
Understanding the Role of Autophagy in Cancer Formation and Progression Is a Real Opportunity to Treat and Cure Human Cancers

 ,
, 

 , ,
, ,  ,
,  ,
,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
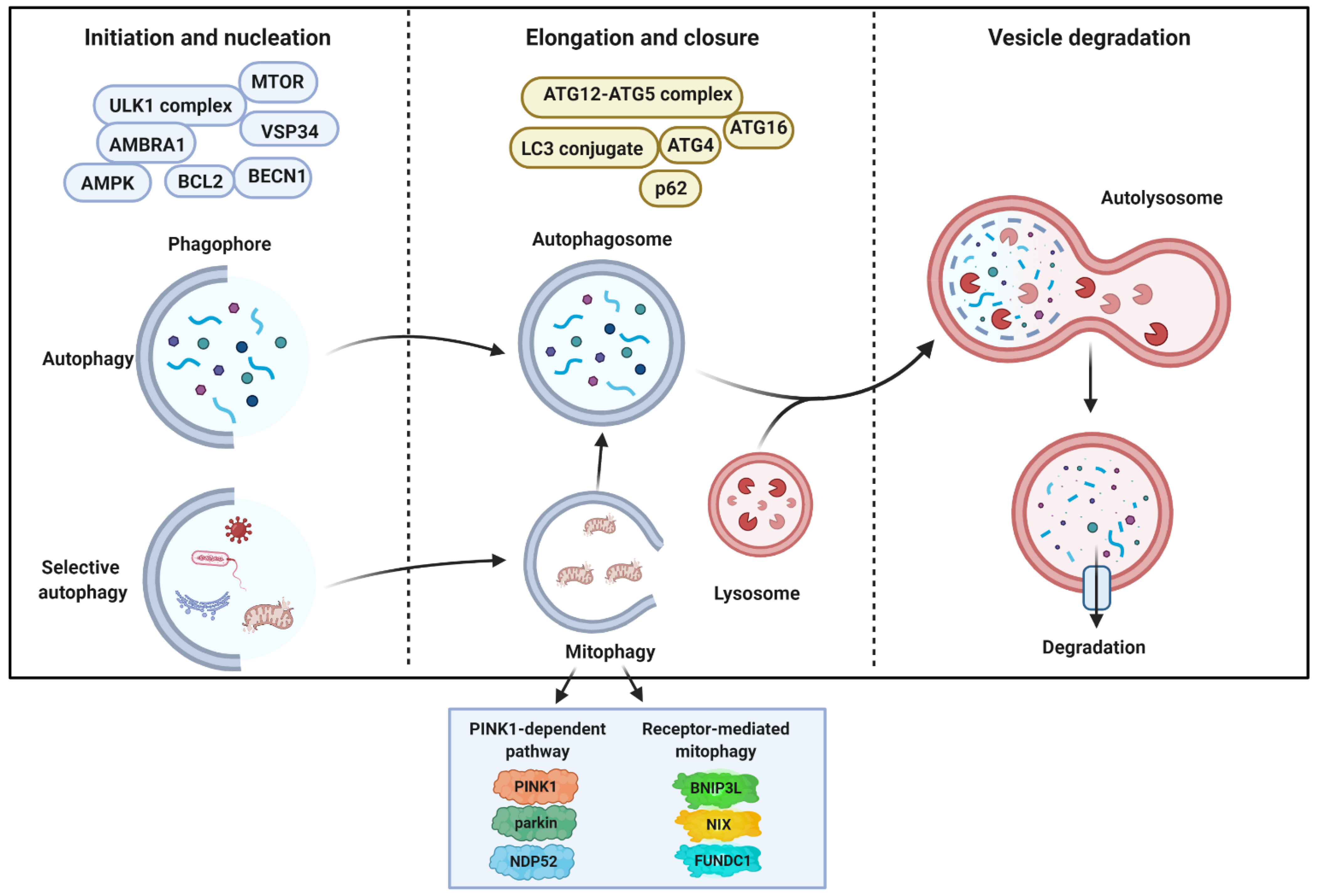
2. General Aspects of Autophagy and Molecular Mechanisms
3. Autophagy in Health and Disease
3.1. Autophagy and Neurodegeneration
3.2. Autophagy and Microbial Adaptations
3.3. Autophagy in Cardiovascular Diseases
3.4. Autophagy in Diabetes and Tissue Metabolism
3.5. Role of Autophagy in Aging
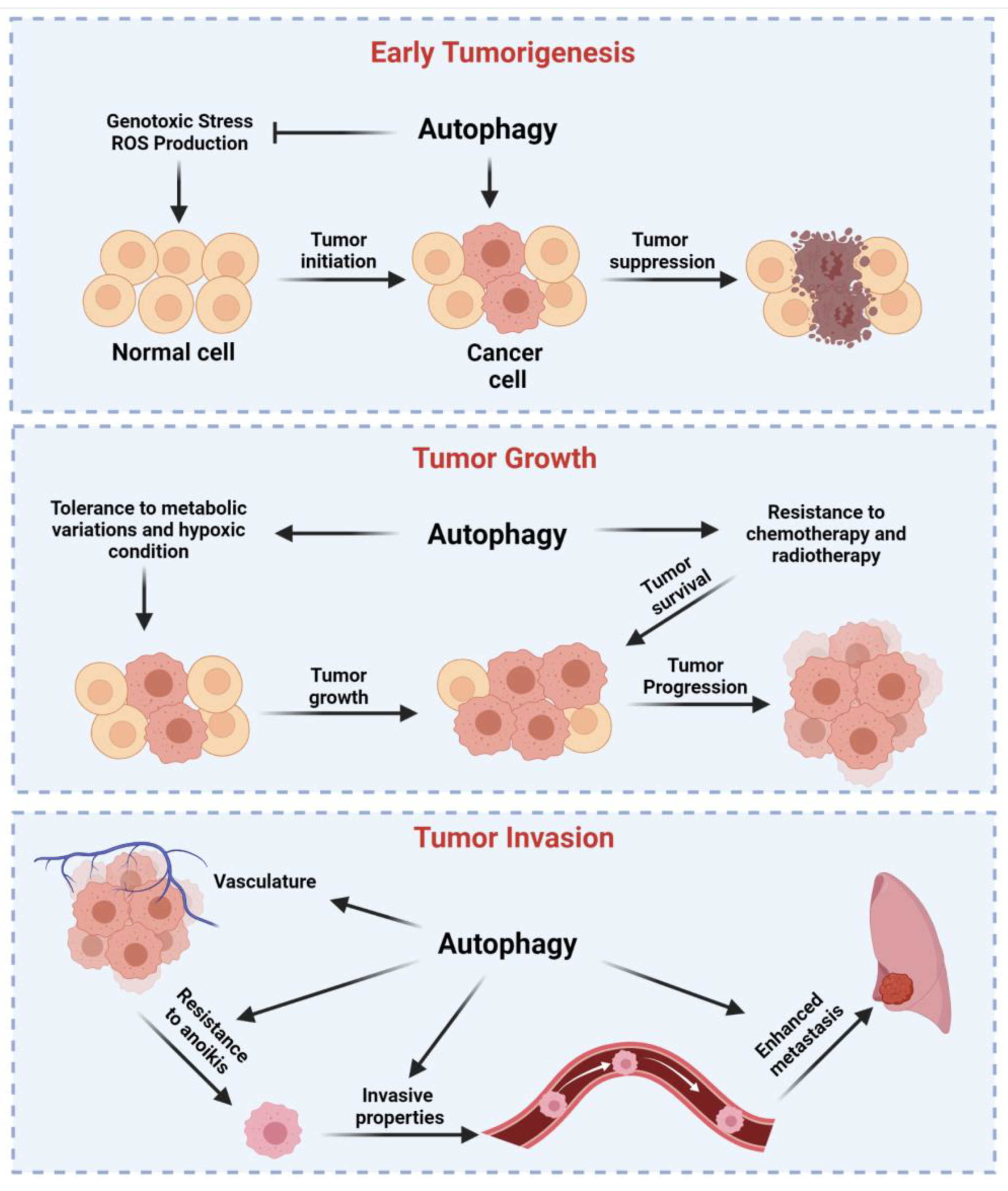
4. Dual Role of Autophagy in Cancer Regulation
4.1. Autophagy as a Tumor Suppressor Mechanism
4.2. Autophagy Drives Tumor Growth of Established Tumors, Metastasis, and Resistance to Therapy
5. Autophagy and Tumor Immune Response
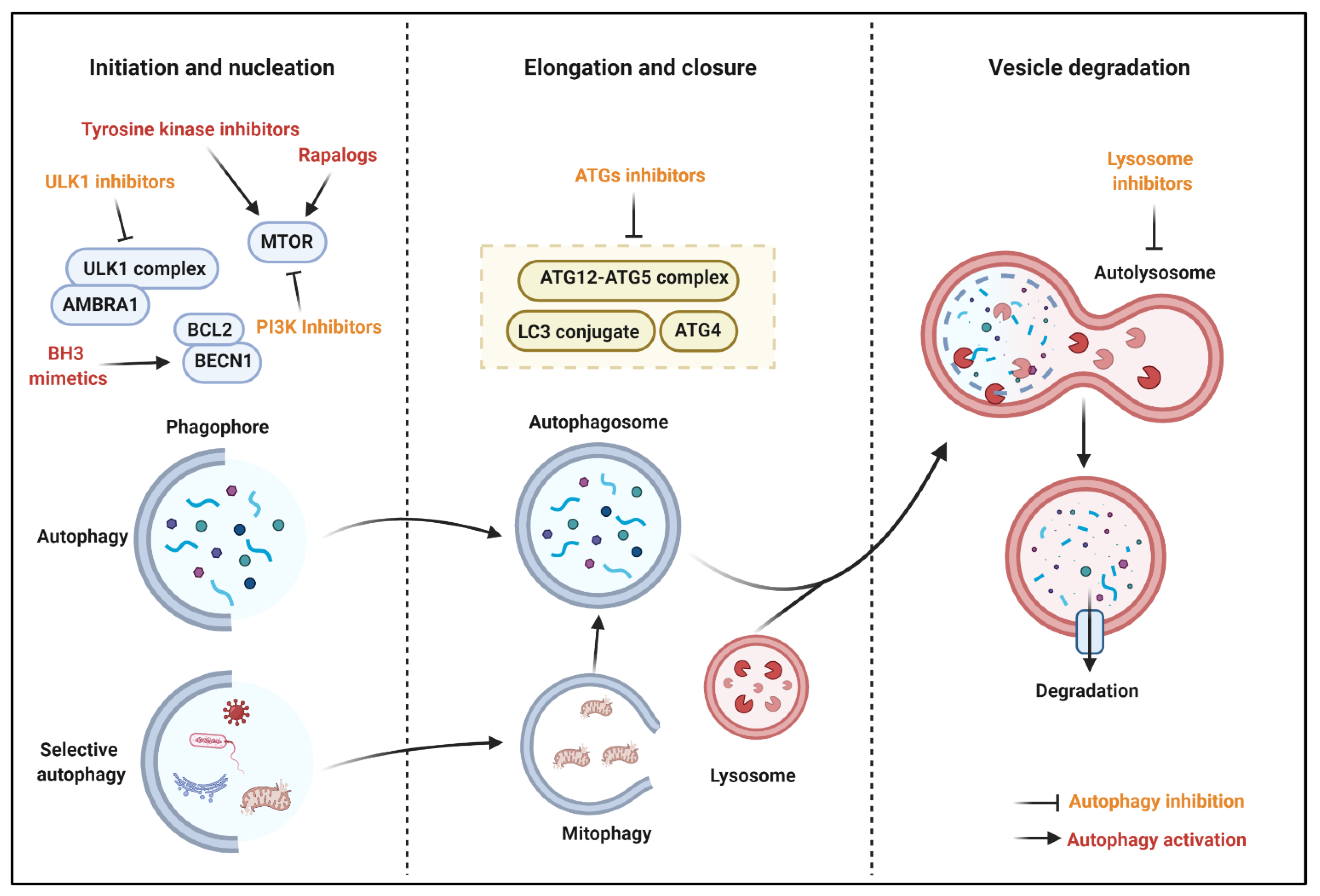
6. Autophagy Modulation for Cancer
6.1. Autophagy Stimulation for Cancer Treatment
6.1.1. MTOR Inhibitors
6.1.2. Histone Deacetylase Inhibitors
6.1.3. BH3 (BCL2 Homology 3) Mimetics
6.1.4. Tyrosine Kinase Inhibitors
6.2. Autophagy Inhibition for Cancer Treatment
6.2.1. ATG Inhibitors
6.2.2. ULK1 Inhibitors
6.2.3. PI3K Inhibitors
6.2.4. Lysosome Inhibitors
7. Cancer Clinical Trials
8. Summary and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Patergnani, S.; Missiroli, S.; Marchi, S.; Giorgi, C. Mitochondria-Associated Endoplasmic Reticulum Membranes Microenvironment: Targeting Autophagic and Apoptotic Pathways in Cancer Therapy. Front. Oncol. 2015, 5, 173. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. USA 2017, 114, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Chipurupalli, S.; Kannan, E.; Tergaonkar, V.; D’Andrea, R.; Robinson, N. Hypoxia Induced ER Stress Response as an Adaptive Mechanism in Cancer. Int. J. Mol. Sci. 2019, 20, 749. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- He, L.; Zhang, J.; Zhao, J.; Ma, N.; Kim, S.W.; Qiao, S.; Ma, X. Autophagy: The Last Defense against Cellular Nutritional Stress. Adv. Nutr. 2018, 9, 493–504. [Google Scholar] [CrossRef]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Park, S.J.; Stamatakou, E.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 2020, 11, 3148. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, D.N.; Chowdhury, R.; Trudel, L.J.; Tee, A.R.; Slack, R.S.; Walker, C.L.; Wogan, G.N. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. USA 2013, 110, E2950–E2957. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef]
- Stjepanovic, G.; Baskaran, S.; Lin, M.G.; Hurley, J.H. Vps34 Kinase Domain Dynamics Regulate the Autophagic PI 3-Kinase Complex. Mol. Cell 2017, 67, 528–534.e523. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Song, Y.; Li, Z.; Wang, Y.; Lin, H.; Cheng, H.; Zhou, R. RAB37 interacts directly with ATG5 and promotes autophagosome formation via regulating ATG5-12-16 complex assembly. Cell Death Differ. 2018, 25, 918–934. [Google Scholar] [CrossRef]
- Arakawa, S.; Honda, S.; Yamaguchi, H.; Shimizu, S. Molecular mechanisms and physiological roles of Atg5/Atg7-independent alternative autophagy. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Wesch, N.; Kirkin, V.; Rogov, V.V. Atg8-Family Proteins-Structural Features and Molecular Interactions in Autophagy and Beyond. Cells 2020, 9, 2008. [Google Scholar] [CrossRef]
- Chowdhury, S.; Otomo, C.; Leitner, A.; Ohashi, K.; Aebersold, R.; Lander, G.C.; Otomo, T. Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. Proc. Natl. Acad. Sci. USA 2018, 115, E9792–E9801. [Google Scholar] [CrossRef]
- Graef, M.; Friedman, J.R.; Graham, C.; Babu, M.; Nunnari, J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell 2013, 24, 2918–2931. [Google Scholar] [CrossRef] [PubMed]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Kriegenburg, F.; Ungermann, C.; Reggiori, F. Coordination of Autophagosome-Lysosome Fusion by Atg8 Family Members. Curr. Biol. CB 2018, 28, R512–R518. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Miao, G.; Xue, X.; Guo, X.; Yuan, C.; Wang, Z.; Zhang, G.; Chen, Y.; Feng, D.; Hu, J.; et al. The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol. Cell 2016, 63, 781–795. [Google Scholar] [CrossRef]
- Patergnani, S.; Bonora, M.; Bouhamida, E.; Danese, A.; Marchi, S.; Morciano, G.; Previati, M.; Pedriali, G.; Rimessi, A.; Anania, G.; et al. Methods to Monitor Mitophagy and Mitochondrial Quality: Implications in Cancer, Neurodegeneration, and Cardiovascular Diseases. Methods Mol. Biol. 2021, 2310, 113–159. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Bonora, M.; Ingusci, S.; Previati, M.; Marchi, S.; Zucchini, S.; Perrone, M.; Wieckowski, M.R.; Castellazzi, M.; Pugliatti, M.; et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2020078118. [Google Scholar] [CrossRef] [PubMed]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Pedriali, G.; Cimaglia, P.; Mikus, E.; Calvi, S.; Albertini, A.; Giorgi, C.; Campo, G.; Ferrari, R.; et al. Impairment of mitophagy and autophagy accompanies calcific aortic valve stenosis favoring cell death and the severity of disease. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.R.; Lewis, W.H. Mitochondria (and other cytoplasmic structures) in tissue cultures. Am. J. Anat. 1915, 17, 339–401. [Google Scholar] [CrossRef]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Randall, M.S.; Loyd, M.R.; Dorsey, F.C.; Kundu, M.; Cleveland, J.L.; Ney, P.A. Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood 2009, 114, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Ferguson, D.J.; Edelmann, M.; Kessler, B.; Morten, K.J.; Komatsu, M.; Simon, A.K. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Aerbajinai, W.; Giattina, M.; Lee, Y.T.; Raffeld, M.; Miller, J.L. The proapoptotic factor Nix is coexpressed with Bcl-xL during terminal erythroid differentiation. Blood 2003, 102, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010, 11, 45–51. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. CB 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e210. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, Y.; Qiu, X.; Wang, G.; Hu, Z.; Chen, S.; Wu, Z.; Yuan, N.; Gao, H.; Wang, J.; et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 2019, 20, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Kolitsida, P.; Zhou, J.; Rackiewicz, M.; Nolic, V.; Dengjel, J.; Abeliovich, H. Phosphorylation of mitochondrial matrix proteins regulates their selective mitophagic degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 20517–20527. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Kesidou, E.; Lagoudaki, R.; Touloumi, O.; Poulatsidou, K.N.; Simeonidou, C. Autophagy and neurodegenerative disorders. Neural Regen. Res. 2013, 8, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef]
- Wang, Y.; Martinez-Vicente, M.; Kruger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef]
- Lucin, K.M.; O’Brien, C.E.; Bieri, G.; Czirr, E.; Mosher, K.I.; Abbey, R.J.; Mastroeni, D.F.; Rogers, J.; Spencer, B.; Masliah, E.; et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 2013, 79, 873–886. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta 2015, 1852, 1665–1675. [Google Scholar] [CrossRef]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Bosi, C.; Brombo, G.; Pugliatti, M.; Seripa, D.; Zuliani, G.; et al. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 2019, 9, 20009. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain J. Neurol. 2017, 140, 3233–3251. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Rimessi, A.; Pozzato, C.; Carparelli, L.; Rossi, A.; Ranucci, S.; De Fino, I.; Cigana, C.; Talarico, A.; Wieckowski, M.R.; Ribeiro, C.M.P.; et al. Pharmacological modulation of mitochondrial calcium uniporter controls lung inflammation in cystic fibrosis. Sci. Adv. 2020, 6, eaax9093. [Google Scholar] [CrossRef]
- Pitale, D.M.; Gendalur, N.S.; Descoteaux, A.; Shaha, C. Leishmania donovani Induces Autophagy in Human Blood-Derived Neutrophils. J. Immunol. 2019, 202, 1163–1175. [Google Scholar] [CrossRef]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008, 455, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, L.A. Regulation of autophagy in the heart in health and disease. J. Cardiovasc. Pharmacol. 2012, 60, 109. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Knaapen, M.W.; Davies, M.J.; De Bie, M.; Haven, A.J.; Martinet, W.; Kockx, M.M. Apoptotic versus autophagic cell death in heart failure. Cardiovasc. Res. 2001, 51, 304–312. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Taneike, M.; Yamaguchi, O.; Nakai, A.; Hikoso, S.; Takeda, T.; Mizote, I.; Oka, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Song, Y.; Liu, L.; Hou, N.; An, X.; Zhan, D.; Li, Y.; Zhou, L.; Li, P.; Yu, L.; et al. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 2017, 24, 1205–1213. [Google Scholar] [CrossRef]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Ono, K.; Nagao, K.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2261–H2271. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W., 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef]
- Diwan, A.; Krenz, M.; Syed, F.M.; Wansapura, J.; Ren, X.; Koesters, A.G.; Li, H.; Kirshenbaum, L.A.; Hahn, H.S.; Robbins, J.; et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J. Clin. Investig. 2007, 117, 2825–2833. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.C.; Kim, K.H.; Kim, G.H.; Kim, S.W.; Kim, H.L.; Lee, M.K.; et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Kruse, R.; Pedersen, A.J.; Kristensen, J.M.; Petersson, S.J.; Wojtaszewski, J.F.; Hojlund, K. Intact initiation of autophagy and mitochondrial fission by acute exercise in skeletal muscle of patients with Type 2 diabetes. Clin. Sci. 2017, 131, 37–47. [Google Scholar] [CrossRef]
- Laurin, N.; Brown, J.P.; Morissette, J.; Raymond, V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 2002, 70, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Droge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining "clean" cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Bartke, A. The key role of growth hormone-insulin-IGF-1 signaling in aging and cancer. Crit. Rev. Oncol./Hematol. 2013, 87, 201–223. [Google Scholar] [CrossRef]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Muller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef]
- Tavera-Mendoza, L.E.; Westerling, T.; Libby, E.; Marusyk, A.; Cato, L.; Cassani, R.; Cameron, L.A.; Ficarro, S.B.; Marto, J.A.; Klawitter, J.; et al. Vitamin D receptor regulates autophagy in the normal mammary gland and in luminal breast cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2186–E2194. [Google Scholar] [CrossRef]
- Rosen, R.D.; Sapra, A. TNM Classification. In StatPearls; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Mulcahy Levy, J.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef]
- Wang, P.; Du, Y.; Wang, J. Indentification of breast cancer subtypes sensitive to HCQ-induced autophagy inhibition. Pathol. Res. Pract. 2019, 215, 152609. [Google Scholar] [CrossRef]
- Grandvallet, C.; Feugeas, J.P.; Monnien, F.; Despouy, G.; Valerie, P.; Michael, G.; Hervouet, E.; Peixoto, P. Autophagy is associated with a robust specific transcriptional signature in breast cancer subtypes. Genes Cancer 2020, 11, 154–168. [Google Scholar] [CrossRef]
- Lauzier, A.; Normandeau-Guimond, J.; Vaillancourt-Lavigueur, V.; Boivin, V.; Charbonneau, M.; Rivard, N.; Scott, M.S.; Dubois, C.M.; Jean, S. Colorectal cancer cells respond differentially to autophagy inhibition in vivo. Sci. Rep. 2019, 9, 11316. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Koukourakis, M.I.; Harris, A.L.; Polychronidis, A.; Gatter, K.C.; Sivridis, E. Prognostic relevance of light chain 3 (LC3A) autophagy patterns in colorectal adenocarcinomas. J. Clin. Pathol. 2010, 63, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Burada, F.; Nicoli, E.R.; Ciurea, M.E.; Uscatu, D.C.; Ioana, M.; Gheonea, D.I. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, L.; Ao, H.; Zhao, M.; Leng, X.; Liu, M.; Ma, J.; Zhu, J. Prognostic implications of autophagy-associated gene signatures in non-small cell lung cancer. Aging 2019, 11, 11440–11462. [Google Scholar] [CrossRef]
- Gu, Y.; Li, P.; Peng, F.; Zhang, M.; Zhang, Y.; Liang, H.; Zhao, W.; Qi, L.; Wang, H.; Wang, C.; et al. Autophagy-related prognostic signature for breast cancer. Mol. Carcinog. 2016, 55, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Du, J.X.; Chen, C.; Luo, Y.H.; Cai, J.L.; Cai, C.Z.; Xu, J.; Ni, X.J.; Zhu, W. Establishment and validation of a novel autophagy-related gene signature for patients with breast cancer. Gene 2020, 762, 144974. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cao, Y.; Yu, X.; Jin, F.; Li, Y. A novel autophagy-related genes prognostic risk model and validation of autophagy-related oncogene VPS35 in breast cancer. Cancer Cell Int. 2021, 21, 265. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, Y.; Wang, B.; Zhang, L.; Su, Y.; Xu, M.; Zhang, M. A signature based on 11 autophagy genes for prognosis prediction of colorectal cancer. PLoS ONE 2021, 16, e0258741. [Google Scholar] [CrossRef]
- Guo, G.F.; Jiang, W.Q.; Zhang, B.; Cai, Y.C.; Xu, R.H.; Chen, X.X.; Wang, F.; Xia, L.P. Autophagy-related proteins Beclin-1 and LC3 predict cetuximab efficacy in advanced colorectal cancer. World J. Gastroenterol. 2011, 17, 4779–4786. [Google Scholar] [CrossRef]
- Xie, K.; Liang, C.; Li, Q.; Yan, C.; Wang, C.; Gu, Y.; Zhu, M.; Du, F.; Wang, H.; Dai, J.; et al. Role of ATG10 expression quantitative trait loci in non-small cell lung cancer survival. Int. J. Cancer 2016, 139, 1564–1573. [Google Scholar] [CrossRef]
- Uberall, I.; Gachechiladze, M.; Joerger, M.; Andel, J.; Smickova, P.; Kolek, V.; Grygarkova, I.; Skarda, J. Tumor autophagy is associated with survival outcomes in patients with resected non-small cell lung cancer. Lung Cancer 2019, 129, 85–91. [Google Scholar] [CrossRef]
- Eissa, S.; Matboli, M.; Awad, N.; Kotb, Y. Identification and validation of a novel autophagy gene expression signature for human bladder cancer patients. Tumour Biol. 2017, 39, 1010428317698360. [Google Scholar] [CrossRef]
- Zhou, C.; Li, A.H.; Liu, S.; Sun, H. Identification of an 11-Autophagy-Related-Gene Signature as Promising Prognostic Biomarker for Bladder Cancer Patients. Biology 2021, 10, 375. [Google Scholar] [CrossRef]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Cai, H.; Teplova, I.; Bowman-Colin, C.; Chen, G.; Price, S.; Barnard, N.; Ganesan, S.; Karantza, V.; White, E.; et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013, 3, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.; Cacheux, W.; Bara, M.A.; L’Hermitte, A.; Lepage, P.; Fraudeau, M.; Trentesaux, C.; Lemarchand, J.; Durand, A.; Crain, A.M.; et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol. 2015, 17, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Aita, V.M.; Liang, X.H.; Murty, V.V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.C.; Levine, B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef]
- Cicchini, M.; Chakrabarti, R.; Kongara, S.; Price, S.; Nahar, R.; Lozy, F.; Zhong, H.; Vazquez, A.; Kang, Y.; Karantza, V. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy 2014, 10, 2036–2052. [Google Scholar] [CrossRef]
- Morciano, G.; Vezzani, B.; Missiroli, S.; Boncompagni, C.; Pinton, P.; Giorgi, C. An Updated Understanding of the Role of YAP in Driving Oncogenic Responses. Cancers 2021, 13, 3100. [Google Scholar] [CrossRef]
- Wijshake, T.; Zou, Z.; Chen, B.; Zhong, L.; Xiao, G.; Xie, Y.; Doench, J.G.; Bennett, L.; Levine, B. Tumor-suppressor function of Beclin 1 in breast cancer cells requires E-cadherin. Proc. Natl. Acad. Sci. USA 2021, 118, e2020478118. [Google Scholar] [CrossRef]
- Li, Z.; Yin, S.; Zhang, L.; Liu, W.; Chen, B. Prognostic value of reduced E-cadherin expression in breast cancer: A meta-analysis. Oncotarget 2017, 8, 16445–16455. [Google Scholar] [CrossRef]
- Berx, G.; Van Roy, F. The E-cadherin/catenin complex: An important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res. 2001, 3, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mule, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.D.; Young, A.R.J.; Young, C.N.J.; Soilleux, E.J.; Fielder, E.; Weigand, B.M.; Lagnado, A.; Brais, R.; Ktistakis, N.T.; Wiggins, K.A.; et al. Temporal inhibition of autophagy reveals segmental reversal of ageing with increased cancer risk. Nat. Commun. 2020, 11, 307. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Di Leo, L.; Bodemeyer, V.; Bosisio, F.M.; Claps, G.; Carretta, M.; Rizza, S.; Faienza, F.; Frias, A.; Khan, S.; Bordi, M.; et al. Loss of Ambra1 promotes melanoma growth and invasion. Nat. Commun. 2021, 12, 2550. [Google Scholar] [CrossRef]
- Maiani, E.; Milletti, G.; Nazio, F.; Holdgaard, S.G.; Bartkova, J.; Rizza, S.; Cianfanelli, V.; Lorente, M.; Simoneschi, D.; Di Marco, M.; et al. AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature 2021, 592, 799–803. [Google Scholar] [CrossRef]
- Hess, A.R.; Postovit, L.M.; Margaryan, N.V.; Seftor, E.A.; Schneider, G.B.; Seftor, R.E.; Nickoloff, B.J.; Hendrix, M.J. Focal adhesion kinase promotes the aggressive melanoma phenotype. Cancer Res. 2005, 65, 9851–9860. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Menck, C.F.M.; Leandro, G.S. Autophagy Roles in the Modulation of DNA Repair Pathways. Int. J. Mol. Sci. 2017, 18, 2351. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef]
- Rodriguez-Vargas, J.M.; Rodriguez, M.I.; Majuelos-Melguizo, J.; Garcia-Diaz, A.; Gonzalez-Flores, A.; Lopez-Rivas, A.; Virag, L.; Illuzzi, G.; Schreiber, V.; Dantzer, F.; et al. Autophagy requires poly(adp-ribosyl)ation-dependent AMPK nuclear export. Cell Death Differ. 2016, 23, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef]
- Bonhoure, A.; Vallentin, A.; Martin, M.; Senff-Ribeiro, A.; Amson, R.; Telerman, A.; Vidal, M. Acetylation of translationally controlled tumor protein promotes its degradation through chaperone-mediated autophagy. Eur. J. Cell Biol. 2017, 96, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Pinton, P. Mitophagy and mitochondrial balance. Methods Mol. Biol. 2015, 1241, 181–194. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Zeh, H.J., 3rd; Lotze, M.T. High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid. Redox Signal. 2011, 15, 2185–2195. [Google Scholar] [CrossRef]
- Kang, R.; Livesey, K.M.; Zeh, H.J.; Loze, M.T.; Tang, D. HMGB1: A novel Beclin 1-binding protein active in autophagy. Autophagy 2010, 6, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Zhang, H.L.; Li, D.D.; Yang, K.L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.Y.; Ravichandran, S.; et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef]
- Brisson, L.; Banski, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.J.; Van Hee, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef]
- Wojtkowiak, J.W.; Rothberg, J.M.; Kumar, V.; Schramm, K.J.; Haller, E.; Proemsey, J.B.; Lloyd, M.C.; Sloane, B.F.; Gillies, R.J. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res. 2012, 72, 3938–3947. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Kong, Q.; Xu, L.H.; Xu, W.; Fang, J.P.; Xu, H.G. HMGB1 translocation is involved in the transformation of autophagy complexes and promotes chemoresistance in leukaemia. Int. J. Oncol. 2015, 47, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Patergnani, S.; Giorgi, C.; Suarez, J.; Goto, K.; Bononi, A.; Tanji, M.; Novelli, F.; Pastorino, S.; Xu, R.; et al. Asbestos induces mesothelial cell transformation via HMGB1-driven autophagy. Proc. Natl. Acad. Sci. USA 2020, 117, 25543–25552. [Google Scholar] [CrossRef]
- Gilmore, A.P. Anoikis. Cell Death Differ. 2005, 12, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Tuloup-Minguez, V.; Greffard, A.; Codogno, P.; Botti, J. Regulation of autophagy by extracellular matrix glycoproteins in HeLa cells. Autophagy 2011, 7, 27–39. [Google Scholar] [CrossRef]
- Debnath, J.; Mills, K.R.; Collins, N.L.; Reginato, M.J.; Muthuswamy, S.K.; Brugge, J.S. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002, 111, 29–40. [Google Scholar] [CrossRef]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Prabavathy, D.; Swarnalatha, Y.; Ramadoss, N. Lung cancer stem cells-origin, characteristics and therapy. Stem Cell Investig 2018, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Kozovska, Z.; Gabrisova, V.; Kucerova, L. Malignant melanoma: Diagnosis, treatment and cancer stem cells. Neoplasma 2016, 63, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, S.G.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Gong, C.; Song, E.; Codogno, P.; Mehrpour, M. The roles of BECN1 and autophagy in cancer are context dependent. Autophagy 2012, 8, 1853–1855. [Google Scholar] [CrossRef]
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Muller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013, 15, R109. [Google Scholar] [CrossRef]
- Chaterjee, M.; van Golen, K.L. Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res. 2011, 2011, 362938. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Delomenie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.J.; Zhang, S.S.; Guo, X.L.; Sun, K.; Han, Z.P.; Li, R.; Zhao, Q.D.; Deng, W.J.; Xie, X.Q.; Zhang, J.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef]
- Rausch, V.; Liu, L.; Apel, A.; Rettig, T.; Gladkich, J.; Labsch, S.; Kallifatidis, G.; Kaczorowski, A.; Groth, A.; Gross, W.; et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J. Pathol. 2012, 227, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, Q.; Sun, H.; Yin, L.; Wu, J.; Xu, J.; He, T.; Yang, C.; Liang, C. Defective autophagy leads to the suppression of stem-like features of CD271(+) osteosarcoma cells. J. Biomed. Sci. 2016, 23, 82. [Google Scholar] [CrossRef]
- Wu, S.; Wang, X.; Chen, J.; Chen, Y. Autophagy of cancer stem cells is involved with chemoresistance of colon cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 898–903. [Google Scholar] [CrossRef] [PubMed]
- Mele, L.; Del Vecchio, V.; Liccardo, D.; Prisco, C.; Schwerdtfeger, M.; Robinson, N.; Desiderio, V.; Tirino, V.; Papaccio, G.; La Noce, M. The role of autophagy in resistance to targeted therapies. Cancer Treat. Rev. 2020, 88, 102043. [Google Scholar] [CrossRef]
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A.L. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Sieuwerts, A.M.; Bussink, J.; Sweep, F.C.; Look, M.P.; Foekens, J.A.; Martens, J.W.; Span, P.N. LAMP3 is involved in tamoxifen resistance in breast cancer cells through the modulation of autophagy. Endocr. Relat. Cancer 2014, 21, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.M.; Bold, G.; Buchdunger, E.; Cozens, R.; Ferrari, S.; Frei, J.; Hofmann, F.; Mestan, J.; Mett, H.; O’Reilly, T.; et al. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2000, 60, 2178–2189. [Google Scholar]
- Zhao, Z.; Xia, G.; Li, N.; Su, R.; Chen, X.; Zhong, L. Autophagy Inhibition Promotes Bevacizumab-induced Apoptosis and Proliferation Inhibition in Colorectal Cancer Cells. J. Cancer 2018, 9, 3407–3416. [Google Scholar] [CrossRef]
- Lu, W.; Ke, H.; Qianshan, D.; Zhen, W.; Guoan, X.; Honggang, Y. Apatinib has anti-tumor effects and induces autophagy in colon cancer cells. Iran. J. Basic Med. Sci. 2017, 20, 990–995. [Google Scholar] [CrossRef]
- Cheng, X.; Feng, H.; Wu, H.; Jin, Z.; Shen, X.; Kuang, J.; Huo, Z.; Chen, X.; Gao, H.; Ye, F.; et al. Targeting autophagy enhances apatinib-induced apoptosis via endoplasmic reticulum stress for human colorectal cancer. Cancer Lett. 2018, 431, 105–114. [Google Scholar] [CrossRef]
- San Jose-Eneriz, E.; Roman-Gomez, J.; Jimenez-Velasco, A.; Garate, L.; Martin, V.; Cordeu, L.; Vilas-Zornoza, A.; Rodriguez-Otero, P.; Calasanz, M.J.; Prosper, F.; et al. MicroRNA expression profiling in Imatinib-resistant Chronic Myeloid Leukemia patients without clinically significant ABL1-mutations. Mol. Cancer 2009, 8, 69. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, L.; Zhao, M.; Zhu, S.; Kang, R.; Vernon, P.; Tang, D.; Cao, L. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 2012, 26, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.; Hopcroft, L.E.M.; Baquero, P.; Allan, E.K.; Hewit, K.; James, D.; Hamilton, G.; Mukhopadhyay, A.; O’Prey, J.; Hair, A.; et al. Targeting BCR-ABL-Independent TKI Resistance in Chronic Myeloid Leukemia by mTOR and Autophagy Inhibition. J. Natl. Cancer Inst. 2018, 110, 467–478. [Google Scholar] [CrossRef]
- Tong, Y.; Liu, Y.Y.; You, L.S.; Qian, W.B. Perifosine induces protective autophagy and upregulation of ATG5 in human chronic myelogenous leukemia cells in vitro. Acta Pharmacol. Sin. 2012, 33, 542–550. [Google Scholar] [CrossRef]
- Amrein, L.; Soulieres, D.; Johnston, J.B.; Aloyz, R. p53 and autophagy contribute to dasatinib resistance in primary CLL lymphocytes. Leuk. Res. 2011, 35, 99–102. [Google Scholar] [CrossRef]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat. Commun. 2019, 10, 1693. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Hao, F.; Zhang, Q.; Qiu, Y. ROS mediated radiotherapy-induced protective autophagy in thyroid cancer. All Life 2021, 14, 58–65. [Google Scholar] [CrossRef]
- Chaachouay, H.; Ohneseit, P.; Toulany, M.; Kehlbach, R.; Multhoff, G.; Rodemann, H.P. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2011, 99, 287–292. [Google Scholar] [CrossRef]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.P.; Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008, 68, 1485–1494. [Google Scholar] [CrossRef]
- Chen, Y.S.; Song, H.X.; Lu, Y.; Li, X.; Chen, T.; Zhang, Y.; Xue, J.X.; Liu, H.; Kan, B.; Yang, G.; et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis. Esophagus 2011, 24, 437–443. [Google Scholar] [CrossRef]
- Jiang, T.; Chen, X.; Ren, X.; Yang, J.M.; Cheng, Y. Emerging role of autophagy in anti-tumor immunity: Implications for the modulation of immunotherapy resistance. Drug Resist. Updat. 2021, 56, 100752. [Google Scholar] [CrossRef]
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Joseph Espat, N.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Xu, Z.; Zhuang, Y.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Hematological Malignancies. J. Cancer 2021, 12, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Burugu, S.; Dancsok, A.R.; Nielsen, T.O. Emerging targets in cancer immunotherapy. Semin. Cancer Biol. 2018, 52, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Tittarelli, A.; Janji, B.; Van Moer, K.; Noman, M.Z.; Chouaib, S. The Selective Degradation of Synaptic Connexin 43 Protein by Hypoxia-induced Autophagy Impairs Natural Killer Cell-mediated Tumor Cell Killing. J. Biol. Chem. 2015, 290, 23670–23679. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, H.; Wang, Z.; Wu, P.; Gong, W. 5-Hydroxytryptamine1a receptors on tumour cells induce immune evasion in lung adenocarcinoma patients with depression via autophagy/pSTAT3. Eur. J. Cancer 2019, 114, 8–24. [Google Scholar] [CrossRef]
- Zhu, J.; Li, Y.; Luo, Y.; Xu, J.; Liufu, H.; Tian, Z.; Huang, C.; Li, J.; Huang, C. A Feedback Loop Formed by ATG7/Autophagy, FOXO3a/miR-145 and PD-L1 Regulates Stem-Like Properties and Invasion in Human Bladder Cancer. Cancers 2019, 11, 349. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, W.K.K.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J. Exp. Clin. Cancer Res. 2019, 38, 140. [Google Scholar] [CrossRef]
- Lin, S.Y.; Hsieh, S.Y.; Fan, Y.T.; Wei, W.C.; Hsiao, P.W.; Tsai, D.H.; Wu, T.S.; Yang, N.S. Necroptosis promotes autophagy-dependent upregulation of DAMP and results in immunosurveillance. Autophagy 2018, 14, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Capan, E.; Zhao, Y.; Zhao, J.; Stolz, D.; Watkins, S.C.; Jin, S.; Lu, B. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J. Immunol. 2006, 177, 5163–5168. [Google Scholar] [CrossRef]
- Pua, H.H.; Dzhagalov, I.; Chuck, M.; Mizushima, N.; He, Y.W. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 2007, 204, 25–31. [Google Scholar] [CrossRef]
- Zhang, T.Y.; Ren, H.Y.; Pan, N.; Dong, H.X.; Zhao, S.M.; Wen, Z.F.; Wang, X.R.; Wang, L.X. Tumor cell-derived autophagosomes (DRibbles)-activated B cells induce specific naive CD8(+) T cell response and exhibit antitumor effect. Cancer Immunol. Immunother. 2021, 70, 463–474. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef]
- Liang, X.; De Vera, M.E.; Buchser, W.J.; Romo de Vivar Chavez, A.; Loughran, P.; Beer Stolz, D.; Basse, P.; Wang, T.; Van Houten, B.; Zeh, H.J., 3rd; et al. Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res. 2012, 72, 2791–2801. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.; Wang, W.; Piao, S.; Zhou, J.; Saiyin, W.; Zheng, C.; Sun, H.; Li, Y. Inhibition of autophagy by 3-MA enhances IL-24-induced apoptosis in human oral squamous cell carcinoma cells. J. Exp. Clin. Cancer Res. 2015, 34, 97. [Google Scholar] [CrossRef]
- Lawson, K.A.; Sousa, C.M.; Zhang, X.; Kim, E.; Akthar, R.; Caumanns, J.J.; Yao, Y.; Mikolajewicz, N.; Ross, C.; Brown, K.R.; et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 2020, 586, 120–126. [Google Scholar] [CrossRef]
- Perez-Hernandez, M.; Arias, A.; Martinez-Garcia, D.; Perez-Tomas, R.; Quesada, R.; Soto-Cerrato, V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef]
- Han, H.; Li, J.; Feng, X.; Zhou, H.; Guo, S.; Zhou, W. Autophagy-related genes are induced by histone deacetylase inhibitor suberoylanilide hydroxamic acid via the activation of cathepsin B in human breast cancer cells. Oncotarget 2017, 8, 53352–53365. [Google Scholar] [CrossRef]
- De, U.; Son, J.Y.; Sachan, R.; Park, Y.J.; Kang, D.; Yoon, K.; Lee, B.M.; Kim, I.S.; Moon, H.R.; Kim, H.S. A New Synthetic Histone Deacetylase Inhibitor, MHY2256, Induces Apoptosis and Autophagy Cell Death in Endometrial Cancer Cells via p53 Acetylation. Int. J. Mol. Sci. 2018, 19, 2743. [Google Scholar] [CrossRef] [PubMed]
- Dupere-Richer, D.; Kinal, M.; Menasche, V.; Nielsen, T.H.; Del Rincon, S.; Pettersson, F.; Miller, W.H., Jr. Vorinostat-induced autophagy switches from a death-promoting to a cytoprotective signal to drive acquired resistance. Cell Death Dis. 2013, 4, e486. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Azab, B.; Quinn, B.A.; Shen, X.; Wang, X.Y.; Das, S.K.; Rahmani, M.; Wei, J.; Hedvat, M.; Dent, P.; et al. Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. Proc. Natl. Acad. Sci. USA 2011, 108, 8785–8790. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, L.; Bornhauser, B.C.; Schmitz, M.; Cario, G.; Ziegler, U.; Niggli, F.K.; Schafer, B.W.; Schrappe, M.; Stanulla, M.; Bourquin, J.P. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J. Clin. Investig. 2010, 120, 1310–1323. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Appelman, C.; Smith, A.R.; Yu, J.; Larsen, S.; Marquez, R.T.; Liu, H.; Wu, X.; Gao, P.; Roy, A.; et al. Natural product (-)-gossypol inhibits colon cancer cell growth by targeting RNA-binding protein Musashi-1. Mol. Oncol. 2015, 9, 1406–1420. [Google Scholar] [CrossRef]
- Benvenuto, M.; Mattera, R.; Sticca, J.I.; Rossi, P.; Cipriani, C.; Giganti, M.G.; Volpi, A.; Modesti, A.; Masuelli, L.; Bei, R. Effect of the BH3 Mimetic Polyphenol (-)-Gossypol (AT-101) on the in vitro and in vivo Growth of Malignant Mesothelioma. Front. Pharmacol. 2018, 9, 1269. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Bauvy, C.; Souquere, S.; Tonelli, G.; Liu, L.; Zhu, Y.; Qiao, Z.; Bakula, D.; Proikas-Cezanne, T.; Pierron, G.; et al. The Bcl-2 homology domain 3 mimetic gossypol induces both Beclin 1-dependent and Beclin 1-independent cytoprotective autophagy in cancer cells. J. Biol. Chem. 2010, 285, 25570–25581. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jerusalem, G.; Longuespee, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Oviedo, L.; Ortega-Muelas, M.; Garcia-Cano, J.; Valero, M.L.; Cimas, F.J.; Pascual-Serra, R.; Fernandez-Aroca, D.M.; Roche, O.; Ruiz-Hidalgo, M.J.; Belandia, B.; et al. Autophagic cell death associated to Sorafenib in renal cell carcinoma is mediated through Akt inhibition in an ERK1/2 independent fashion. PLoS ONE 2018, 13, e0200878. [Google Scholar] [CrossRef]
- Kharaziha, P.; Chioureas, D.; Baltatzis, G.; Fonseca, P.; Rodriguez, P.; Gogvadze, V.; Lennartsson, L.; Bjorklund, A.C.; Zhivotovsky, B.; Grander, D.; et al. Sorafenib-induced defective autophagy promotes cell death by necroptosis. Oncotarget 2015, 6, 37066–37082. [Google Scholar] [CrossRef]
- Li, Y.Y.; Lam, S.K.; Mak, J.C.; Zheng, C.Y.; Ho, J.C. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer 2013, 81, 354–361. [Google Scholar] [CrossRef]
- Zou, Y.; Ling, Y.H.; Sironi, J.; Schwartz, E.L.; Perez-Soler, R.; Piperdi, B. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J. Thorac. Oncol. 2013, 8, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Du, T.; Dong, X.; Li, Z.; Wu, G.; Zhang, R. Autophagy inhibition facilitates erlotinib cytotoxicity in lung cancer cells through modulation of endoplasmic reticulum stress. Int. J. Oncol. 2016, 48, 2558–2566. [Google Scholar] [CrossRef]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.M.; Kim, J.S.; Horenstein, N.; Dunn, W.A., Jr. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef]
- Kurdi, A.; Cleenewerck, M.; Vangestel, C.; Lyssens, S.; Declercq, W.; Timmermans, J.P.; Stroobants, S.; Augustyns, K.; De Meyer, G.R.Y.; Van Der Veken, P.; et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem. Pharmacol. 2017, 138, 150–162. [Google Scholar] [CrossRef]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2019, 15, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Dower, C.M.; Bhat, N.; Gebru, M.T.; Chen, L.; Wills, C.A.; Miller, B.A.; Wang, H.G. Targeted Inhibition of ULK1 Promotes Apoptosis and Suppresses Tumor Growth and Metastasis in Neuroblastoma. Mol. Cancer Ther. 2018, 17, 2365–2376. [Google Scholar] [CrossRef]
- Tang, F.; Hu, P.; Yang, Z.; Xue, C.; Gong, J.; Sun, S.; Shi, L.; Zhang, S.; Li, Z.; Yang, C.; et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 2017, 37, 3449–3458. [Google Scholar] [CrossRef]
- Dite, T.A.; Langendorf, C.G.; Hoque, A.; Galic, S.; Rebello, R.J.; Ovens, A.J.; Lindqvist, L.M.; Ngoei, K.R.W.; Ling, N.X.Y.; Furic, L.; et al. AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J. Biol. Chem. 2018, 293, 8874–8885. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.; Celano, S.L.; Solitro, A.R.; Gunaydin, H.; Scott, M.; O’Hagan, R.C.; Shumway, S.D.; Fuller, P.; MacKeigan, J.P. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. iScience 2018, 8, 74–84. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Novotny, C.J.; Shokat, K.M. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem. Biol. 2015, 10, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. [Google Scholar] [CrossRef]
- Skah, S.; Richartz, N.; Duthil, E.; Gilljam, K.M.; Bindesboll, C.; Naderi, E.H.; Eriksen, A.B.; Ruud, E.; Dirdal, M.M.; Simonsen, A.; et al. cAMP-mediated autophagy inhibits DNA damage-induced death of leukemia cells independent of p53. Oncotarget 2018, 9, 30434–30449. [Google Scholar] [CrossRef][Green Version]
- Chen, Y.; Xie, X.; Wang, C.; Hu, Y.; Zhang, H.; Zhang, L.; Tu, S.; He, Y.; Li, Y. Dual targeting of NUAK1 and ULK1 using the multitargeted inhibitor MRT68921 exerts potent antitumor activities. Cell Death Dis. 2020, 11, 712. [Google Scholar] [CrossRef]
- Akter, R.; Hossain, M.Z.; Kleve, M.G.; Gealt, M.A. Wortmannin induces MCF-7 breast cancer cell death via the apoptotic pathway, involving chromatin condensation, generation of reactive oxygen species, and membrane blebbing. Breast Cancer 2012, 4, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Ryabaya, O.O.; Inshakov, A.N.; Egorova, A.V.; Emelyanova, M.A.; Nasedkina, T.V.; Zasedatelev, A.S.; Khochenkov, D.A.; Stepanova, E.V. Autophagy inhibitors chloroquine and LY294002 enhance temozolomide cytotoxicity on cutaneous melanoma cell lines in vitro. Anti-Cancer Drugs 2017, 28, 307–315. [Google Scholar] [CrossRef]
- Feng, Y.; Gao, Y.; Wang, D.; Xu, Z.; Sun, W.; Ren, P. Autophagy Inhibitor (LY294002) and 5-fluorouracil (5-FU) Combination-Based Nanoliposome for Enhanced Efficacy Against Esophageal Squamous Cell Carcinoma. Nanoscale Res. Lett. 2018, 13, 325. [Google Scholar] [CrossRef]
- Qin, A.C.; Li, Y.; Zhou, L.N.; Xing, C.G.; Lu, X.S. Dual PI3K-BRD4 Inhibitor SF1126 Inhibits Colorectal Cancer Cell Growth in Vitro and in Vivo. Cell. Physiol. Biochem. 2019, 52, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Thomas, S.; Golden, E.B.; Hofman, F.M.; Chen, T.C.; Petasis, N.A.; Schonthal, A.H.; Louie, S.G. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012, 326, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.H.; Yao, C.J.; Hsiao, C.H.; Lin, K.H.; Lin, Y.W.; Wen, Y.C.; Liu, C.C.; Yan, M.D.; Chuang, S.E.; Lai, G.M.; et al. Mefloquine exerts anticancer activity in prostate cancer cells via ROS-mediated modulation of Akt, ERK, JNK and AMPK signaling. Oncol. Lett. 2013, 5, 1541–1545. [Google Scholar] [CrossRef] [PubMed]
- Lam Yi, H.; Than, H.; Sng, C.; Cheong, M.A.; Chuah, C.; Xiang, W. Lysosome Inhibition by Mefloquine Preferentially Enhances the Cytotoxic Effects of Tyrosine Kinase Inhibitors in Blast Phase Chronic Myeloid Leukemia. Transl. Oncol. 2019, 12, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- McAfee, Q.; Zhang, Z.; Samanta, A.; Levi, S.M.; Ma, X.H.; Piao, S.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E.; et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Nicastri, M.C.; McLaughlin, N.; Fennelly, C.; McAfee, Q.; Ronghe, A.; Nofal, M.; Lim, C.Y.; Witze, E.; Chude, C.I.; et al. A Unified Approach to Targeting the Lysosome’s Degradative and Growth Signaling Roles. Cancer Discov. 2017, 7, 1266–1283. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Na, K.; Chen, D.; Wang, Y.; Pan, H.; Wang, X. Effects of non-steroidal anti-inflammatory drug-activated gene-1 on Ganoderma lucidum polysaccharides-induced apoptosis of human prostate cancer PC-3 cells. Int. J. Oncol. 2018, 53, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Wang, Y.; Na, K.; Wang, Y.; Wang, L.; Li, Z.; Guo, C.; Guo, D.; Wang, X. Autophagic flux disruption contributes to Ganoderma lucidum polysaccharide-induced apoptosis in human colorectal cancer cells via MAPK/ERK activation. Cell Death Dis. 2019, 10, 456. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Guardia, C.M.; Roy, A.; Vassilev, A.; Saric, A.; Griner, L.N.; Marugan, J.; Ferrer, M.; Bonifacino, J.S.; DePamphilis, M.L. A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis. Autophagy 2019, 15, 1694–1718. [Google Scholar] [CrossRef]
- Bongiorno-Borbone, L.; Giacobbe, A.; Compagnone, M.; Eramo, A.; De Maria, R.; Peschiaroli, A.; Melino, G. Anti-tumoral effect of desmethylclomipramine in lung cancer stem cells. Oncotarget 2015, 6, 16926–16938. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhu, L.; Zhao, Y.; Jiang, Y.; Chen, L.; Yu, Y.; Ouyang, L. Fluoxetine induces autophagic cell death via eEF2K-AMPK-mTOR-ULK complex axis in triple negative breast cancer. Cell Prolif. 2018, 51, e12402. [Google Scholar] [CrossRef]
- Khing, T.M.; Po, W.W.; Sohn, U.D. Fluoxetine Enhances Anti-tumor Activity of Paclitaxel in Gastric Adenocarcinoma Cells by Triggering Apoptosis and Necroptosis. Anticancer Res. 2019, 39, 6155–6163. [Google Scholar] [CrossRef]
- Yin, Y.C.; Lin, C.C.; Chen, T.T.; Chen, J.Y.; Tsai, H.J.; Wang, C.Y.; Chen, S.Y. Clozapine induces autophagic cell death in non-small cell lung cancer cells. Cell. Physiol. Biochem. 2015, 35, 945–956. [Google Scholar] [CrossRef]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafa, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Giorgi, C. Novel function of the tumor suppressor PML at ER-mitochondria sites in the control of autophagy. Oncotarget 2017, 8, 81723–81724. [Google Scholar] [CrossRef] [PubMed]
- Selvakumaran, M.; Amaravadi, R.K.; Vasilevskaya, I.A.; O’Dwyer, P.J. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin. Cancer Res. 2013, 19, 2995–3007. [Google Scholar] [CrossRef]
- Xie, X.; White, E.P.; Mehnert, J.M. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS ONE 2013, 8, e55096. [Google Scholar] [CrossRef]
- Dragowska, W.H.; Weppler, S.A.; Wang, J.C.; Wong, L.Y.; Kapanen, A.I.; Rawji, J.S.; Warburton, C.; Qadir, M.A.; Donohue, E.; Roberge, M.; et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS ONE 2013, 8, e76503. [Google Scholar] [CrossRef]
- Cook, K.L.; Warri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ breast cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Q.; Pan, D.; Zhang, S.W.; Zheng, X.L.; Chen, H. Autophagy regulates chemoresistance of gastric cancer stem cells via the Notch signaling pathway. Eur. Rev. Med. Pharm. Sci. 2018, 22, 3402–3407. [Google Scholar] [CrossRef]
- Sun, R.; Shen, S.; Zhang, Y.J.; Xu, C.F.; Cao, Z.T.; Wen, L.P.; Wang, J. Nanoparticle-facilitated autophagy inhibition promotes the efficacy of chemotherapeutics against breast cancer stem cells. Biomaterials 2016, 103, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Singh, S.K.; Bhattacharyya, S. JAK-mediated autophagy regulates stemness and cell survival in cisplatin resistant bladder cancer cells. Biochim. Biophys. Acta 2016, 1860, 2484–2497. [Google Scholar] [CrossRef]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Hamai, A.; Tonelli, G.; Bauvy, C.; Nicolas, V.; Tharinger, H.; Codogno, P.; Mehrpour, M. Inhibition of the autophagic flux by salinomycin in breast cancer stem-like/progenitor cells interferes with their maintenance. Autophagy 2013, 9, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.; Wolf, D.; Ruocco, V. Antimalarials: Unapproved uses or indications. Clin. Dermatol. 2000, 18, 17–35. [Google Scholar] [CrossRef]
- Sotelo, J.; Briceno, E.; Lopez-Gonzalez, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.B.; Supko, J.G.; Neal, J.W.; Muzikansky, A.; Digumarthy, S.; Fidias, P.; Temel, J.S.; Heist, R.S.; Shaw, A.T.; McCarthy, P.O.; et al. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J. Thorac. Oncol. 2012, 7, 1602–1608. [Google Scholar] [CrossRef]
- El-Chemaly, S.; Taveira-Dasilva, A.; Goldberg, H.J.; Peters, E.; Haughey, M.; Bienfang, D.; Jones, A.M.; Julien-Williams, P.; Cui, Y.; Villalba, J.A.; et al. Sirolimus and Autophagy Inhibition in Lymphangioleiomyomatosis: Results of a Phase I Clinical Trial. Chest 2017, 151, 1302–1310. [Google Scholar] [CrossRef]
- Chi, M.S.; Lee, C.Y.; Huang, S.C.; Yang, K.L.; Ko, H.L.; Chen, Y.K.; Chung, C.H.; Liao, K.W.; Chi, K.H. Double autophagy modulators reduce 2-deoxyglucose uptake in sarcoma patients. Oncotarget 2015, 6, 29808–29817. [Google Scholar] [CrossRef]
- Chi, K.H.; Ko, H.L.; Yang, K.L.; Lee, C.Y.; Chi, M.S.; Kao, S.J. Addition of rapamycin and hydroxychloroquine to metronomic chemotherapy as a second line treatment results in high salvage rates for refractory metastatic solid tumors: A pilot safety and effectiveness analysis in a small patient cohort. Oncotarget 2015, 6, 16735–16745. [Google Scholar] [CrossRef]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Puentes, L.L.; Gonzalez-Pinedo, M.; Crismatt, A.; Ortega-Gomez, A.; Gamboa-Vignolle, C.; Nunez-Gomez, R.; Dorantes-Gallareta, Y.; Arce-Salinas, C.; Arrieta, O. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol. 2013, 8, 209. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [PubMed]
- Horne, G.A.; Stobo, J.; Kelly, C.; Mukhopadhyay, A.; Latif, A.L.; Dixon-Hughes, J.; McMahon, L.; Cony-Makhoul, P.; Byrne, J.; Smith, G.; et al. A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia 2020, 34, 1775–1786. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Autophagy-Related Gene Identified | Function | References |
|---|---|---|---|
| Breast | BCL2, BIRC5, EIF4EBP1, ERO1L, FOS, GAPDH, ITPR1, and VEGFA | Upregulated levels have been associated with increased survival | [118] |
| EIF4EBP1 and ATG4A | Risk associated genes in advanced breast cancer subgroups (stage III–IV) | [119] | |
| BAG1, MAP1LC3A, and SERPINA1 | Protective genes in advanced breast cancer subgroups (III–IV) | [119] | |
| VPS35 | Oncogenic and prognostic factor | [120] | |
| Colorectal | CX3CL1, ULK3, CDKN2A, NRG1, ATG4B, GAA, RGS19, DDIT3, GRID1, DAPK1, and SERPINA1 | Associated with the immune microenvironment of CRC | [121] |
| LC3 and BECN1 | Low value has been associated with a good response to treatment and a good survival prognosis | [122] | |
| LC3A | Increased expression was linked to metastasis and a worse prognosis in patients with stage IIA-III colorectal adenocarcinomas | [115] | |
| Lung | ATG10 | High expression levels were associated with unfavorable prognosis in non-small cell lung cancer | [123] |
| LC3A | Increased levels associated with a poor overall survival | [124] | |
| ATG16, DRAM, ATG12 | High expression in lung adenocarcinoma | [117] | |
| LAMP2, ATG5, LC3 | High expression in squamous cell lung cancer | [117] | |
| Bladder cancer | ATG12, FYCO1, TECPR1, and ULK1 | Reduced expression levels in bilharzial bladder cancer | [125] |
| ATG4B, DRAM, ATG5, PTEN, and ULK | High expression associated with higher survival | [126] |
| Treatment | Tumor Type | Study Phase | Results | Clinical Trials.gov Identifier | Publication |
|---|---|---|---|---|---|
| HCQ 200 mg twice daily with docetaxel 75 mg/m2 intravenously (IV) every 21 days on day 1 of the treatment cycle. A cycle is defined as 21 days. | Metastatic prostate cancer | Phase II | Lack of efficacy | NCT00786682 | NONE |
| Erlotinib 150 mg per day with HCQ given at escalating doses of 400, 600, 800, and 1000 mg per day. | Advanced non-small cell lung cancer (NSCLC) | Phase II | Low efficacy | NCT01026844 | [287] |
| Daily administration of HCQ (400 mg) and rapamycin (2 mg) in combination with metronomic chemotherapy. | Refractory metastatic solid tumors | Phase I | Benefit rate of 84% in cohort patients | NCT00909831 | NONE |
| HCQ (200 and 400 mg daily) with rapamycin 2 mg twice a week. | Lymphangioleiomyomatosis | Phase I | Limited response | NCT01687179 | [288] |
| HCQ (1200 mg daily) with rapamycin 2 mg twice a week. | Pancreatic cancer | Phase II | Moderate response | NCT01978184 | NONE |
| HCQ (400 mg) with rapamycin 2 mg daily for 2 treatment cycles composed of 28 days each. | Soft tissue sarcoma | Phase II | Partial response | NCT01842594 | [289] |
| HCQ (200 mg daily) with temozolomide (150–200 mg/m2 IV every 28 days) on 1st day of radiotherapy. After 6 weeks, 4 weeks of HCQ alone daily. | Brain and central nervous system tumors | Phase I | Efficacious to block autophagy | NCT00486603 | [286] |
| HCQ (200 mg daily) with temozolomide (75 mg/m2 IV every 28 days) on 1st day of radiotherapy. After 6 weeks, 4 weeks of HCQ alone daily. | Brain and central nervous system tumors | Phase II | Limited response | NCT00486603 | [286] |
| Daily HCQ (200, 400, 600, 800, 1000, or 1200 mg) after first dose of gemcitabine (10 mg/m2 IV) on days 1 and 15, prior to surgical resection. | Subjects with high risk stage IIb or III adenocarcinoma of the pancreas | Phase I/II | Safe and well tolerated with encouraging oncologic outcomes | NCT01128296 | [284] |
| HCQ 200–600 every day. Bortezomib 1.0–1.3 mg/m2 IV at days 1, 4, 8, and 11 of each 21 day cycle. | Multiple myeloma and plasma cell neoplasm | Phase I | Partial response | NCT00568880 | [290] |
| Vorinostat (400 mg daily) with gemcitabine (1000 mg/m2) and abraxane (125 mg/m2) IV on days 3, 10, 17, 31, 38, and 45 as an intravenous infusion. | Breast cancer | Phase II | Moderate response | NCT00365599 | [291] |
| CQ 150 mg 1 h prior to the radiation treatment (30 Gy in 10 daily fractions from Monday to Friday). CQ treatment continues for 28 days. | Brain metastasis | Phase II | Limited response | NCT01894633 | [292] |
| HCQ low (400 mg twice per day) and high (600 mg twice per day) dose for 1 month prior to surgical removal of the ductal carcinoma in situ lesion. | Metastatic pancreatic cancer | Phase II | Lack of efficacy | NCT01273805 | [293] |
| Oral imatinib mesylate (IM, 400–600 mg) daily and oral HCQ (400 mg) twice daily. Treatment repeats every 4 weeks for up to 12 months. | Chronic myeloid leukemia | Phase II | Well tolerated; clinical advantage for 48 weeks in IM/HCQ group | NCT01227135 | [294] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patergnani, S.; Missiroli, S.; Morciano, G.; Perrone, M.; Mantovani, C.M.; Anania, G.; Fiorica, F.; Pinton, P.; Giorgi, C. Understanding the Role of Autophagy in Cancer Formation and Progression Is a Real Opportunity to Treat and Cure Human Cancers. Cancers 2021, 13, 5622. https://doi.org/10.3390/cancers13225622
Patergnani S, Missiroli S, Morciano G, Perrone M, Mantovani CM, Anania G, Fiorica F, Pinton P, Giorgi C. Understanding the Role of Autophagy in Cancer Formation and Progression Is a Real Opportunity to Treat and Cure Human Cancers. Cancers. 2021; 13(22):5622. https://doi.org/10.3390/cancers13225622
Chicago/Turabian StylePatergnani, Simone, Sonia Missiroli, Giampaolo Morciano, Mariasole Perrone, Cristina M. Mantovani, Gabriele Anania, Francesco Fiorica, Paolo Pinton, and Carlotta Giorgi. 2021. "Understanding the Role of Autophagy in Cancer Formation and Progression Is a Real Opportunity to Treat and Cure Human Cancers" Cancers 13, no. 22: 5622. https://doi.org/10.3390/cancers13225622
APA StylePatergnani, S., Missiroli, S., Morciano, G., Perrone, M., Mantovani, C. M., Anania, G., Fiorica, F., Pinton, P., & Giorgi, C. (2021). Understanding the Role of Autophagy in Cancer Formation and Progression Is a Real Opportunity to Treat and Cure Human Cancers. Cancers, 13(22), 5622. https://doi.org/10.3390/cancers13225622