [CrossRef]
- Salih, B.A.; Gucin, Z.; Bayyurt, N. A study on the effect of Helicobacter pylori infection on p53 expression in gastric cancer and gastritis tissues. J. Infect. Dev. Ctries. 2013, 7, 651–657. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Satoh, K.; Kihira, K.; Kawata, H.; Tokumaru, K.; Kumakura, Y.; Ishino, Y.; Kawakami, S.; Inoue, K.; Kojima, T.; Satoh, Y.; et al. p53 Expression in the Gastric Mucosa Before and After Eradication of Helicobacter pylori. Helicobacter 2001, 6, 31–36. [Google Scholar] [CrossRef]
- Teh, M.; Tan, K.B.; Seet, B.L.; Yeoh, K.G. Study of p53 immunostaining in the gastric epithelium of cagA-positive and cagA-negativeHelicobacter pylori gastritis. Cancer 2002, 95, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; El Zaatari, M.; Merchant, J.L. Recapitulating Human Gastric Cancer Pathogenesis: Experimental Models of Gastric Cancer. Adv. Exp. Med. Biol. 2016, 908, 441–478. [Google Scholar] [CrossRef]
- Sethi, N.S.; Kikuchi, O.; Duronio, G.N.; Stachler, M.D.; McFarland, J.M.; Ferrer-Luna, R.; Zhang, Y.; Bao, C.; Bronson, R.; Patil, D.; et al. Early TP53 alterations engage environmental exposures to promote gastric premalignancy in an integrative mouse model. Nat. Genet. 2020, 52, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Nanki, K.; Toshimitsu, K.; Takano, A.; Fujii, M.; Shimokawa, M.; Ohta, Y.; Matano, M.; Seino, T.; Nishikori, S.; Ishikawa, K.; et al. Divergent Routes toward Wnt and R-spondin Niche Independency during Human Gastric Carcinogenesis. Cell 2018, 174, 856–869.e817. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Bandla, S.; Reveiller, M.; Toia, L.; Zhou, Z.; Gooding, W.E.; Kalatskaya, I.; Stein, L.; D’Souza, M.; Litle, V.R.; et al. Early G1 Cyclin-Dependent Kinases as Prognostic Markers and Potential Therapeutic Targets in Esophageal Adenocarcinoma. Clin. Cancer Res. 2011, 17, 4513–4522. [Google Scholar] [CrossRef]
- Yaeger, R.; Yao, Z.; Hyman, D.M.; Hechtman, J.F.; Vakiani, E.; Zhao, H.; Su, W.; Wang, L.; Joelson, A.; Cercek, A.; et al. Mechanisms of Acquired Resistance to BRAF V600E Inhibition in Colon Cancers Converge on RAF Dimerization and Are Sensitive to Its Inhibition. Cancer Res. 2017, 77, 6513–6523. [Google Scholar] [CrossRef]
- Wong, G.S.; Zhou, J.; Bin Liu, J.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J.; et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018, 24, 968–977. [Google Scholar] [CrossRef]
- McClelland, S.E. Role of chromosomal instability in cancer progression. Endocr.-Relat. Cancer 2017, 24, T23–T31. [Google Scholar] [CrossRef]
- Chen, Z.; Xiao, Z.; Gu, W.-Z.; Xue, J.; Bui, M.H.; Kovar, P.; Li, G.; Wang, G.; Tao, Z.-F.; Tong, Y.; et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int. J. Cancer 2006, 119, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012, 61, 673–684. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.-B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Färkkilä, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.-J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.-C.; Hendrickson, A.E.W.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Hendrickson, A.E.W.; et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, M.A.; Hodgson, D.; Harbron, C.; Wellings, R.; O’Connor, M.J.; Womack, C.; Yin, X.; Bang, Y.-J.; Im, S.-A.; et al. Concordance of ATM (Ataxia Telangiectasia Mutated) Immunohistochemistry between Biopsy or Metastatic Tumor Samples and Primary Tumors in Gastric Cancer Patients. Pathobiology 2013, 80, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Xu, R.-H.; Chin, K.; Lee, K.-W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.-A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| High Level Focal Amplification | Prevalence in GE Adenocarcinoma | Potential Therapeutic Agent | Trial References |
|---|---|---|---|
| ERBB2 | 24–32% | trastuzumab 3,*, lapatinib 3, neratinib 1, tucatinib 0, trastuzumab deruxtecan 2,* | [22,23,24,25] |
| VEGFA | 28% | VEGF inhibitors (ramucirumab 3,*, Lenvatinib 2) | [26,27,28,29,30,31,32,33,34] |
| KRAS | 13–17% | MEK inhibitors (binimetinib 0, cobimetinib 0), ERK inhibitors 0, RAF inhibitors 0 | - |
| CDK6 | 14% | palbociclib 2, abemaciclib 0, ribociclib 0 | [35] |
| EGFR | 10% | cetuximab 2, panitumumab 3, ABT-806 2 | [36,37,38,39] |
| FGFR1/2 | 8–10% | bemarituzumab 2 | [40,41] |
| MET | 8% | crizotinib 1, capmatinib 1, tepotinib 1 | [42,43,44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahgal, P.; Huffman, B.M.; Patil, D.T.; Chatila, W.K.; Yaeger, R.; Cleary, J.M.; Sethi, N.S. Early TP53 Alterations Shape Gastric and Esophageal Cancer Development. Cancers 2021, 13, 5915. https://doi.org/10.3390/cancers13235915
Sahgal P, Huffman BM, Patil DT, Chatila WK, Yaeger R, Cleary JM, Sethi NS. Early TP53 Alterations Shape Gastric and Esophageal Cancer Development. Cancers. 2021; 13(23):5915. https://doi.org/10.3390/cancers13235915
Chicago/Turabian StyleSahgal, Pranshu, Brandon M. Huffman, Deepa T. Patil, Walid K. Chatila, Rona Yaeger, James M. Cleary, and Nilay S. Sethi. 2021. "Early TP53 Alterations Shape Gastric and Esophageal Cancer Development" Cancers 13, no. 23: 5915. https://doi.org/10.3390/cancers13235915
APA StyleSahgal, P., Huffman, B. M., Patil, D. T., Chatila, W. K., Yaeger, R., Cleary, J. M., & Sethi, N. S. (2021). Early TP53 Alterations Shape Gastric and Esophageal Cancer Development. Cancers, 13(23), 5915. https://doi.org/10.3390/cancers13235915