
The Potential of Induced Pluripotent Stem Cells to Advance the Treatment of Pancreatic Ductal Adenocarcinoma

 ,
, 
Simple Summary
Abstract
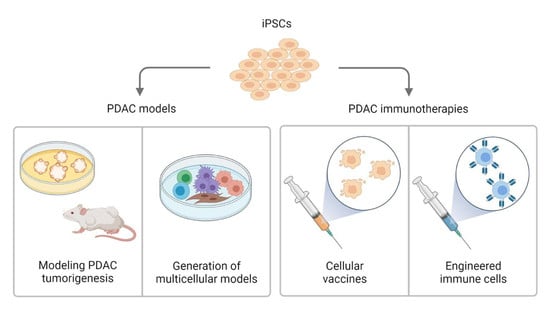
1. Introduction
2. The Potential of Induced Pluripotent Stem Cells (iPSCs)
3. iPSC-Based PDAC Models and Their Potential for Disease Modeling
4. iPSCs as a Cell-Based Immunotherapy
5. iPSC-Derived Immune Cells for Cancer Immunotherapies
6. The Importance of Controlled Reprogramming and Differentiation of iPSCs
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. Cancer Research for Cancer Prevention World Cancer Report; World Health Organization: Lyon, France, 2020. [Google Scholar]
- National Cancer Institute. SEER Cancer Stat Facts: Pancreatic Cancer. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 1 July 2021).
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Adsay, N.V.; Albores–Saavedra, J.; Compton, C.; Garrett, E.S.; Goodman, S.N.; Kern, S.E.; Klimstra, D.S.; Klöppel, G.; Longnecker, D.S.; et al. Pancreatic Intraepithelial Neoplasia. Am. J. Surg. Pathol. 2001, 25, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Bardeesy, N.; Mizukami, Y. Diversity of Precursor Lesions For Pancreatic Cancer: The Genetics and Biology of Intraductal Papillary Mucinous Neoplasm. Clin. Transl. Gastroenterol. 2017, 8, e86. [Google Scholar] [CrossRef]
- Kopp, J.L.; Von Figura, G.; Mayes, E.; Liu, F.-F.; Dubois, C.L.; Morris, J.P.; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef]
- Wagner, M.; Lührs, H.; Klöppel, G.; Adler, G.; Schmid, R.M. Malignant transformation of duct-like cells originating from acini in transforming growth factor α transgenic mice. Gastroenterology 1998, 115, 1254–1262. [Google Scholar] [CrossRef]
- Zhang, L.; Sanagapalli, S.; Stoita, A. Challenges in diagnosis of pancreatic cancer. World J. Gastroenterol. 2018, 24, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Viqueira, B.; Jimeno, A.; Cusatis, G.; Zhang, X.; Iacobuzio-Donahue, C.; Karikari, C.; Shi, C.; Danenberg, K.; Danenberg, P.V.; Kuramochi, H.; et al. An In vivo Platform for Translational Drug Development in Pancreatic Cancer. Clin. Cancer Res. 2006, 12, 4652–4661. [Google Scholar] [CrossRef]
- Kim, M.P.; Evans, D.B.; Wang, H.; Abbruzzese, J.L.; Fleming, J.B.; E Gallick, G. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat. Protoc. 2009, 4, 1670–1680. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Ishizawa, K.; Rasheed, Z.A.; Karisch, R.; Wang, Q.; Kowalski, J.; Susky, E.; Pereira, K.; Karamboulas, C.; Moghal, N.; Rajeshkumar, N.; et al. Tumor-Initiating Cells Are Rare in Many Human Tumors. Cell Stem Cell 2010, 7, 279–282. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; Brink, S.V.D.; Schumacher, T.; Voest, E.E. Tumor organoid–T-cell coculture systems. Nat. Protoc. 2020, 15, 15–39. [Google Scholar] [CrossRef]
- Dijkstra, K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; Van De Haar, J.; Fanchi, L.F.; Slagter, M.; Van Der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef]
- De Miranda, N.F.C.C.; Trajanoski, Z. Advancing cancer immunotherapy: A vision for the field. Genome Med. 2019, 11, 1–3. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.; Cuevas, C.; Chang, A.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.; Nywening, T.M.; Hawkins, T.M.N.W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Nywening, T.M.; A Belt, B.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; E Sanford, D.; Jacobs, R.C.; Ye, J.; A Patel, A.; E Gillanders, W.; et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef]
- Mitchem, J.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Bulk, J.V.D.; Verdegaal, E.M.E.; Ruano, D.; Ijsselsteijn, M.E.; Visser, M.; Van Der Breggen, R.; Duhen, T.; Van Der Ploeg, M.; De Vries, N.L.; Oosting, J.; et al. Neoantigen-specific immunity in low mutation burden colorectal cancers of the consensus molecular subtype 4. Genome Med. 2019, 11, 1–15. [Google Scholar] [CrossRef]
- Bobisse, S.; Genolet, R.; Roberti, A.; Tanyi, J.L.; Racle, J.; Stevenson, B.J.; Iseli, C.; Michel, A.; Le Bitoux, M.-A.; Guillaume, P.; et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8 + T cells in immunotherapy-naive ovarian cancer. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Ivey, G.; Li, Y.F.; El-Gamil, M.; Lalani, A.; et al. Unique Neoantigens Arise from Somatic Mutations in Patients with Gastrointestinal Cancers. Cancer Discov. 2019, 9, 1022–1035. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Tran, E.; Parkhurst, M.R.; Ilyas, S.; Pasetto, A.; Groh, E.M.; Robbins, P.F.; Yossef, R.; Garcia-Garijo, A.; Fajardo, C.A.; et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J. Clin. Investig. 2019, 129, 4992–5004. [Google Scholar] [CrossRef]
- Brouwer, T.P.; Vahrmeijer, A.L.; de Miranda, N.F.C.C. Immunotherapy for pancreatic cancer: Chasing the light at the end of the tunnel. Cell. Oncol. 2021, 44, 261–278. [Google Scholar] [CrossRef]
- Tabatabai, R.; Linhares, Y.; Bolos, D.; Mita, M.; Mita, A. Targeting the Wnt Pathway in Cancer: A Review of Novel Therapeutics. Target. Oncol. 2017, 12, 623–641. [Google Scholar] [CrossRef]
- Argentiero, A.; De Summa, S.; Di Fonte, R.; Iacobazzi, R.M.; Porcelli, L.; Da Vià, M.; Brunetti, O.; Azzariti, A.; Silvestris, N.; Solimando, A.G. Gene Expression Comparison between the Lymph Node-Positive and -Negative Reveals a Peculiar Immune Microenvironment Signature and a Theranostic Role for WNT Targeting in Pancreatic Ductal Adenocarcinoma: A Pilot Study. Cancers 2019, 11, 942. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nat. Cell Biol. 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- González, F.; Boue, S.; Belmonte, J.C.I. Methods for making induced pluripotent stem cells: Reprogramming à la carte. Nat. Rev. Genet. 2011, 12, 231–242. [Google Scholar] [CrossRef]
- Malik, N.; Rao, M.S. A Review of the Methods for Human iPSC Derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Shao, L.; Feng, W.; Sun, Y.; Bai, H.; Liu, J.; Currie, C.; Kim, J.; Gama, R.; Wang, Z.; Qian, Z.; et al. Generation of iPS cells using defined factors linked via the self-cleaving 2A sequences in a single open reading frame. Cell Res. 2009, 19, 296–306. [Google Scholar] [CrossRef][Green Version]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Nishimura, K.; Sano, M.; Ohtaka, M.; Furuta, B.; Umemura, Y.; Nakajima, Y.; Ikehara, Y.; Kobayashi, T.; Segawa, H.; Takayasu, S.; et al. Development of Defective and Persistent Sendai Virus Vector. J. Biol. Chem. 2011, 286, 4760–4771. [Google Scholar] [CrossRef]
- Woltjen, K.; Michael, I.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nat. Cell Biol. 2009, 458, 766–770. [Google Scholar] [CrossRef]
- Anokye-Danso, F.; Trivedi, C.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly Efficient miRNA-Mediated Reprogramming of Mouse and Human Somatic Cells to Pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.; Smith, Z.D.; Meissner, A.; et al. Highly Efficient Reprogramming to Pluripotency and Directed Differentiation of Human Cells with Synthetic Modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef]
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of Induced Pluripotent Stem Cells Using Recombinant Proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.-I.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of Mouse Induced Pluripotent Stem Cells Without Viral Vectors. science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.; Amoroso, M.W.; Oakley, D.; et al. Reference Maps of Human ES and iPS Cell Variation Enable High-Throughput Characterization of Pluripotent Cell Lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef]
- Mallon, B.S.; Hamilton, R.S.; Kozhich, O.A.; Johnson, K.R.; Fann, Y.C.; Rao, M.S.; Robey, P. Comparison of the molecular profiles of human embryonic and induced pluripotent stem cells of isogenic origin. Stem Cell Res. 2014, 12, 376–386. [Google Scholar] [CrossRef]
- Mallon, B.S.; Chenoweth, J.G.; Johnson, K.R.; Hamilton, R.S.; Tesar, P.J.; Yavatkar, A.S.; Tyson, L.J.; Park, K.; Chen, K.; Fann, Y.C.; et al. StemCellDB: The Human Pluripotent Stem Cell Database at the National Institutes of Health. Stem Cell Res. 2013, 10, 57–66. [Google Scholar] [CrossRef]
- Choi, J.; Lee, S.; Mallard, W.; Clement, K.; Tagliazucchi, G.M.; Lim, H.; Choi, I.Y.; Ferrari, F.; Tsankov, A.M.; Pop, R.; et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015, 33, 1173–1181. [Google Scholar] [CrossRef]
- Breunig, M.; Merkle, J.; Wagner, M.; Melzer, M.K.; Barth, T.F.; Engleitner, T.; Krumm, J.; Wiedenmann, S.; Cohrs, C.M.; Perkhofer, L.; et al. Modeling plasticity and dysplasia of pancreatic ductal organoids derived from human pluripotent stem cells. Cell Stem Cell 2021, 28, 1105–1124. [Google Scholar] [CrossRef] [PubMed]
- Calle, A.S.; Nair, N.; Oo, A.K.K.; Prieto-Vila, M.; Koga, M.; Khayrani, A.C.; Seno, M. A New PDAC Mouse Model Originated from iPSCs-Converted Pancreatic Cancer Stem Cells (CSCcm). Am. J. Cancer Res. 2016, 6, 2799–2815. [Google Scholar] [PubMed]
- Huang, L.; Desai, R.; Conrad, D.N.; Leite, N.C.; Akshinthala, D.; Lim, C.M.; Gonzalez, R.; Muthuswamy, L.B.; Gartner, Z.; Muthuswamy, S.K. Commitment and oncogene-induced plasticity of human stem cell-derived pancreatic acinar and ductal organoids. Cell Stem Cell 2021, 28, 1090–1104. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hoffman, J.P.; Alpaugh, R.K.; Rhim, A.D.; Reichert, M.; Stanger, B.Z.; Furth, E.E.; Sepulveda, A.R.; Yuan, C.-X.; Won, K.J.; et al. An iPSC Line from Human Pancreatic Ductal Adenocarcinoma Undergoes Early to Invasive Stages of Pancreatic Cancer Progression. Cell Rep. 2013, 3, 2088–2099. [Google Scholar] [CrossRef]
- Hassan, G.; Afify, S.M.; Nair, N.; Kumon, K.; Osman, A.; Du, J.; Mansour, H.; A Abu Quora, H.; Nawara, H.M.; Satoh, A.; et al. Hematopoietic Cells Derived from Cancer Stem Cells Generated from Mouse Induced Pluripotent Stem Cells. Cancers 2019, 12, 82. [Google Scholar] [CrossRef]
- Matsuda, S.; Yan, T.; Mizutani, A.; Sota, T.; Hiramoto, Y.; Prieto-Vila, M.; Chen, L.; Satoh, A.; Kudoh, T.; Kasai, T.; et al. Cancer stem cells maintain a hierarchy of differentiation by creating their niche. Int. J. Cancer 2014, 135, 27–36. [Google Scholar] [CrossRef]
- Nair, N.; Calle, A.S.; Zahra, M.H.; Prieto-Vila, M.; Oo, A.; Hurley, L.; Vaidyanath, A.; Seno, A.; Masuda, J.; Iwasaki, Y.; et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti–PD-1 therapy. Sci. Transl. Med. 2020, 12, 1–15. [Google Scholar] [CrossRef]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Kooreman, N.G.; Kim, Y.; de Almeida, P.E.; Termglinchan, V.; Diecke, S.; Shao, N.-Y.; Wei, T.-T.; Yi, H.; Dey, D.; Nelakanti, R.; et al. Autologous iPSC-Based Vaccines Elicit Anti-tumor Responses In Vivo. Cell Stem Cell 2018, 22, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, Z.; Du, P.; Chard, L.S.; Yan, W.; El Khouri, M.; Wang, Z.; Zhang, Z.; Chu, Y.; Gao, D.; et al. A Virus-Infected, Reprogrammed Somatic Cell–Derived Tumor Cell (VIReST) Vaccination Regime Can Prevent Initiation and Progression of Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 465–476. [Google Scholar] [CrossRef]
- Ouyang, X.; Liu, Y.; Zhou, Y.; Guo, J.; Wei, T.-T.; Liu, C.; Lee, B.; Chen, B.; Zhang, A.; Casey, K.M.; et al. Antitumor effects of iPSC-based cancer vaccine in pancreatic cancer. Stem Cell Rep. 2021, 16, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 1–5. [Google Scholar] [CrossRef]
- Chen, L.; Kasai, T.; Li, Y.; Sugii, Y.; Jin, G.; Okada, M.; Vaidyanath, A.; Mizutani, A.; Satoh, A.; Kudoh, T.; et al. A Model of Cancer Stem Cells Derived from Mouse Induced Pluripotent Stem Cells. PLoS ONE 2012, 7, e33544. [Google Scholar] [CrossRef]
- Bailey, P.; Initiative, A.P.C.G.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nat. Cell Biol. 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Biankin, A.V.; Initiative, A.P.C.G.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nat. Cell Biol. 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.C.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.C.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, e6744. [Google Scholar] [CrossRef]
- Tu, Q.; Hao, J.; Zhou, X.; Yan, L.; Dai, H.; Sun, B.; Yang, D.; An, S.; Lv, L.; Jiao, B.; et al. CDKN2B deletion is essential for pancreatic cancer development instead of unmeaningful co-deletion due to juxtaposition to CDKN2A. Oncogene 2017, 37, 128–138. [Google Scholar] [CrossRef]
- Waddell, N.; Initiative, A.P.C.G.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nat. Cell Biol. 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, M.C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell– and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Hohwieler, M.; Illing, A.; Hermann, P.C.; Mayer, T.; Stockmann, M.; Perkhofer, L.; Eiseler, T.; Antony, J.S.; Müller, M.; Renz, S.; et al. Human pluripotent stem cell-derived acinar/ductal organoids generate human pancreas upon orthotopic transplantation and allow disease modelling. Gut 2016, 66, 473–486. [Google Scholar] [CrossRef]
- Ferreira, R.; Sancho, R.; Messal, H.A.; Nye, E.; Spencer-Dene, B.; Stone, R.K.; Stamp, G.; Rosewell, I.; Quaglia, A.; Behrens, A. Duct- and Acinar-Derived Pancreatic Ductal Adenocarcinomas Show Distinct Tumor Progression and Marker Expression. Cell Rep. 2017, 21, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nat. Cell Biol. 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Lee, C.; Kozaki, T.; Ginhoux, F. Studying tissue macrophages in vitro: Are iPSC-derived cells the answer? Nat. Rev. Immunol. 2018, 18, 716–725. [Google Scholar] [CrossRef]
- Gutbier, S.; Wanke, F.; Dahm, N.; Rümmelin, A.; Zimmermann, S.; Christensen, K.; Köchl, F.; Rautanen, A.; Hatje, K.; Geering, B.; et al. Large-Scale Production of Human iPSC-Derived Macrophages for Drug Screening. Int. J. Mol. Sci. 2020, 21, 4808. [Google Scholar] [CrossRef] [PubMed]
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 1–8. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, N.M.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Sunami, Y.; Böker, V.; Kleeff, J. Targeting and Reprograming Cancer-Associated Fibroblasts and the Tumor Microenvironment in Pancreatic Cancer. Cancers 2021, 13, 697. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Yan, T.; Calle, A.S.; Nair, N.; Hurley, L.; Kasai, T.; Seno, M. iPSC-Derived Cancer Stem Cells Provide a Model of Tumor Vasculature. Am. J. Cancer Res. 2016, 6, 1906–1921. [Google Scholar]
- Brewer, B.G.; Mitchell, R.A.; Harandi, A.; Eaton, J.W. Embryonic vaccines against cancer: An early history. Exp. Mol. Pathol. 2009, 86, 192–197. [Google Scholar] [CrossRef]
- Ghosh, Z.; Huang, M.; Hu, S.; Wilson, K.D.; Dey, D.; Wu, J.C. Dissecting the Oncogenic and Tumorigenic Potential of Differentiated Human Induced Pluripotent Stem Cells and Human Embryonic Stem Cells. Cancer Res. 2011, 71, 5030–5039. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; A Weinberg, R. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Zhao, T.; Zhang, Z.-N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nat. Cell Biol. 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, P.E.; Meyer, E.H.; Kooreman, N.G.; Diecke, S.; Dey, D.; Sanchez-Freire, V.; Hu, S.; Ebert, A.D.; I Odegaard, J.; Mordwinkin, N.M.; et al. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat. Commun. 2014, 5, 3903. [Google Scholar] [CrossRef] [PubMed]
- de Jesus, B.B.; Neves, B.M.; Ferreira, M.; Nóbrega-Pereira, S. Strategies for Cancer Immunotherapy Using Induced Pluripoten-cy Stem Cells-Based Vaccines. Cancers 2020, 12, 3581. [Google Scholar] [CrossRef] [PubMed]
- Gąbka-Buszek, A.; Kwiatkowska-Borowczyk, E.; Jankowski, J.; Kozłowska, A.K.; Mackiewicz, A. Novel Genetic Melanoma Vaccines Based on Induced Pluripotent Stem Cells or Melanosphere-Derived Stem-Like Cells Display High Efficacy in a murine Tumor Rejection Model. Vaccines 2020, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shao, L.; Wu, L.; Ma, W.; Zheng, Y.; Hu, C.; Li, F. Expression levels of a gene signature in hiPSC associated with lung adenocarcinoma stem cells and its capability in eliciting specific antitumor immune-response in a humanized mice model. Thorac. Cancer 2020, 11, 1603–1612. [Google Scholar] [CrossRef]
- Li, Y.; Zeng, H.; Xu, R.-H.; Liu, B.; Li, Z. Vaccination with Human Pluripotent Stem Cells Generates a Broad Spectrum of Immunological and Clinical Response against Colon Cancer. Stem Cells 2009, 27, 3103–3111. [Google Scholar] [CrossRef]
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013, 19, 998–1004. [Google Scholar] [CrossRef]
- Kooreman, N.G.; Wu, J.C. Tumorigenicity of pluripotent stem cells: Biological insights from molecular imaging. J. R. Soc. Interface 2010, 7, S753–S763. [Google Scholar] [CrossRef]
- Koyanagi-Aoi, M.; Ohnuki, M.; Takahashi, K.; Okita, K.; Noma, H.; Sawamura, Y.; Teramoto, I.; Narita, M.; Sato, Y.; Ichisaka, T.; et al. Differentiation-defective phenotypes revealed by large-scale analyses of human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 20569–20574. [Google Scholar] [CrossRef]
- Kawamura, A.; Miyagawa, S.; Fukushima, S.; Kawamura, T.; Kashiyama, N.; Ito, E.; Watabe, T.; Masuda, S.; Toda, K.; Hatazawa, J.; et al. Teratocarcinomas Arising from Allogeneic Induced Pluripotent Stem Cell-Derived Cardiac Tissue Constructs Provoked Host Immune Rejection in Mice. Sci. Rep. 2016, 6, 19464. [Google Scholar] [CrossRef]
- Hochedlinger, K.; Yamada, Y.; Beard, C.; Jaenisch, R. Ectopic Expression of Oct-4 Blocks Progenitor-Cell Differentiation and Causes Dysplasia in Epithelial Tissues. Cell 2005, 121, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, G.-R.; Hu, P.; Xi, Z.; Guang-Rong, H.; Pin, H. Over-expression of Oct4 in human esophageal squamous cell carcinoma. Mol. Cells 2011, 32, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Suvà, M.-L.; De Vito, C.; Provero, P.; Stehle, J.-C.; Baumer, K.; Cironi, L.; Janiszewska, M.; Petricevic, T.; Suvà, D.; et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010, 24, 916–932. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.-Q.; Misra, Y.; Li, D.-B.; Wadsworth, M.P.; Krag, D.; Weaver, D.; Tessitore, J.; Zhang, G.; Tian, Q.; Buss, K. Differential expression of Oct3/4 in human breast cancer and normal tissues. Int. J. Oncol. 2018, 52, 2069–2078. [Google Scholar] [CrossRef]
- Bae, K.-M.; Su, Z.; Frye, C.; McClellan, S.; Allan, R.W.; Andrejewski, J.T.; Kelley, V.; Jorgensen, M.; Steindler, D.A.; Vieweg, J.; et al. Expression of Pluripotent Stem Cell Reprogramming Factors by Prostate Tumor Initiating Cells. J. Urol. 2010, 183, 2045–2053. [Google Scholar] [CrossRef]
- Sarkar, A.; Hochedlinger, K. The Sox Family of Transcription Factors: Versatile Regulators of Stem and Progenitor Cell Fate. Cell Stem Cell 2013, 12, 15–30. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Clemente-Periván, S.I.; Gómez-Gómez, Y.; Leyva-Vázquez, M.A.; Lagunas-Martínez, A.; Organista-Nava, J.; Illades-Aguiar, B. Role of Oct3/4 in Cervical Cancer Tumorigenesis. Front. Oncol. 2020, 10, 247. [Google Scholar] [CrossRef]
- Themeli, M.; Rivière, I.; Sadelain, M. New Cell Sources for T Cell Engineering and Adoptive Immunotherapy. Cell Stem Cell 2015, 16, 357–366. [Google Scholar] [CrossRef]
- Leone, P.; Shin, E.-C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC Class I Antigen Processing and Presenting Machinery: Organization, Function, and Defects in Tumor Cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.-P.; Kros, J.M.; Li, J. Approved CAR T cell therapies: Ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov. Today 2018, 23, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Kim, C.H. Evolution of chimeric antigen receptor (CAR) T cell therapy: Current status and future perspectives. Arch. Pharmacal Res. 2019, 42, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, e1109. [Google Scholar] [CrossRef]
- Senju, S.; Koba, C.; Haruta, M.; Matsunaga, Y.; Matsumura, K.; Haga, E.; Sasaki, Y.; Ikeda, T.; Takamatsu, K.; Nishimura, Y. Application of iPS cell-derived macrophages to cancer therapy. OncoImmunology 2014, 3, e27927. [Google Scholar] [CrossRef][Green Version]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef]
- Nishizawa, M.; Chonabayashi, K.; Nomura, M.; Tanaka, A.; Nakamura, M.; Inagaki, A.; Nishikawa, M.; Takei, I.; Oishi, A.; Tanabe, K.; et al. Epigenetic Variation between Human Induced Pluripotent Stem Cell Lines Is an Indicator of Differentiation Capacity. Cell Stem Cell 2016, 19, 341–354. [Google Scholar] [CrossRef]
- Newman, A.M.; Cooper, J.B. Lab-Specific Gene Expression Signatures in Pluripotent Stem Cells. Cell Stem Cell 2010, 7, 258–262. [Google Scholar] [CrossRef]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- The International Stem Cell Initiative Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [CrossRef]
- Rouhani, F.J.; Nik-Zainal, S.; Wuster, A.; Li, Y.; Conte, N.; Koike-Yusa, H.; Kumasaka, N.; Vallier, L.; Yusa, K.; Bradley, A. Mutational History of a Human Cell Lineage from Somatic to Induced Pluripotent Stem Cells. PLoS Genet. 2016, 12, e1005932. [Google Scholar] [CrossRef] [PubMed]
- Sougawa, N.; Miyagawa, S.; Fukushima, S.; Kawamura, A.; Yokoyama, J.; Ito, E.; Harada, A.; Okimoto, K.; Mochizuki-Oda, N.; Saito, A.; et al. Immunologic targeting of CD30 eliminates tumourigenic human pluripotent stem cells, allowing safer clinical application of hiPSC-based cell therapy. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, R.; Ishikawa, Y.; Katayama, T.; Quantock, A.J.; Nishida, K. CD200 facilitates the isolation of corneal epithelial cells derived from human pluripotent stem cells. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nat. Cell Biol. 2017, 548, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Nudel, N.; Benvenisty, N. Immunologic and chemical targeting of the tight-junction protein Claudin-6 eliminates tumorigenic human pluripotent stem cells. Nat. Commun. 2013, 4, e1992. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Gan, Q.-F.; Golan-Lev, T.; Arora, P.; Yanuka, O.; Oren, Y.S.; Leikin-Frenkel, A.; Graf, M.; Garippa, R.; Boehringer, M.; et al. Selective Elimination of Human Pluripotent Stem Cells by an Oleate Synthesis Inhibitor Discovered in a High-Throughput Screen. Cell Stem Cell 2013, 12, 167–179. [Google Scholar] [CrossRef]
- Lee, M.-O.; Moon, S.H.; Jeong, H.-C.; Yi, J.-Y.; Lee, T.-H.; Shim, S.H.; Rhee, Y.-H.; Lee, S.-H.; Oh, S.-J.; Han, M.-J.; et al. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, 3281–3290. [Google Scholar] [CrossRef]
- Richards, M.; Phoon, C.W.; Goh, G.T.W.; Seng, E.K.; Guo, X.M.; Tan, C.M.F.; Chan, W.-K.; Lee, J.M.K. A New Class of Pluripotent Stem Cell Cytotoxic Small Molecules. PLoS ONE 2014, 9, e85039. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Miyoshi, H.; Nagoshi, N.; Kohyama, J.; Itakura, G.; Kawabata, S.; Ozaki, M.; Iida, T.; Sugai, K.; Ito, S.; et al. Selective Ablation of Tumorigenic Cells Following Human Induced Pluripotent Stem Cell-Derived Neural Stem/Progenitor Cell Transplantation in Spinal Cord Injury. Stem Cells Transl. Med. 2019, 8, 260–270. [Google Scholar] [CrossRef]
- Miyagawa, S.; Fukushima, S.; Imanishi, Y.; Kawamura, T.; Oda, N.M.-; Masuda, S.; Sawa, Y. Building A New Treatment For Heart Failure-Transplantation of Induced Pluripotent Stem Cell-derived Cells into the Heart. Curr. Med. Chem. 2016, 16, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Horton, B.L.; Morgan, D.M.; Momin, N.; Zagorulya, M.; Torres-Mejia, E.; Bhandarkar, V.; Wittrup, K.D.; Love, J.C.; Spranger, S. Lack of CD8 + T cell effector differentiation during priming mediates checkpoint blockade resistance in non–small cell lung cancer. Sci. Immunol. 2021, 6, e8800. [Google Scholar] [CrossRef] [PubMed]
- Connolly, K.A.; Kuchroo, M.; Venkat, A.; Khatun, A.; Wang, J.; William, I.; Hornick, N.I.; Fitzgerald, B.L.; Damo, M.; Kasmani, M.Y.; et al. A reservoir of stem-like CD8 + T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci. Immunol. 2021, 6, e7836. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Pros + | Cons − | |||
|---|---|---|---|---|
| Models [55,56,57,58,59,60,61] | + | Insight into disease onset and development | − | Risk of transcriptional and genetic alterations during iPSC induction |
| + | Reliable cell source for multicellular models | − | Heterogeneity within iPSC populations | |
| Immunotherapeutic approaches [62,63,64,65,66,67,68,69,70] (ClincalTrials.gov, Identifier: NCT03841110 jrct.niph.go.jp, Trial ID: jRCT2033200116) | + | iPSCs are immunogenic and express tumor-associated antigens | − | Oncogenic potential of iPSCs reprogramming factors |
| + | Reliable cell source for off-the-shelf cellular therapies | − | Risk of transcriptional and genetic alterations during iPSC induction | |
| + | Large-scale cell production | − | Heterogeneity within iPSC populations | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krog, R.T.; de Miranda, N.F.C.C.; Vahrmeijer, A.L.; Kooreman, N.G. The Potential of Induced Pluripotent Stem Cells to Advance the Treatment of Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 5789. https://doi.org/10.3390/cancers13225789
Krog RT, de Miranda NFCC, Vahrmeijer AL, Kooreman NG. The Potential of Induced Pluripotent Stem Cells to Advance the Treatment of Pancreatic Ductal Adenocarcinoma. Cancers. 2021; 13(22):5789. https://doi.org/10.3390/cancers13225789
Chicago/Turabian StyleKrog, Ricki T., Noel F. C. C. de Miranda, Alexander L. Vahrmeijer, and Nigel G. Kooreman. 2021. "The Potential of Induced Pluripotent Stem Cells to Advance the Treatment of Pancreatic Ductal Adenocarcinoma" Cancers 13, no. 22: 5789. https://doi.org/10.3390/cancers13225789
APA StyleKrog, R. T., de Miranda, N. F. C. C., Vahrmeijer, A. L., & Kooreman, N. G. (2021). The Potential of Induced Pluripotent Stem Cells to Advance the Treatment of Pancreatic Ductal Adenocarcinoma. Cancers, 13(22), 5789. https://doi.org/10.3390/cancers13225789