
Single-Cell RNA-Seq Analysis Reveals the Acquisition of Cancer Stem Cell Traits and Increase of Cell–Cell Signaling during EMT Progression



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Preprocessing of Single-Cell Datasets
2.2. Clustering and Trajectory Inference with QuanTC
2.3. E-M Scoring with AUCell
2.4. Calculation of Transcriptional Diversity and Developmental Potential
2.5. Quantification of EMT Circuit Energy
2.6. Analysis of Cell–Cell Communication
2.7. Data Availability
3. Results
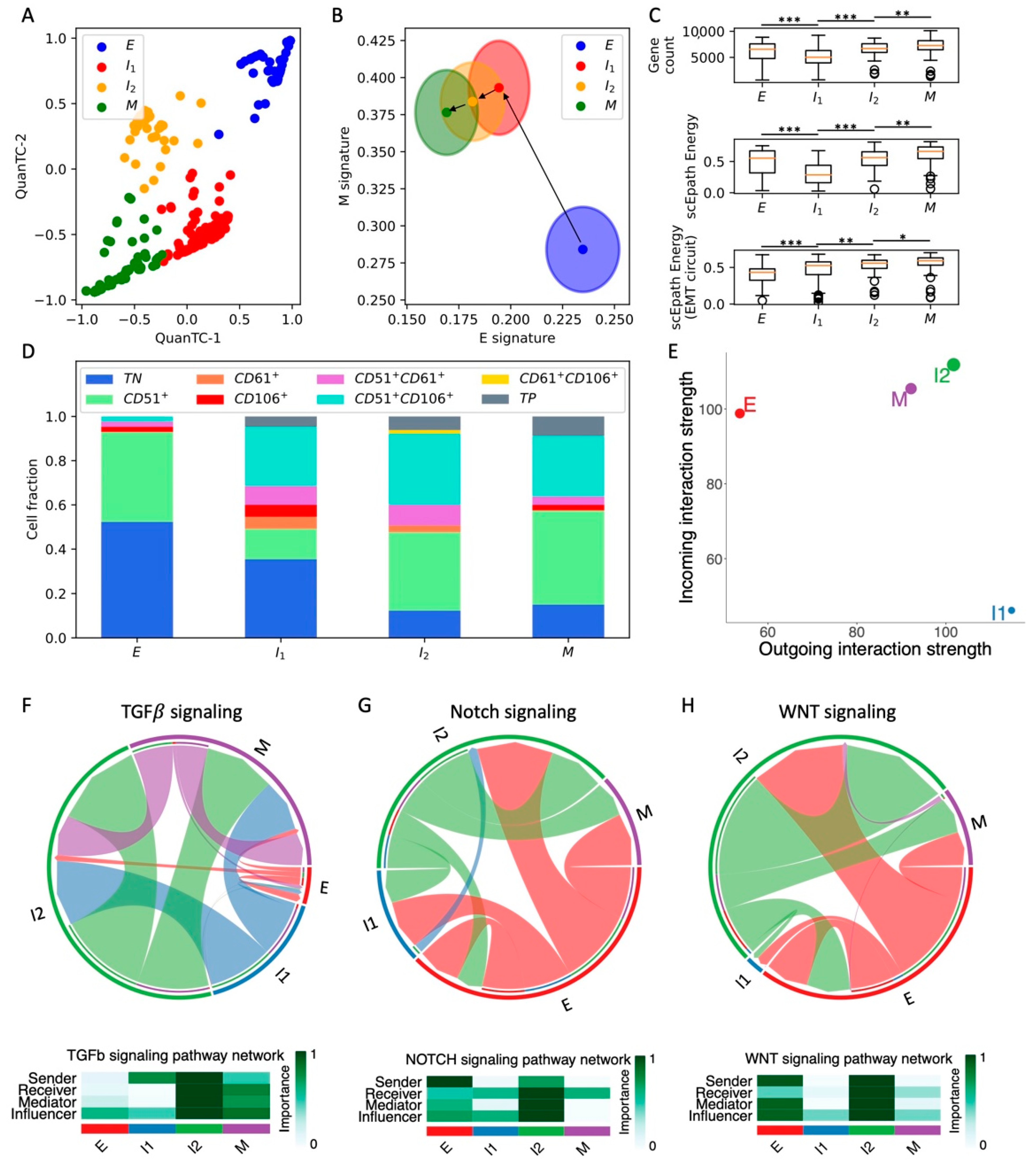
3.1. Plasticity of Intermediate EMT States from In Vivo Squamous Cell Carcinoma CTCs
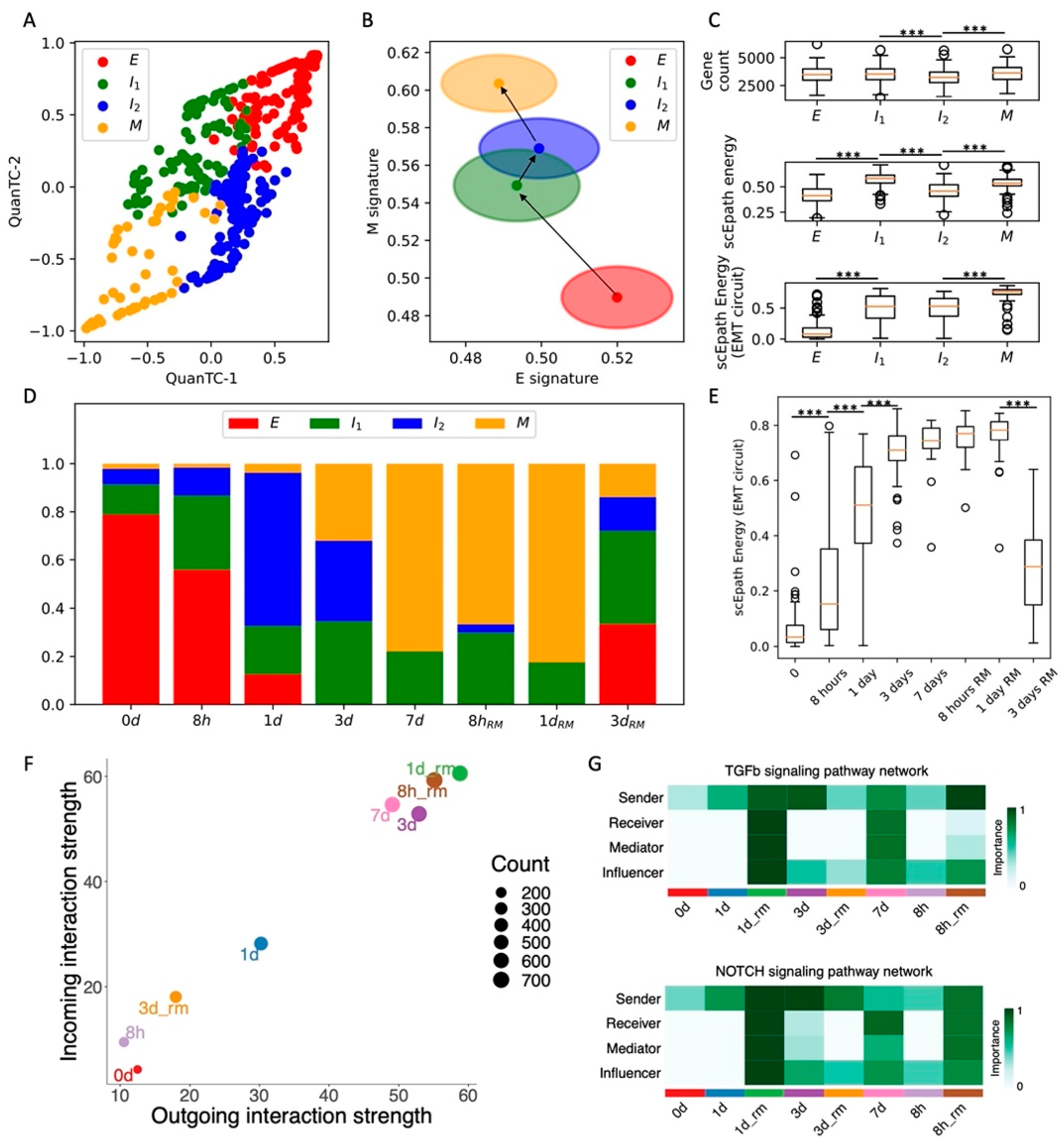
3.2. EMT Time Course of an Ovarian Cancer Cell Line Highlights Transient Activation of EMT Plasticity and Cell–Cell Signaling
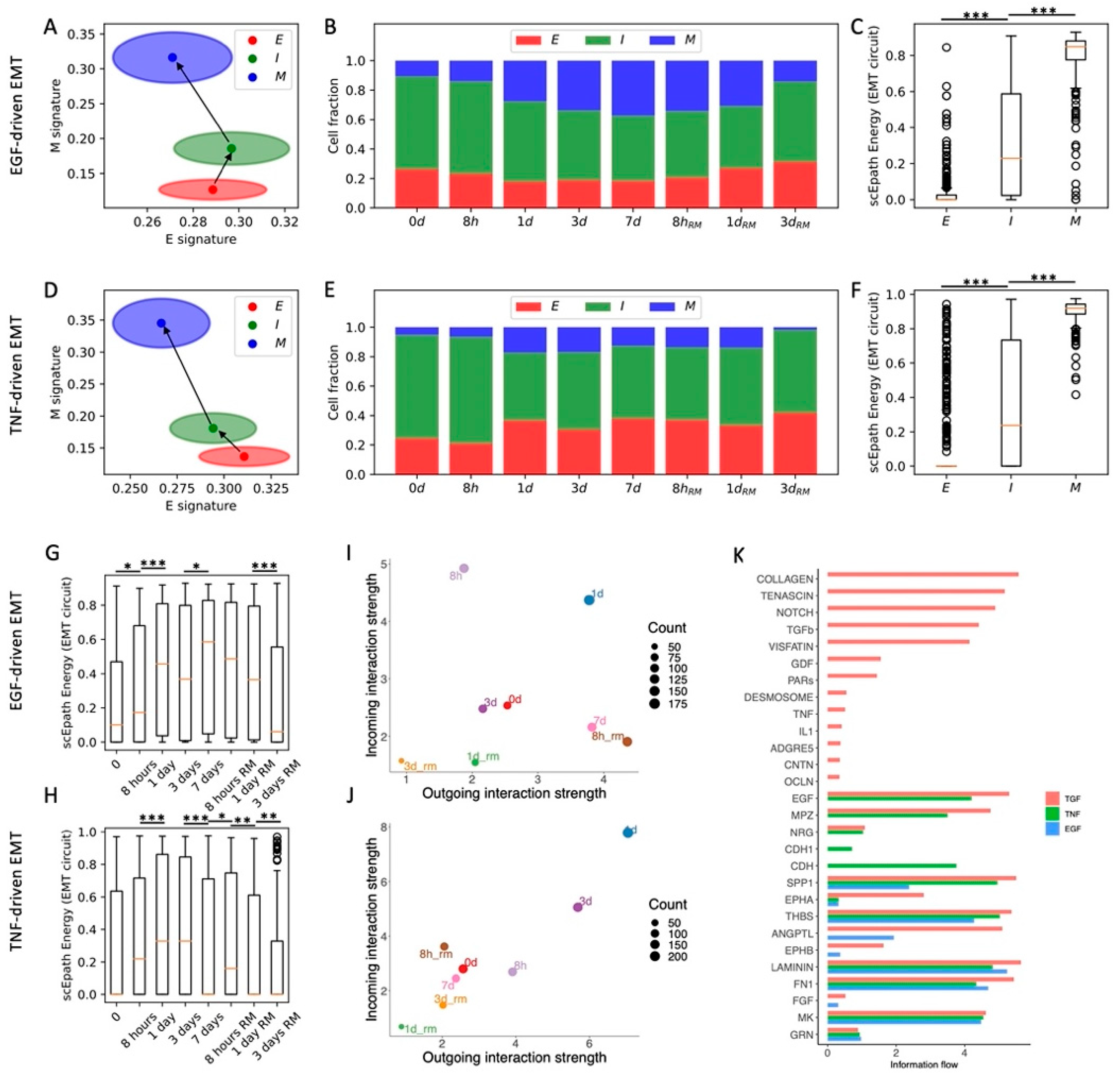
3.3. Comparison of Different EMT-Inducing Signals in OVCA420 Cells Confirms the Relationship between EMT Progression and Activation of Cell–Cell Signaling
3.4. Space-Dependent EMT Phenotype Fractions in MCF10A Cells
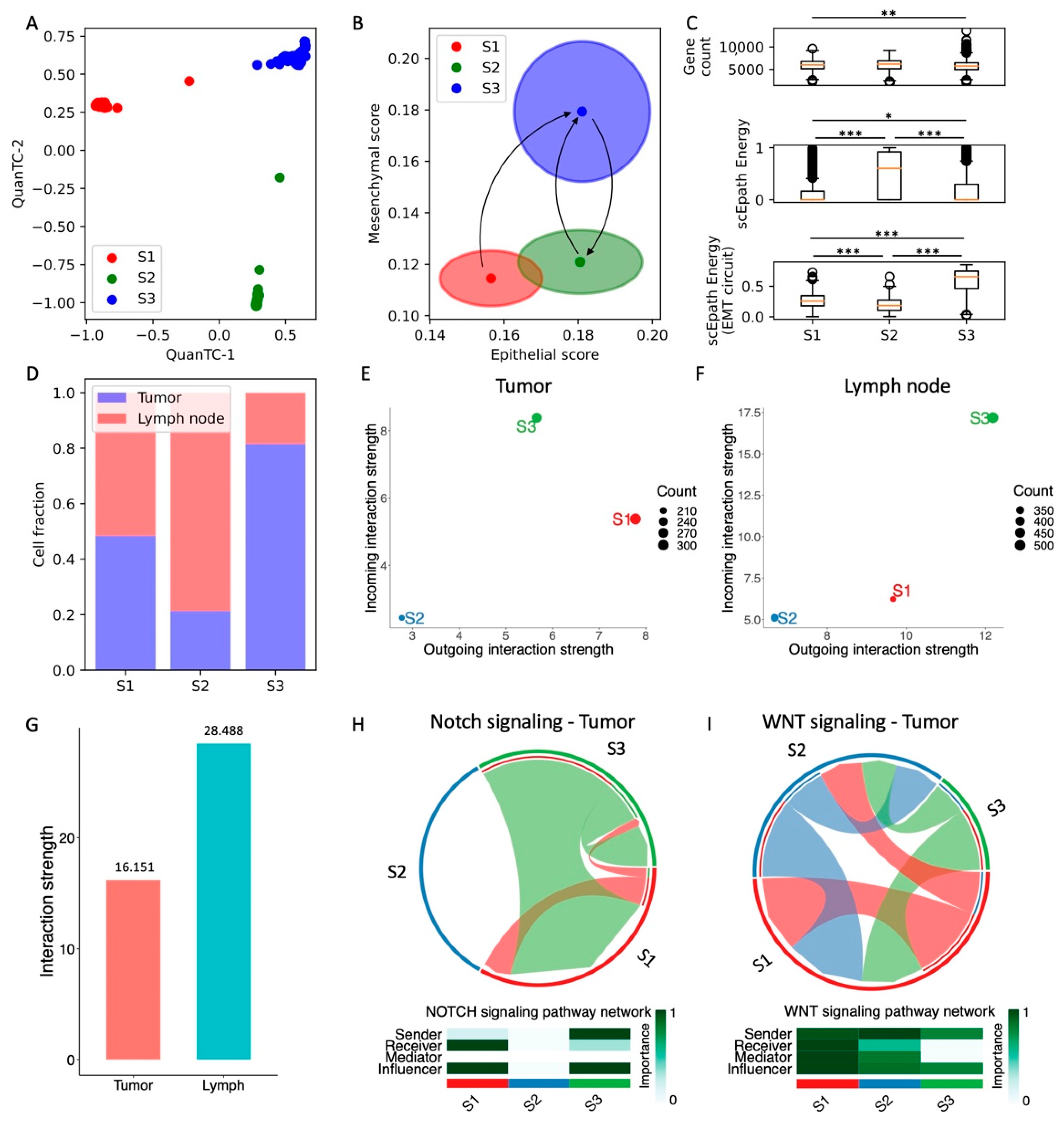
3.5. Simultaneous Acquisition of Epithelial and Mesenchymal Traits in Head and Neck SCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jia, D.; Li, X.; Bocci, F.; Tripathi, S.; Deng, Y.; Jolly, M.K.; Onuchic, J.N.; Levine, H. Quantifying cancer epithelial-mesenchymal plasticity and its association with stemness and immune response. J. Clin. Med. 2019, 8, 725. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Sha, Y.; Haensel, D.; Gutierrez, G.; Du, H.; Dai, X.; Nie, Q. Intermediate cell states in epithelial-to-mesenchymal transition. Phys. Biol. 2018, 16, 021001. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2018, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef] [Green Version]
- Fabisiewicz, A.; Grzybowska, E.; Grybowska, E. CTC clusters in cancer progression and metastasis. Med. Oncol. 2017, 34, 12. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.P.; Vanderhyden, B.C. Context specificity of the EMT transcriptional response. Nat. Commun. 2020, 11, 2142. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.; Wang, S.; Zhou, P.; Nie, Q. Inference and multiscale model of epithelial-to-mesenchymal transition via single-cell transcriptomic data. Nucleic Acids Res. 2020, 48, 9505–9520. [Google Scholar] [CrossRef]
- Sha, Y.; Wang, S.; Bocci, F.; Zhou, P.; Nie, Q. Inference of Intercellular Communications and Multilayer Gene-Regulations of Epithelial–Mesenchymal Transition From Single-Cell Transcriptomic Data. Front. Genet. 2021, 11, 604585. [Google Scholar] [CrossRef] [PubMed]
- Foroutan, M.; Cursons, J.; Hediyeh-Zadeh, S.; Thompson, E.W.; Davis, M.J. A transcriptional program for detecting TGFβ-induced EMT in Cancer. Mol. Cancer Res. 2017, 15, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Celià-Terrassa, T.; Jolly, M.K. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition in Cancer Metastasis. Cold Spring Harb. Perspect. Med. 2019, 10, a036905. [Google Scholar] [CrossRef]
- Tanabe, S.; Quader, S.; Cabral, H.; Ono, R. Interplay of EMT and CSC in Cancer and the Potential Therapeutic Strategies. Front. Pharmacol. 2020, 11, 904. [Google Scholar] [CrossRef]
- Thankamony, A.P.; Saxena, K.; Murali, R.; Jolly, M.K.; Nair, R. Cancer Stem Cell Plasticity—A Deadly Deal. Front. Mol. Biosci. 2020, 7, 79. [Google Scholar] [CrossRef]
- Gulati, G.S.; Sikandar, S.S.; Wesche, D.J.; Manjunath, A.; Bharadwaj, A.; Berger, M.J.; Ilagan, F.; Kuo, A.H.; Hsieh, R.W.; Cai, S.; et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 2020, 367, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; MacLean, A.L.; Peng, T.; Nie, Q. scEpath: Energy landscape-based inference of transition probabilities and cellular trajectories from single-cell transcriptomic data. Bioinformatics 2018, 34, 2077–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Onuchic, J.N.; Jolly, M.K. Understanding the principles of pattern formation driven by Notch signaling by integrating experiments and theoretical models. Front. Physiol. 2020, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2016, 36, 1461–1473. [Google Scholar] [CrossRef]
- Costanza, B.; Umelo, I.; Bellier, J.; Castronovo, V.; Turtoi, A. Stromal Modulators of TGF-β in Cancer. J. Clin. Med. 2017, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Almet, A.A.; Cang, Z.; Jin, S.; Nie, Q. The landscape of cell-cell communication through single-cell transcriptomics. Curr. Opin. Syst. Biol. 2021, 26, 12–23. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, V.Y.; Kirschner, K.; Schaub, M.T.; Andrews, T.; Yiu, A.; Chandra, T.; Natarajan, K.N.; Reik, W.; Barahona, M.; Green, A.R.; et al. SC3—Consensus clustering of single-cell RNA-Seq data. Nat. Methods 2017, 14, 483–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-Cell Regulatory Network Inference And Clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- McFaline-Figueroa, J.L.; Hill, A.J.; Qiu, X.; Jackson, D.; Shendure, J.; Trapnell, C. A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nat. Genet. 2019, 51, pages1389–1398. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef] [Green Version]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Kristy Horan, J.; Cursons, O.; Davis, M.J. Single sample scoring of molecular phenotypes. BMC Bioinform. 2018, 19, 404. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Masiero, M.; Banham, A.H.; Harris, A.L. The notch ligand JAGGED1 as a target for anti-tumor therapy. Front. Oncol. 2014, 4, 254. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Jolly, M.K.; George, J.T.; Levine, H.; Onuchic, J.N. A mechanism-based computational model to capture the interconnections among epithelial-mesenchymal transition, cancer stem cells and Notch-Jagged signaling. Oncotarget 2018, 9, 29906–29920. [Google Scholar] [CrossRef] [Green Version]
- Celià-Terrassa, T.; Bastian, C.; Liu, D.D.; Ell, B.; Aiello, N.M.; Wei, Y.; Zamalloa, J.; Blanco, A.M.; Hang, X.; Kunisky, D.; et al. Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat. Commun. 2018, 9, 5005. [Google Scholar] [CrossRef] [PubMed]
- Pasello, M.; Manara, M.C.; Scotlandi, K. CD99 at the crossroads of physiology and pathology. J. Cell Commun. Signal. 2018, 12, 55–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arechavaleta-Velasco, F.; Perez-Juarez, C.E.; Gerton, G.L.; Diaz-Cueto, L. Progranulin and Its Biological Effects in Cancer. Med. Oncol. 2018, 34, 194. [Google Scholar] [CrossRef]
- Liang, L.-Y.; Patel, O.; Janes, P.W.; Murphy, J.M.; Lucet, I.S. Eph receptor signalling: From catalytic to non-catalytic functions. Oncogene 2019, 38, 6567–6584. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Tripathi, S.C.; Mercedes, S.V.; George, J.T.; Casabar, J.; Wong, P.K.; Hanash, S.; Levine, H.; Onuchic, J.N.; Jolly, M.K. NRF2 activates a partial Epithelial-Mesenchymal Transition and is maximally present in a hybrid Epithelial/Mesenchymal phenotype. Integr. Biol. 2019, 11, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Mercedes, S.A.V.; Bocci, F.; Zhu, N.; Levine, H.; Onuchic, J.N.; Jolly, M.K.; Wong, P.K. Nrf2 regulates collective cancer migration by modulating the hybrid epithelial/mesenchymal phenotype. bioRxiv 2021, 100, 440858. [Google Scholar] [CrossRef]
- Font-Clos, F.; Zapperi, S.; La Porta, C.A.M. Classification of triple-negative breast cancers through a Boolean network model of the epithelial-mesenchymal transition. Cell Syst. 2021, 12, 457–462. [Google Scholar] [CrossRef]
- May, C.D.; Sphyris, N.; Evans, K.W.; Werden, S.J.; Guo, W.; Mani, S.A. Epithelial-mesenchymal transition and cancer stem cells: A dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011, 13, 202. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Deciphering the Dynamics of Epithelial-Mesenchymal Transition and Cancer Stem Cells in Tumor Progression. Curr. Stem Cell Rep. 2019, 5, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Youssef, G.; Gammon, L.; Ambler, L.; Wicker, B.; Patel, S.; Cottom, H.; Piper, K.; Mackenzie, I.C.; Philpott, M.P.; Biddle, A. Disseminating cells in human tumours acquire an EMT stem cell state that is predictive of metastasis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; Den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef] [PubMed]
- Simeonov, K.P.; Byrns, C.N.; Clark, M.L.; Norgard, R.J.; Martin, B.; Stanger, B.Z.; Shendure, J.; McKenna, A.; Lengner, C.J. Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell 2021, 39, 1150–1162. [Google Scholar] [CrossRef]
- Nam, A.S.; Chaligne, R.; Landau, D.A. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat. Rev. Genet. 2020, 22, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sfakianos, J.P.; Beaumont, K.G.; Akturk, G.; Horowitz, A.; Sebra, R.P.; Farkas, A.M.; Gnjatic, S.; Hake, A.; Izadmehr, S.; et al. Myeloid Cell–associated Resistance to PD-1/PD-L1 Blockade in Urothelial Cancer Revealed Through Bulk and Single-cell RNA Sequencing. Clin. Cancer Res. 2021, 27, 4287–4300. [Google Scholar] [CrossRef]
- Colacino, J.A.; Azizi, E.; Brooks, M.D.; Harouaka, R.; Fouladdel, S.; McDermott, S.P.; Lee, M.; Hill, D.; Madden, J.; Boerner, J.; et al. Heterogeneity of Human Breast Stem and Progenitor Cells as Revealed by Transcriptional Profiling. Stem Cell Rep. 2018, 10, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.; Blick, T.; Thompson, E.W.; Williams, E.D. Diversity of Epithelial-Mesenchymal Phenotypes in Circulating Tumour Cells from Prostate Cancer Patient-Derived Xenograft Models. Cancers 2021, 13, 2750. [Google Scholar] [CrossRef]
- Bocci, F.; Mandal, S.; Tejaswi, T.; Jolly, M.K. Investigating epithelial-mesenchymal heterogeneity of tumors and circulating tumor cells with transcriptomic analysis and biophysical modeling. Comput. Syst. Oncol. 2021, 1, e1015. [Google Scholar] [CrossRef]
- Burgess, D.J. Spatial transcriptomics coming of age. Nat. Rev. Genet. 2019, 20, 317. [Google Scholar] [CrossRef]
- Bocci, F.; Gaerhart-Serna, L.; Ribeiro, M.; Boareto, M.; Ben-Jacob, E.; Devi, G.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Towards understaning Cancer Stem Cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, Z.; Wu, Q.; Li, C.; Li, J.; Zhang, Y.; Wang, C.; Sun, S.; Sun, S. DNER promotes epithelial–mesenchymal transition and prevents chemosensitivity through the Wnt/β-catenin pathway in breast cancer. Cell Death Dis. 2020, 11, 642. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bocci, F.; Zhou, P.; Nie, Q. Single-Cell RNA-Seq Analysis Reveals the Acquisition of Cancer Stem Cell Traits and Increase of Cell–Cell Signaling during EMT Progression. Cancers 2021, 13, 5726. https://doi.org/10.3390/cancers13225726
Bocci F, Zhou P, Nie Q. Single-Cell RNA-Seq Analysis Reveals the Acquisition of Cancer Stem Cell Traits and Increase of Cell–Cell Signaling during EMT Progression. Cancers. 2021; 13(22):5726. https://doi.org/10.3390/cancers13225726
Chicago/Turabian StyleBocci, Federico, Peijie Zhou, and Qing Nie. 2021. "Single-Cell RNA-Seq Analysis Reveals the Acquisition of Cancer Stem Cell Traits and Increase of Cell–Cell Signaling during EMT Progression" Cancers 13, no. 22: 5726. https://doi.org/10.3390/cancers13225726
APA StyleBocci, F., Zhou, P., & Nie, Q. (2021). Single-Cell RNA-Seq Analysis Reveals the Acquisition of Cancer Stem Cell Traits and Increase of Cell–Cell Signaling during EMT Progression. Cancers, 13(22), 5726. https://doi.org/10.3390/cancers13225726