
Detection of VAR2CSA-Captured Colorectal Cancer Cells from Blood Samples by Real-Time Reverse Transcription PCR


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Production and Biotinylation of rVAR2
2.2. Cell Culture
2.3. Magnetic Bead Preparation
2.4. Cancer Cell Capture from Blood
2.5. Cancer Cell Detection by Immunocytochemistry
2.6. Cell Lysis and RNA Extraction
2.7. Validation of Organ-Specific RT-qPCR Assays
2.8. Cancer Cell Detection by Target Enriched RT-qPCR
2.9. Analysis of qPCR Data Using the LightCycler® 480 Software
3. Results
3.1. Cytokeratin Antibodies Provide a Sensitive Strategy for Cancer Cell Detection after rVAR2 Capture
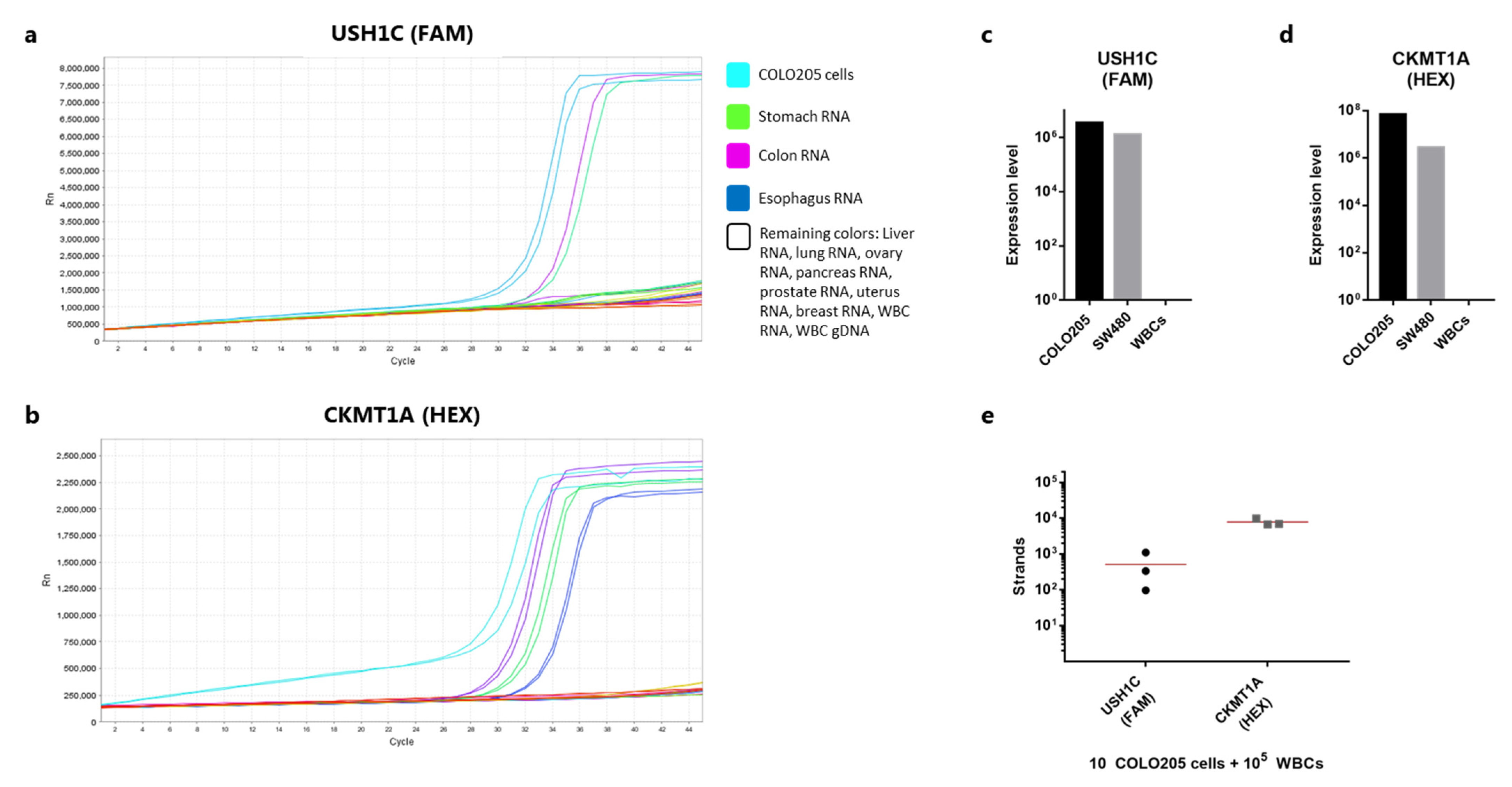
3.2. USH1C and CKMT1A Are Specifically Expressed in Intestinal-Related Tissues and Cancer Cell Lines
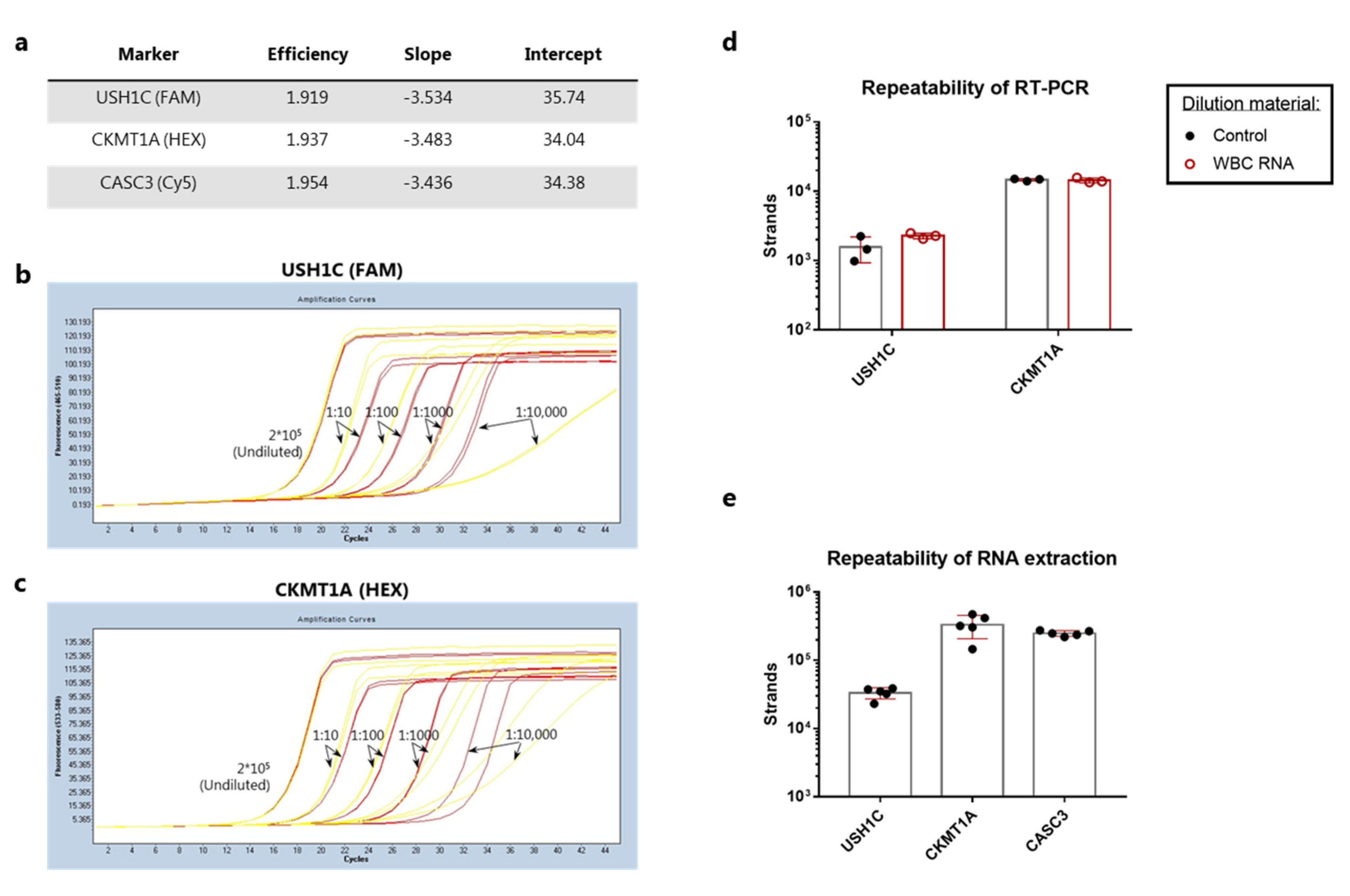
3.3. Analytical Validation of RT-qPCR Assay Performance for Detection of Colorectal Cancer Cells
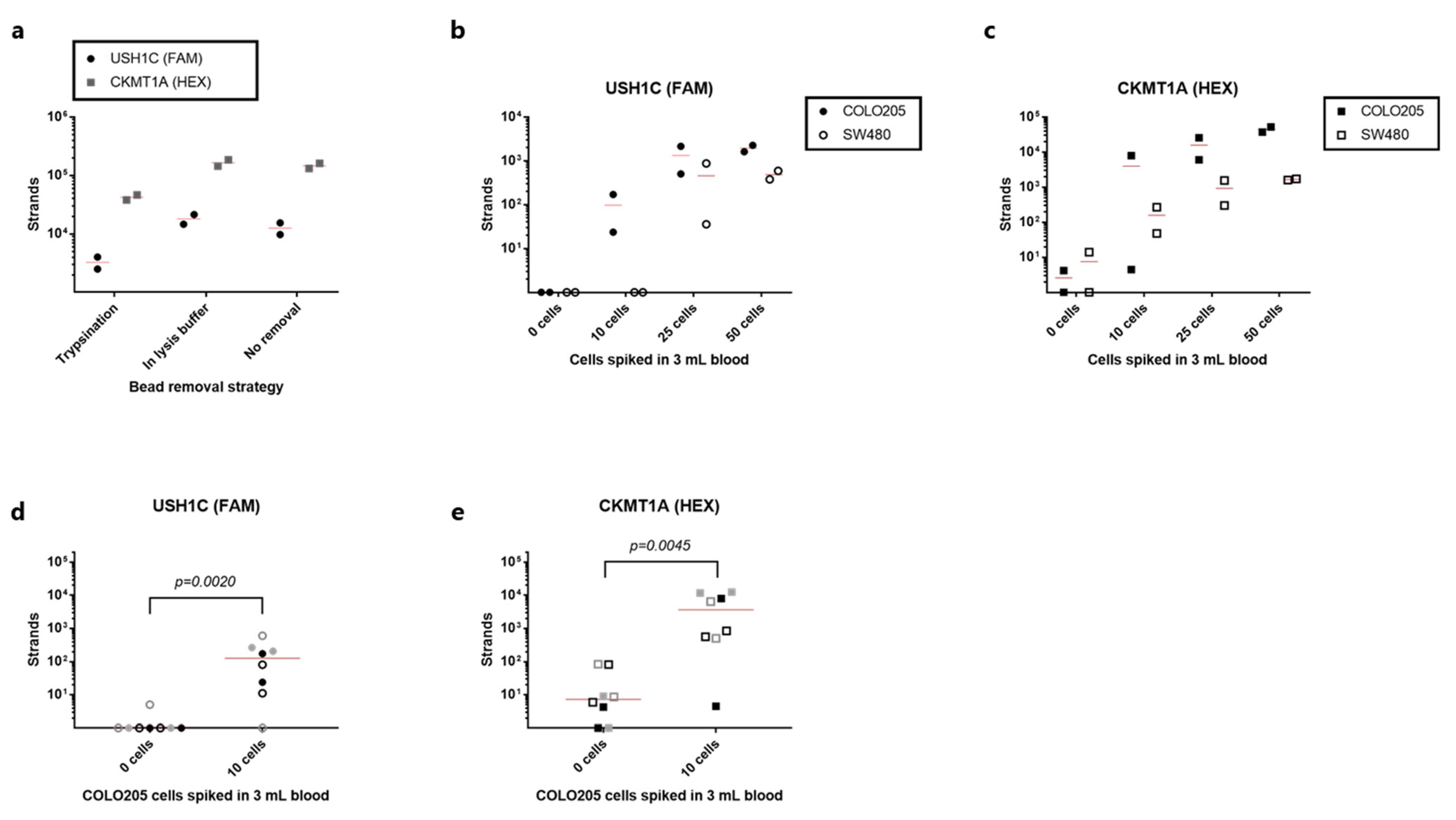
3.4. Detection of rVAR2-Captured Colorectal Cancer Cells by RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Marcuello, M.; Vymetalkova, V.; Neves, R.P.L.; Duran-Sanchon, S.; Vedeld, H.M.; Tham, E.; van Dalum, G.; Flugen, G.; Garcia-Barberan, V.; Fijneman, R.J.; et al. Circulating biomarkers for early detection and clinical management of colorectal cancer. Mol. Asp. Med. 2019, 69, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Husemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmuller, G.; et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stott, S.L.; Lee, R.J.; Nagrath, S.; Yu, M.; Miyamoto, D.T.; Ulkus, L.; Inserra, E.J.; Ulman, M.; Springer, S.; Nakamura, Z.; et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci. Transl. Med. 2010, 2, 25ra23. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, J.E.; Hewett, P.J.; Sage, R.E.; Finch, J.L.; Nuttall, J.D.; Kotasek, D.; Dobrovic, A. Molecular detection of blood-borne epithelial cells in colorectal cancer patients and in patients with benign bowel disease. Int. J. Cancer 2000, 89, 8–13. [Google Scholar] [CrossRef]
- Tsai, W.S.; Chen, J.S.; Shao, H.J.; Wu, J.C.; Lai, J.M.; Lu, S.H.; Hung, T.F.; Chiu, Y.C.; You, J.F.; Hsieh, P.S.; et al. Circulating Tumor Cell Count Correlates with Colorectal Neoplasm Progression and Is a Prognostic Marker for Distant Metastasis in Non-Metastatic Patients. Sci. Rep. 2016, 6, 24517. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.; Uhr, J.W.; Terstappen, L.W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Alix-Panabieres, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Salanti, A.; Clausen, T.M.; Agerbaek, M.O.; Al Nakouzi, N.; Dahlback, M.; Oo, H.Z.; Lee, S.; Gustavsson, T.; Rich, J.R.; Hedberg, B.J.; et al. Targeting Human Cancer by a Glycosaminoglycan Binding Malaria Protein. Cancer Cell 2015, 28, 500–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, T.M.; Pereira, M.A.; Al Nakouzi, N.; Oo, H.Z.; Agerbaek, M.O.; Lee, S.; Orum-Madsen, M.S.; Kristensen, A.R.; El-Naggar, A.; Grandgenett, P.M.; et al. Oncofetal Chondroitin Sulfate Glycosaminoglycans Are Key Players in Integrin Signaling and Tumor Cell Motility. Mol. Cancer Res. 2016, 14, 1288–1299. [Google Scholar] [CrossRef] [Green Version]
- Agerbaek, M.O.; Bang-Christensen, S.R.; Yang, M.H.; Clausen, T.M.; Pereira, M.A.; Sharma, S.; Ditlev, S.B.; Nielsen, M.A.; Choudhary, S.; Gustavsson, T.; et al. The VAR2CSA malaria protein efficiently retrieves circulating tumor cells in an EpCAM-independent manner. Nat. Commun. 2018, 9, 3279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang-Christensen, S.R.; Pedersen, R.S.; Pereira, M.A.; Clausen, T.M.; Loppke, C.; Sand, N.T.; Ahrens, T.D.; Jorgensen, A.M.; Lim, Y.C.; Goksoyr, L.; et al. Capture and Detection of Circulating Glioma Cells Using the Recombinant VAR2CSA Malaria Protein. Cells 2019, 8, 998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sand, N.T.; Petersen, T.B.; Bang-Christensen, S.R.; Ahrens, T.D.; Loppke, C.; Jorgensen, A.M.; Gustavsson, T.; Choudhary, S.; Theander, T.G.; Salanti, A.; et al. Optimization of rVAR2-Based Isolation of Cancer Cells in Blood for Building a Robust Assay for Clinical Detection of Circulating Tumor Cells. Int. J. Mol. Sci. 2020, 21, 2401. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Allawi, H.; Cao, X.; Domanico, M.; Harrington, J.; Taylor, W.R.; Yab, T.; Ahlquist, D.A.; Lidgard, G. Quantification of methylated markers with a multiplex methylation-specific technology. Clin. Chem. 2012, 58, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef] [Green Version]
- Eis, P.S.; Olson, M.C.; Takova, T.; Curtis, M.L.; Olson, S.M.; Vener, T.I.; Ip, H.S.; Vedvik, K.L.; Bartholomay, C.T.; Allawi, H.T.; et al. An invasive cleavage assay for direct quantitation of specific RNAs. Nat. Biotechnol. 2001, 19, 673–676. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559–1582. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willipinski-Stapelfeldt, B.; Riethdorf, S.; Assmann, V.; Woelfle, U.; Rau, T.; Sauter, G.; Heukeshoven, J.; Pantel, K. Changes in cytoskeletal protein composition indicative of an epithelial-mesenchymal transition in human micrometastatic and primary breast carcinoma cells. Clin. Cancer Res. 2005, 11, 8006–8014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosse, S.A.; Hannemann, J.; Spotter, J.; Bauche, A.; Andreas, A.; Muller, V.; Pantel, K. Changes in keratin expression during metastatic progression of breast cancer: Impact on the detection of circulating tumor cells. Clin. Cancer Res. 2012, 18, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Raba, K.; Guglielmi, R.; Behrens, B.; Van Dalum, G.; Flugen, G.; Koch, A.; Patel, S.; Knoefel, W.T.; Stoecklein, N.H.; et al. Magnetic-Based Enrichment of Rare Cells from High Concentrated Blood Samples. Cancers 2020, 12, 933. [Google Scholar] [CrossRef] [Green Version]
- Lianidou, E.S. Gene expression profiling and DNA methylation analyses of CTCs. Mol. Oncol. 2016, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Lampignano, R.; Neumann, M.H.D.; Weber, S.; Kloten, V.; Herdean, A.; Voss, T.; Groelz, D.; Babayan, A.; Tibbesma, M.; Schlumpberger, M.; et al. Multicenter Evaluation of Circulating Cell-Free DNA Extraction and Downstream Analyses for the Development of Standardized (Pre)analytical Work Flows. Clin. Chem. 2020, 66, 149–160. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bang-Christensen, S.R.; Katerov, V.; Jørgensen, A.M.; Gustavsson, T.; Choudhary, S.; Theander, T.G.; Salanti, A.; Allawi, H.T.; Agerbæk, M.Ø. Detection of VAR2CSA-Captured Colorectal Cancer Cells from Blood Samples by Real-Time Reverse Transcription PCR. Cancers 2021, 13, 5881. https://doi.org/10.3390/cancers13235881
Bang-Christensen SR, Katerov V, Jørgensen AM, Gustavsson T, Choudhary S, Theander TG, Salanti A, Allawi HT, Agerbæk MØ. Detection of VAR2CSA-Captured Colorectal Cancer Cells from Blood Samples by Real-Time Reverse Transcription PCR. Cancers. 2021; 13(23):5881. https://doi.org/10.3390/cancers13235881
Chicago/Turabian StyleBang-Christensen, Sara R., Viatcheslav Katerov, Amalie M. Jørgensen, Tobias Gustavsson, Swati Choudhary, Thor G. Theander, Ali Salanti, Hatim T. Allawi, and Mette Ø. Agerbæk. 2021. "Detection of VAR2CSA-Captured Colorectal Cancer Cells from Blood Samples by Real-Time Reverse Transcription PCR" Cancers 13, no. 23: 5881. https://doi.org/10.3390/cancers13235881
APA StyleBang-Christensen, S. R., Katerov, V., Jørgensen, A. M., Gustavsson, T., Choudhary, S., Theander, T. G., Salanti, A., Allawi, H. T., & Agerbæk, M. Ø. (2021). Detection of VAR2CSA-Captured Colorectal Cancer Cells from Blood Samples by Real-Time Reverse Transcription PCR. Cancers, 13(23), 5881. https://doi.org/10.3390/cancers13235881