
Venetoclax in Acute Myeloid Leukemia: Molecular Basis, Evidences for Preclinical and Clinical Efficacy and Strategies to Target Resistance

Abstract
:Simple Summary
Abstract
1. Introduction
2. Molecular Basis for BCL-2 Homology 3 (BH3)-Mimetics Drugs in Cancer Therapy
2.1. Apoptotic Pathway
2.2. The BH3-Mimetics Class in Cancer Therapy
2.3. Rationale for Targeting BCL-2 in AML
2.4. BCL-2 Inhibitors in AML, Preclinical Studies
3. Clinical Studies of VEN in AML
3.1. VEN Monotherapy
3.2. Venetoclax in Combination with HMA
3.2.1. Phase I Study
3.2.2. Phase II Study
3.2.3. Phase III Study (VIALE-A Trial)
3.3. Combination with LDAC
3.3.1. Phase I/II Study
3.3.2. Phase III Study (VIALE-C)
3.4. HMA or LDAC-Based Combinations in Patients with RR AML
3.5. Combination with Intensive Chemotherapy
3.5.1. High Dose Cytarabine + Idarubicin-Based Regimens
3.5.2. FLAG-IDA Regimen
3.5.3. CPX-351
3.5.4. Cladribine-Based Regimen
3.5.5. Other Studies
3.6. Combination with Targeted Agents
3.6.1. IDH Inhibitors
3.6.2. FLT3 Inhibitors
4. Molecular Factors Driving Resistance
4.1. Genetic Factors
4.1.1. TP53
4.1.2. Other Genes
4.2. Non-Genetic Factors
4.2.1. BH3 Protein Expression and Occupation
4.2.2. Cellular Differentiation
4.2.3. Metabolic Factors
4.2.4. Mitochondrial Structure and Machinery
5. Future Directions
5.1. Clinical Studies
5.2. Preclinical Studies
5.3. In Vitro Preclinical Screening
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Röllig, C.; Ossenkoppele, G.J. (Eds.) Acute Myeloid Leukemia; Hematologic Malignancies; Springer International Publishing: Berlin/Heidelberg, Germany, 2021. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International Phase 3 Study of Azacitidine vs Conventional Care Regimens in Older Patients with Newly Diagnosed AML with >30% Blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.-P.; Chou, W.-C.; Buckstein, R.; Cermak, J.; et al. Multicenter, Randomized, Open-Label, Phase III Trial of Decitabine versus Patient Choice, with Physician Advice, of Either Supportive Care or Low-Dose Cytarabine for the Treatment of Older Patients with Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, A.K.; Milligan, D.; Prentice, A.G.; Goldstone, A.H.; McMullin, M.F.; Hills, R.K.; Wheatley, K. A Comparison of Low-Dose Cytarabine and Hydroxyurea with or without All-Trans Retinoic Acid for Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome in Patients Not Considered Fit for Intensive Treatment. Cancer 2007, 109, 1114–1124. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the Chromosome Breakpoint of Neoplastic B Cells with the t(14;18) Chromosome Translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Bakhshi, A.; Jensen, J.P.; Goldman, P.; Wright, J.J.; McBride, O.W.; Epstein, A.L.; Korsmeyer, S.J. Cloning the Chromosomal Breakpoint of t(14;18) Human Lymphomas: Clustering around JH on Chromosome 14 and near a Transcriptional Unit on 18. Cell 1985, 41, 899–906. [Google Scholar] [CrossRef]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 Gene Promotes Haemopoietic Cell Survival and Cooperates with c-Myc to Immortalize Pre-B Cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- Hockenbery, D.; Nuñez, G.; Milliman, C.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 Is an Inner Mitochondrial Membrane Protein That Blocks Programmed Cell Death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of Apoptosis in Health and Disease: The Balancing Act of BCL-2 Family Proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Sorcinelli, M.D.; Beard, C.; Korsmeyer, S.J. Antiapoptotic BCL-2 Is Required for Maintenance of a Model Leukemia. Cancer Cell 2004, 6, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Marcucci, G.; Byrd, J.C.; Dai, G.; Klisovic, M.I.; Kourlas, P.J.; Young, D.C.; Cataland, S.R.; Fisher, D.B.; Lucas, D.; Chan, K.K.; et al. Phase 1 and Pharmacodynamic Studies of G3139, a Bcl-2 Antisense Oligonucleotide, in Combination with Chemotherapy in Refractory or Relapsed Acute Leukemia. Blood 2003, 101, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An Inhibitor of Bcl-2 Family Proteins Induces Regression of Solid Tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Del Gaizo Moore, V.; Brown, J.R.; Certo, M.; Love, T.M.; Novina, C.D.; Letai, A. Chronic Lymphocytic Leukemia Requires BCL2 to Sequester Prodeath BIM, Explaining Sensitivity to BCL2 Antagonist ABT-737. J. Clin. Investig. 2007, 117, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Tsao, T.; Shi, Y.; Kornblau, S.; Lu, H.; Konoplev, S.; Antony, A.; Ruvolo, V.; Qiu, Y.H.; Zhang, N.; Coombes, K.R.; et al. Concomitant Inhibition of DNA Methyltransferase and BCL-2 Protein Function Synergistically Induce Mitochondrial Apoptosis in Acute Myelogenous Leukemia Cells. Ann. Hematol. 2012, 91, 1861–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Nimmer, P.M.; Tahir, S.K.; Chen, J.; Fryer, R.M.; Hahn, K.R.; Iciek, L.A.; Morgan, S.J.; Nasarre, M.C.; Nelson, R.; et al. Bcl-2 Family Proteins Are Essential for Platelet Survival. Cell Death Differ. 2007, 14, 943–951. [Google Scholar] [CrossRef]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.-L.; et al. Navitoclax, a Targeted High-Affinity Inhibitor of BCL-2, in Lymphoid Malignancies: A Phase 1 Dose-Escalation Study of Safety, Pharmacokinetics, Pharmacodynamics, and Antitumour Activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.S.; Xiong, H.; et al. Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients with Relapsed or Refractory Disease. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity While Sparing Platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Coutre, S.; Seymour, J.F.; Munir, T.; Puvvada, S.D.; Wendtner, C.-M.; Roberts, A.W.; Jurczak, W.; et al. Venetoclax in Relapsed or Refractory Chronic Lymphocytic Leukaemia with 17p Deletion: A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef]
- Inoue-Yamauchi, A.; Jeng, P.S.; Kim, K.; Chen, H.-C.; Han, S.; Ganesan, Y.T.; Ishizawa, K.; Jebiwott, S.; Dong, Y.; Pietanza, M.C.; et al. Targeting the Differential Addiction to Anti-Apoptotic BCL-2 Family for Cancer Therapy. Nat. Commun. 2017, 8, 16078. [Google Scholar] [CrossRef]
- Konopleva, M.; Letai, A. BCL-2 Inhibition in AML: An Unexpected Bonus? Blood 2018, 132, 1007–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria Primed by Death Signals Determine Cellular Addiction to Antiapoptotic BCL-2 Family Members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, S.; Pioso, M.S.; Olesinski, E.A.; Yilma, B.; Ryan, J.A.; Mashaka, T.; Leutz, B.; Adamia, S.; Zhu, H.; Kuang, Y.; et al. Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 872–890.e6. [Google Scholar] [CrossRef]
- Vo, T.-T.; Ryan, J.; Carrasco, R.; Neuberg, D.; Rossi, D.J.; Stone, R.M.; Deangelo, D.J.; Frattini, M.G.; Letai, A. Relative Mitochondrial Priming of Myeloblasts and Normal HSCs Determines Chemotherapeutic Success in AML. Cell 2012, 151, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Carlson, N.; Takeyama, K.; Dal Cin, P.; Shipp, M.; Letai, A. BH3 Profiling Identifies Three Distinct Classes of Apoptotic Blocks to Predict Response to ABT-737 and Conventional Chemotherapeutic Agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Leverson, J.D.; Sampath, D.; Souers, A.J.; Rosenberg, S.H.; Fairbrother, W.J.; Amiot, M.; Konopleva, M.; Letai, A. Found in Translation: How Preclinical Research Is Guiding the Clinical Development of the BCL2-Selective Inhibitor Venetoclax. Cancer Discov. 2017, 7, 1376–1393. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.-X.; Sneed, T.; et al. Mechanisms of Apoptosis Sensitivity and Resistance to the BH3 Mimetic ABT-737 in Acute Myeloid Leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurlet, S.; Omidvar, N.; Gorombei, P.; Krief, P.; Le Pogam, C.; Setterblad, N.; de la Grange, P.; Leboeuf, C.; Janin, A.; Noguera, M.-E.; et al. BCL-2 Inhibition with ABT-737 Prolongs Survival in an NRAS/BCL-2 Mouse Model of AML by Targeting Primitive LSK and Progenitor Cells. Blood 2013, 122, 2864–2876. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 Inhibition by ABT-199 Causes on-Target Cell Death in Acute Myeloid Leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogenberger, J.M.; Kornblau, S.M.; Pierceall, W.E.; Lena, R.; Chow, D.; Shi, C.-X.; Mantei, J.; Ahmann, G.; Gonzales, I.M.; Choudhary, A.; et al. BCL-2 Family Proteins as 5-Azacytidine-Sensitizing Targets and Determinants of Response in Myeloid Malignancies. Leukemia 2014, 28, 1657–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and Preliminary Efficacy of Venetoclax with Decitabine or Azacitidine in Elderly Patients with Previously Untreated Acute Myeloid Leukaemia: A Non-Randomised, Open-Label, Phase 1b Study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax Combined with Decitabine or Azacitidine in Treatment-Naive, Elderly Patients with Acute Myeloid Leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Maiti, A.; Rausch, C.R.; Pemmaraju, N.; Naqvi, K.; Daver, N.G.; Kadia, T.M.; Borthakur, G.; Ohanian, M.; Alvarado, Y.; et al. 10-Day Decitabine with Venetoclax for Newly Diagnosed Intensive Chemotherapy Ineligible, and Relapsed or Refractory Acute Myeloid Leukaemia: A Single-Centre, Phase 2 Trial. Lancet Haematol. 2020, 7, e724–e736. [Google Scholar] [CrossRef]
- Maiti, A.; DiNardo, C.D.; Wang, S.A.; Jorgensen, J.; Kadia, T.M.; Daver, N.G.; Short, N.J.; Yilmaz, M.; Pemmaraju, N.; Borthakur, G.; et al. Prognostic Value of Measurable Residual Disease after Venetoclax and Decitabine in Acute Myeloid Leukemia. Blood Adv. 2021, 5, 1876–1883. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Strickland, S.A.; Hou, J.-Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for Newly Diagnosed AML Ineligible for Intensive Chemotherapy: A Phase 3 Randomized Placebo-Controlled Trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; DeAngelo, D.J.; Galinsky, I.; Kokulis, C.; Stewart, J.M.; McGinnis, M.; Werner, L.; Letai, A.G.; Konopleva, M.Y.; Luskin, M. Phase I Trial of Escalating Doses of the Bcl-2 Inhibitor Venetoclax in Combination with Daunorubicin/Cytarabine Induction and High Dose Cytarabine Consolidation in Previously Untreated Adults with Acute Myeloid Leukemia (AML). Blood 2019, 134 (Suppl. S1), 3908. [Google Scholar] [CrossRef]
- Chua, C.C.; Roberts, A.W.; Reynolds, J.; Fong, C.Y.; Ting, S.B.; Salmon, J.M.; MacRaild, S.; Ivey, A.; Tiong, I.S.; Fleming, S.; et al. Chemotherapy and Venetoclax in Elderly Acute Myeloid Leukemia Trial (CAVEAT): A Phase Ib Dose-Escalation Study of Venetoclax Combined With Modified Intensive Chemotherapy. J. Clin. Oncol. 2020, 38, 3506–3517. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Xiao, L.; Kadia, T.; Daver, N.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax Combined with FLAG-IDA Induction and Consolidation in Newly Diagnosed and Relapsed or Refractory Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, JCO2003736. [Google Scholar]
- Kadia, T.M.; Borthakur, G.; Takahashi, K.; DiNardo, C.D.; Daver, N.; Pemmaraju, N.; Jabbour, E.; Jain, N.; Short, N.J.; Qiao, W.; et al. Phase II Study of CPX-351 Plus Venetoclax in Patients with Acute Myeloid Leukemia (AML). Blood 2020, 136 (Suppl. S1), 20–22. [Google Scholar] [CrossRef]
- Kadia, T.M.; Reville, P.K.; Borthakur, G.; Yilmaz, M.; Kornblau, S.; Alvarado, Y.; Dinardo, C.D.; Daver, N.; Jain, N.; Pemmaraju, N.; et al. Venetoclax plus Intensive Chemotherapy with Cladribine, Idarubicin, and Cytarabine in Patients with Newly Diagnosed Acute Myeloid Leukaemia or High-Risk Myelodysplastic Syndrome: A Cohort from a Single-Centre, Single-Arm, Phase 2 Trial. Lancet Haematol. 2021, 8, e552–e561. [Google Scholar] [CrossRef]
- Kadia, T. Proceedings of the Phase II Study of Venetoclax Added to Cladribine + Low Dose AraC (LDAC) Alternating with 5-Azacytidine Demonstrates High Rates of Minimal Residual Disease (MRD) Negative Complete Remissions (CR) and Excellent Tolerability in Older Patients with Newly Diagnosed Acute Myeloid Leukemia (AML), ASH (Virtual meeting), 5 December 2020.
- Lachowiez, C. Proceedings of the A Phase Ib/Ii Study of Ivosidenib with Venetoclax +/− Azacitidine in Idh1-Mutated Myeloid Malignancies, EHA (Virtual meeting), 6 September 2021.
- Daver, N. Proceedings of the Efficacy and Safety of Venetoclax in Combination with Gilteritinib for Relapsed/Refractory FLT3-Mutated Acute Myeloid Leukemia in the Expansion Cohort of a Phase 1b Study, ASH (Virtual meeting), 6 December 2020. [CrossRef]
- Maiti, A.; DiNardo, C.D.; Daver, N.G.; Rausch, C.R.; Ravandi, F.; Kadia, T.M.; Pemmaraju, N.; Borthakur, G.; Bose, P.; Issa, G.C.; et al. Triplet Therapy with Venetoclax, FLT3 Inhibitor and Decitabine for FLT3-Mutated Acute Myeloid Leukemia. Blood Cancer J. 2021, 11, 25. [Google Scholar] [CrossRef]
- Issa, J.-P.J.; Garcia-Manero, G.; Giles, F.J.; Mannari, R.; Thomas, D.; Faderl, S.; Bayar, E.; Lyons, J.; Rosenfeld, C.S.; Cortes, J.; et al. Phase 1 Study of Low-Dose Prolonged Exposure Schedules of the Hypomethylating Agent 5-Aza-2’-Deoxycytidine (Decitabine) in Hematopoietic Malignancies. Blood 2004, 103, 1635–1640. [Google Scholar] [CrossRef] [Green Version]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Pratz, K.W. Proceedings of the Measurable Residual Disease Response in Acute Myeloid Leukemia Treated with Venetoclax and Azacitidine, EHA (Virtual meeting), 6 September 2020.
- DiNardo, C.D.; Wei, A.H. How I Treat Acute Myeloid Leukemia in the Era of New Drugs. Blood 2020, 135, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Rausch, C.R.; DiNardo, C.D.; Maiti, A.; Jammal, N.J.; Kadia, T.M.; Marx, K.R.; Borthakur, G.; Savoy, J.M.; Pemmaraju, N.; DiPippo, A.J.; et al. Duration of Cytopenias with Concomitant Venetoclax and Azole Antifungals in Acute Myeloid Leukemia. Cancer 2021, 127, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Samra, B.; Konopleva, M.; Isidori, A.; Daver, N.; DiNardo, C. Venetoclax-Based Combinations in Acute Myeloid Leukemia: Current Evidence and Future Directions. Front. Oncol. 2020, 10, 562558. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Rausch, C.R.; Benton, C.; Kadia, T.; Jain, N.; Pemmaraju, N.; Daver, N.; Covert, W.; Marx, K.R.; Mace, M.; et al. Clinical Experience with the BCL2-Inhibitor Venetoclax in Combination Therapy for Relapsed and Refractory Acute Myeloid Leukemia and Related Myeloid Malignancies. Am. J. Hematol. 2018, 93, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Aldoss, I.; Yang, D.; Aribi, A.; Ali, H.; Sandhu, K.; Al Malki, M.M.; Mei, M.; Salhotra, A.; Khaled, S.; Nakamura, R.; et al. Efficacy of the Combination of Venetoclax and Hypomethylating Agents in Relapsed/Refractory Acute Myeloid Leukemia. Haematologica 2018, 103, e404–e407. [Google Scholar] [CrossRef]
- Winters, A.C.; Gutman, J.A.; Purev, E.; Nakic, M.; Tobin, J.; Chase, S.; Kaiser, J.; Lyle, L.; Boggs, C.; Halsema, K.; et al. Real-World Experience of Venetoclax with Azacitidine for Untreated Patients with Acute Myeloid Leukemia. Blood Adv. 2019, 3, 2911–2919. [Google Scholar] [CrossRef] [Green Version]
- Ni Chonghaile, T.; Sarosiek, K.A.; Vo, T.-T.; Ryan, J.A.; Tammareddi, A.; Moore, V.D.G.; Deng, J.; Anderson, K.C.; Richardson, P.; Tai, Y.-T.; et al. Pretreatment Mitochondrial Priming Correlates with Clinical Response to Cytotoxic Chemotherapy. Science 2011, 334, 1129–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, J.; Sarosiek, K.A.; DeAngelo, J.D.; Maertens, O.; Ryan, J.; Ercan, D.; Piao, H.; Horowitz, N.S.; Berkowitz, R.S.; Matulonis, U.; et al. Drug-Induced Death Signaling Strategy Rapidly Predicts Cancer Response to Chemotherapy. Cell 2015, 160, 977–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Hunter, A.E.; Kjeldsen, L.; Yin, J.; Gibson, B.E.S.; Wheatley, K.; Milligan, D. Optimization of Chemotherapy for Younger Patients with Acute Myeloid Leukemia: Results of the Medical Research Council AML15 Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; DiNardo, C.D.; Nogueras-Gonzalez, G.M.; Kadia, T.M.; Jabbour, E.; Bueso-Ramos, C.E.; O’Brien, S.M.; Konopleva, M.; Jain, N.B.; Daver, N.G.; et al. Results of Second Salvage Therapy in 673 Adults with Acute Myelogenous Leukemia Treated at a Single Institution since 2000. Cancer 2018, 124, 2534–2540. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (Cytarabine and Daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Kadia, T.M.; Cortes, J.; Ravandi, F.; Jabbour, E.; Konopleva, M.; Benton, C.B.; Burger, J.; Sasaki, K.; Borthakur, G.; DiNardo, C.D.; et al. Cladribine and Low-Dose Cytarabine Alternating with Decitabine as Front-Line Therapy for Elderly Patients with Acute Myeloid Leukaemia: A Phase 2 Single-Arm Trial. Lancet Haematol. 2018, 5, e411–e421. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Döhner, H.; Campbell, P.J. Genomic Classification in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 375, 900–901. [Google Scholar] [CrossRef]
- Tommasini-Ghelfi, S.; Murnan, K.; Kouri, F.M.; Mahajan, A.S.; May, J.L.; Stegh, A.H. Cancer-Associated Mutation and beyond: The Emerging Biology of Isocitrate Dehydrogenases in Human Disease. Sci. Adv. 2019, 5, eaaw4543. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.-J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate Dehydrogenase 1 and 2 Mutations Induce BCL-2 Dependence in Acute Myeloid Leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chyla, B.J.; Harb, J.; Mantis, C.; Riehm, J.J.; Ross, J.A.; Sun, Y.; Huang, X.; Jiang, Q.; Dail, M.; Peale, F.V., Jr.; et al. Response to Venetoclax in Combination with Low Intensity Therapy (LDAC or HMA) in Untreated Patients with Acute Myeloid Leukemia Patients with IDH, FLT3 and Other Mutations and Correlations with BCL2 Family Expression. Blood 2019, 134 (Suppl. S1), 546. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, A.S.; Stein, E.M.; Fathi, A.T.; Frankfurt, O.; Schuh, A.C.; Döhner, H.; Martinelli, G.; Patel, P.A.; Raffoux, E.; et al. Mutant Isocitrate Dehydrogenase 1 Inhibitor Ivosidenib in Combination With Azacitidine for Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Dinardo, C.D.; Schuh, A.C.; Stein, E.M.; Montesinos, P.; Wei, A.; De Botton, S.; Zeidan, A.M.; Fathi, A.T.; Quek, L.; Kantarjian, H.M.; et al. Effect of Enasidenib (ENA) plus Azacitidine (AZA) on Complete Remission and Overall Response versus AZA Monotherapy in Mutant-IDH2 (MIDH2) Newly Diagnosed Acute Myeloid Leukemia (ND-AML). J. Clin. Oncol. 2020, 38, 7501. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Krämer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus Salvage Chemotherapy in Relapsed or Refractory FLT3-ITD Acute Myeloid Leukaemia (QuANTUM-R): A Multicentre, Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, S.; Qiao, X.; Knight, T.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Wang, G.; et al. Inhibition of Bcl-2 Synergistically Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6815–6826. [Google Scholar] [CrossRef]
- Singh Mali, R.; Zhang, Q.; DeFilippis, R.A.; Cavazos, A.; Kuruvilla, V.M.; Raman, J.; Mody, V.; Choo, E.F.; Dail, M.; Shah, N.P.; et al. Venetoclax Combines Synergistically with FLT3 Inhibition to Effectively Target Leukemic Cells in FLT3-ITD+ Acute Myeloid Leukemia Models. Haematologica 2021, 106, 1034–1046. [Google Scholar] [CrossRef]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C.; Ferreira-Gonzalez, A.; Grant, S. Inhibition of Bcl-2 Antiapoptotic Members by Obatoclax Potently Enhances Sorafenib-Induced Apoptosis in Human Myeloid Leukemia Cells through a Bim-Dependent Process. Blood 2012, 119, 6089–6098. [Google Scholar] [CrossRef]
- Ohanian, M.; Garcia-Manero, G.; Levis, M.; Jabbour, E.; Daver, N.; Borthakur, G.; Kadia, T.; Pierce, S.; Burger, J.; Richie, M.A.; et al. Sorafenib Combined with 5-Azacytidine in Older Patients with Untreated FLT3-ITD Mutated Acute Myeloid Leukemia. Am. J. Hematol. 2018, 93, 1136–1141. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, M.; Kantarjian, H.M.; Levis, M.; Guerra, V.; Borthakur, G.; Alvarado, Y.; DiNardo, C.D.; Kadia, T.; Garcia-Manero, G.; Ohanian, M.; et al. A Phase I/II Study of the Combination of Quizartinib with Azacitidine or Low-Dose Cytarabine for the Treatment of Patients with Acute Myeloid Leukemia and Myelodysplastic Syndrome. Haematologica 2021, 106, 2121–2130. [Google Scholar] [PubMed]
- Ravandi, F.; Alattar, M.L.; Grunwald, M.R.; Rudek, M.A.; Rajkhowa, T.; Richie, M.A.; Pierce, S.; Daver, N.; Garcia-Manero, G.; Faderl, S.; et al. Phase 2 Study of Azacytidine plus Sorafenib in Patients with Acute Myeloid Leukemia and FLT-3 Internal Tandem Duplication Mutation. Blood 2013, 121, 4655–4662. [Google Scholar] [CrossRef]
- Gilteritinib Plus Azacitidine Combination Shows Promise in Newly Diagnosed FLT3-Mutated AML. Oncologist 2021, 26 (Suppl. S1), S10.
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef]
- Thijssen, R.; Diepstraten, S.T.; Moujalled, D.; Chew, E.; Flensburg, C.; Shi, M.X.; Dengler, M.A.; Litalien, V.; MacRaild, S.; Chen, M.; et al. Intact TP-53 Function Is Essential for Sustaining Durable Responses to BH3-Mimetic Drugs in Leukemias. Blood 2021, 137, 2721–2735. [Google Scholar] [CrossRef]
- Kim, K.; Maiti, A.; Kadia, T.M.; Ravandi, F.; Daver, N.; Pemmaraju, N.; Borthakur, G.; Bose, P.; Issa, G.C.; Short, N.J.; et al. Outcomes of TP53-Mutant Acute Myeloid Leukemia with Venetoclax and Decitabine. Blood 2020, 136 (Suppl. S1), 33–36. [Google Scholar] [CrossRef]
- Moujalled, D. Proceedings of the Acquired Mutations in BAX Confer Resistance to BH3 Mimetics in Acute Myeloid Leukemia, ASH (Virtual meeting), 5 December 2020.
- Blombery, P.; Anderson, M.A.; Gong, J.-N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL-1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [Green Version]
- Hormi, M.; Birsen, R.; Belhadj, M.; Huynh, T.; Cantero Aguilar, L.; Grignano, E.; Haddaoui, L.; Guillonneau, F.; Mayeux, P.; Hunault, M.; et al. Pairing MCL-1 Inhibition with Venetoclax Improves Therapeutic Efficiency of BH3-Mimetics in AML. Eur. J. Haematol. 2020, 105, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Kuusanmäki, H.; Leppä, A.-M.; Pölönen, P.; Kontro, M.; Dufva, O.; Deb, D.; Yadav, B.; Brück, O.; Kumar, A.; Everaus, H.; et al. Phenotype-Based Drug Screening Reveals Association between Venetoclax Response and Differentiation Stage in Acute Myeloid Leukemia. Haematologica 2020, 105, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with Azacitidine Disrupts Energy Metabolism and Targets Leukemia Stem Cells in Patients with Acute Myeloid Leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Stuani, L.; Sabatier, M.; Sarry, J.-E. Exploiting Metabolic Vulnerabilities for Personalized Therapy in Acute Myeloid Leukemia. BMC Biol. 2019, 17, 57. [Google Scholar] [CrossRef] [Green Version]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty Acid Metabolism Underlies Venetoclax Resistance in Acute Myeloid Leukemia Stem Cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Sharon, D.; Cathelin, S.; Mirali, S.; Di Trani, J.M.; Yanofsky, D.J.; Keon, K.A.; Rubinstein, J.L.; Schimmer, A.D.; Ketela, T.; Chan, S.M. Inhibition of Mitochondrial Translation Overcomes Venetoclax Resistance in AML through Activation of the Integrated Stress Response. Sci. Transl. Med. 2019, 11, eaax2863. [Google Scholar] [CrossRef]
- Lin, K.H.; Xie, A.; Rutter, J.C.; Ahn, Y.-R.; Lloyd-Cowden, J.M.; Nichols, A.G.; Soderquist, R.S.; Koves, T.R.; Muoio, D.M.; MacIver, N.J.; et al. Systematic Dissection of the Metabolic-Apoptotic Interface in AML Reveals Heme Biosynthesis to Be a Regulator of Drug Sensitivity. Cell Metab. 2019, 29, 1217–1231.e7. [Google Scholar] [CrossRef]
- Cojocari, D.; Smith, B.N.; Purkal, J.J.; Arrate, M.P.; Huska, J.D.; Xiao, Y.; Gorska, A.; Hogdal, L.J.; Ramsey, H.E.; Boghaert, E.R.; et al. Pevonedistat and Azacitidine Upregulate NOXA (PMAIP1) to Increase Sensitivity to Venetoclax in Preclinical Models of Acute Myeloid Leukemia. Haematologica 2021. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Chung, H.; Kim, J.S.; Cheong, J.-W.; Min, Y.H. Arsenic Trioxide Synergistically Promotes the Antileukaemic Activity of Venetoclax by Downregulating Mcl-1 in Acute Myeloid Leukaemia Cells. Exp. Hematol. Oncol. 2021, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Surka, C.; Jin, L.; Mbong, N.; Lu, C.-C.; Jang, I.S.; Rychak, E.; Mendy, D.; Clayton, T.; Tindall, E.; Hsu, C.; et al. CC-90009, a Novel Cereblon E3 Ligase Modulator, Targets Acute Myeloid Leukemia Blasts and Leukemia Stem Cells. Blood 2021, 137, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Gareau, Y.; Lehnertz, B.; Gingras, S.; Spinella, J.-F.; Corneau, S.; Mayotte, N.; Girard, S.; Frechette, M.; Blouin-Chagnon, V.; et al. Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell 2019, 36, 84–99.e8. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.A.; et al. An Inhibitor of Oxidative Phosphorylation Exploits Cancer Vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting Mitochondrial Oxidative Phosphorylation Eradicates Therapy-Resistant Chronic Myeloid Leukemia Stem Cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [Green Version]
- Skrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, S.U.; Hurren, R.; Voisin, V.; Bridon, G.; Wang, X.; Xu, C.; MacLean, N.; Siriwardena, T.P.; Gronda, M.; Yehudai, D.; et al. Leveraging Increased Cytoplasmic Nucleoside Kinase Activity to Target MtDNA and Oxidative Phosphorylation in AML. Blood 2017, 129, 2657–2666. [Google Scholar] [CrossRef]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e9. [Google Scholar] [CrossRef]
- Collignon, A.; Hospital, M.A.; Montersino, C.; Courtier, F.; Charbonnier, A.; Saillard, C.; D’Incan, E.; Mohty, B.; Guille, A.; Adelaïde, J.; et al. A Chemogenomic Approach to Identify Personalized Therapy for Patients with Relapse or Refractory Acute Myeloid Leukemia: Results of a Prospective Feasibility Study. Blood Cancer J. 2020, 10, 64. [Google Scholar] [CrossRef] [PubMed]
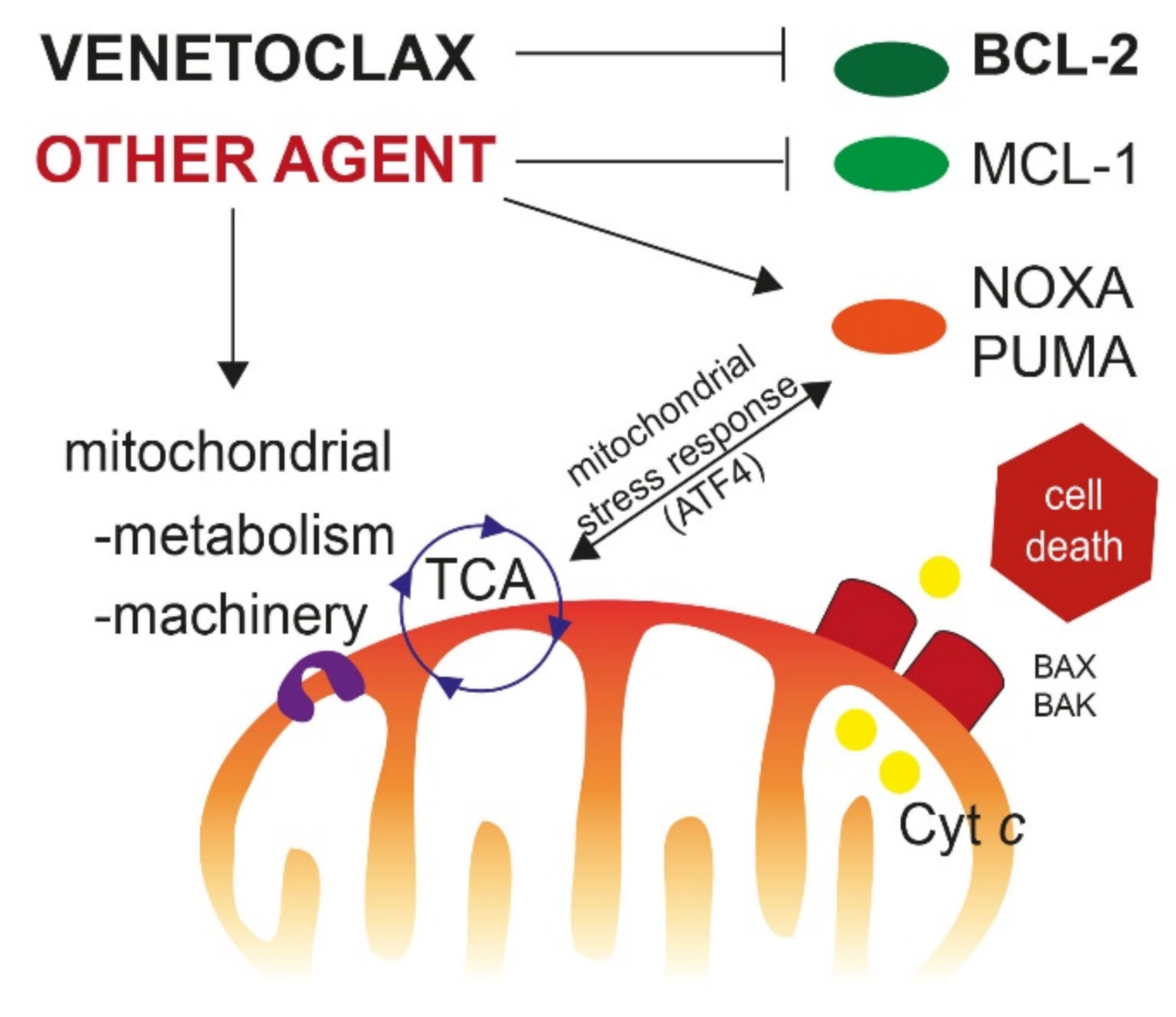

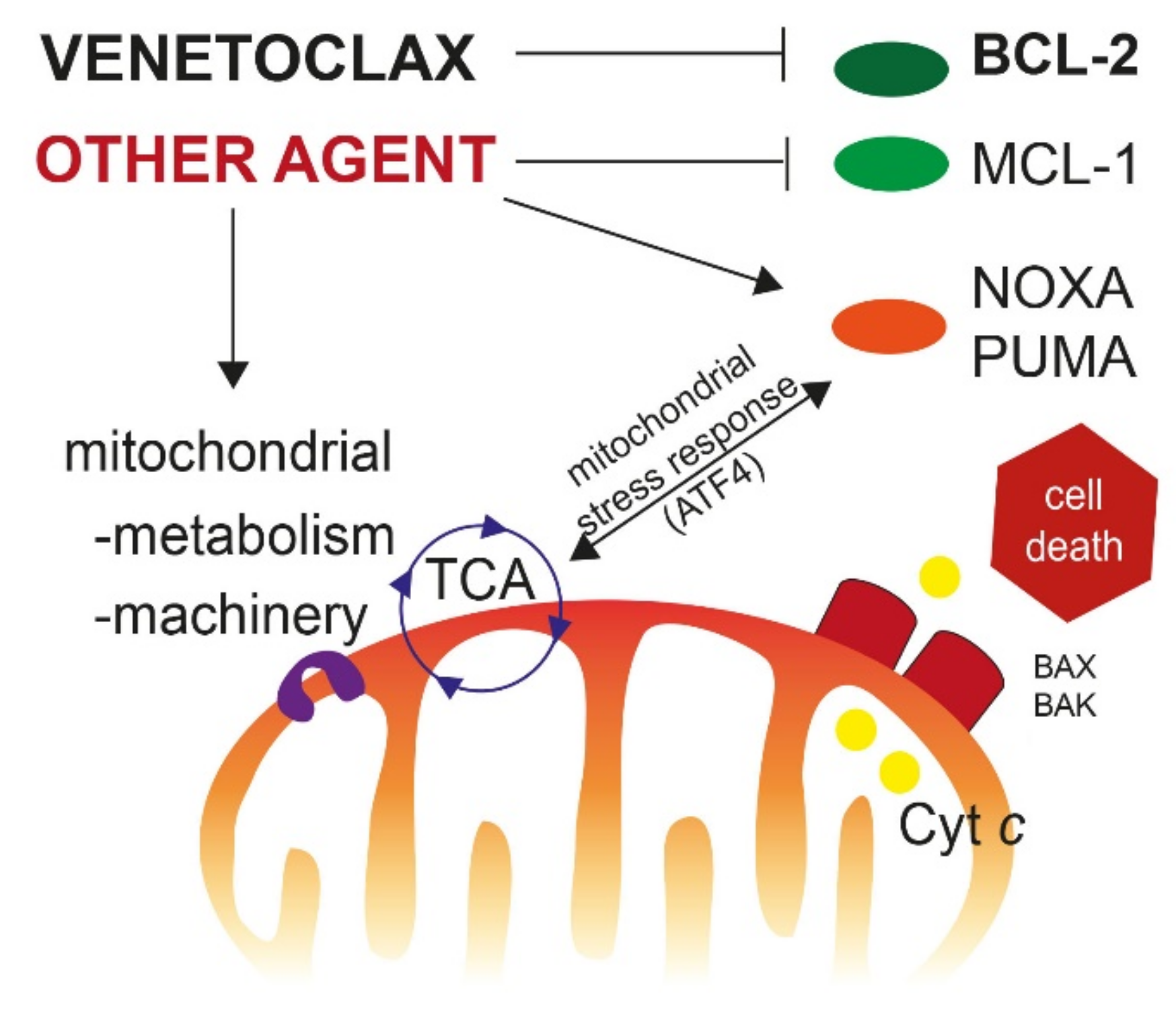{kind=link}
| Combination Therapy | Phase; Name; ID | Study Population | Venetoclax Doses | Other Agents Administration Regimens | Response Rate | Survival Rate | Ref |
|---|---|---|---|---|---|---|---|
| no | I; NCT01994837 | RR AML | 800 mg D1 to D28 | no | 19% (ORR) | 4.7 months (median OS) 2.3 months (median LFS) | [39] |
| HMA | I; (M14-358); NCT02203773 | ND AML ineligible for chemotherapy | 400–800–1200 mg D1 to D28 | AZA 75 mg/m2, D1 to D7 or DEC 20 mg/m2, D1 to D5 | 37% (CR), 68% (ORR) 83% (cCR) | 17.5 months (median OS) | [40,41] |
| II; NCT03404193 | ND patients with AML > 60 yrs RR patients > 18 years | 400 mg D1 to D28 (D1 to D21 if blasts < 5%) | DEC 20 mg/m2, D1 to D10 (induction) DEC 20 mg/m2, D1 to D5 (consolidation) | 63% (CR + CRi), 74% (ORR) | 18.1 months (median OS) | [42,43] | |
| III; (VIALE-A); NCT02993523 | ND AML ineligible for chemotherapy | 400 mg D1 to D28 vs. placebo | AZA 75 mg/m2, D1 to D7 | 36.7% vs. 17.9% (CR) 64.7% vs. 22.8% (CR + CRi) | 14.7 months vs. 9.6 months (median OS) | [44] | |
| LDAC | I/II; NCT02287233 | ND AML ineligible for chemotherapy | 600 mg D1 to D28 | LDAC 20 mg/m2 days D1 to D10 | 26% (CR) 28% (CRi) | 10.1 months (median OS) | [45] |
| III; (VIALE-C); NCT03069352 | ND AML ineligible for chemotherapy | 600 mg D1 to D28 vs. placebo | LDAC 20 mg/m2 D1 to D10 | 27% vs. 7% (CR) 21% vs. 6% (CRi) | 7.2 months vs. 4.1 months (median OS) | [46] | |
| Anthracyclin-based chemotherapy | I | ND AML <60 years | 200 to 600 mg D1 to D11 | DAUNO 60 mg/m2/d D2 to D4 CYTA 200 mg/m2/d D2 to D8 | ND | ND | [47] |
| Ib; (CAVEAT); ACTRN12616000445471 | ND AML ≥ 65 years (>60 years if monosomal karyotype) | Induction 50 to 600 mg D-6 to D7 | CYTA 100 mg/m2/d D1 to D5 IDA 12 mg/m2/d D2 to D3 | 72% (ORR) 41% (CR) 31% (CRi) | 11.2 months (median OS) | [48] | |
| Consolidation 50 to 600 mg (4 cycles) D-6 to D7 | CYTA (bolus) 100 mg/m2/d D1 to D2 IDA 12 mg/m2/d D1 | ||||||
| Maintenance (7 cycles) 50 to 600 mg D1 to D14 | no | ||||||
| Ib/II; NCT03214562 | ND, RR AML or high-risk MDS with > 10% blasts | Induction (1 or 2) 400 mg D1 to D14 | modified FLAG-IDA FLUDA 30 mg/m2 D1 to D14 CYTA 1.5 g/m2 D2 to D6 IDA 8 mg/m2 D4 to D6 (ND AML) or 6 mg/m2 D5 to D6 (RR AML) | 82 % (ORR) 37% (CR) 15% (CRh) 7% (CRi) | NR (median OS), 18 months (median EFS) | [49] | |
| II; NCT03629171 | 18–65 years ND >18 years RR AML | 400 mg D2 to D22 (decreased to 300 mg D2 to D8 due to toxicities) | induction, CPX-351 D1, D3, D5 | 44% (ORR) 6% (CR) 33% (CRi) 6% (MLFS) | 6.1 months (median OS) | [50] | |
| Cladribin-based chemotherapy | II; NCT02115295 | ND AML < 65 years | 400 mg D2 to D8 | Induction: Cladribine 5 mg/m2 D1 to D5 CYTA (1.5 g/m2 for pts < 60 years and 1 g/m2 for pts aged ≥ 60 years) D1 to D5 IDA 10 mg/m2/d D1 to D3 | 84% (CR) 94% (ORR) | NR (median OS and EFS) | [51] |
| Consolidation, Cladribine 5 mg/m2 D1 to D3 CYTA 1 g/m2 (for patients aged <60 yrs) and 0.75 g/m2 (for patients aged ≥60 yrs) D1 to D5, IDA 8 mg/m2/d D1 to D2 | |||||||
| II; NCT03586609 | ND AML > 60 years or <60 years unfit for conventional chemotherapy | Induction 100–400 mg D1 to D21 | Cladribine 5 mg/m2 D1 to D5 LDAC 20 mg/m2 D1 to D10 | 78% (CR) 15% (CRi) | NR (median OS) | [52] | |
| consolidation VEN 100–400 mg D1 to D14 (Patients with MRD negativity received only 7 days of VEN) | Alternance of 2 cycles of A and B A, Cladribine 5 mg/m2 D1 to D3; LDAC 20 mg/m2 D1 to D10 B, AZA 75 mg/m2 D1 to D7 | ||||||
| consolidation VEN 100–400 mg D1 to D14 (Patients with MRD negativity received only 7 days of VEN) | Alternance of 2 cycles of A and B A, Cladribine 5 mg/m2 D1 to D3; LDAC 20 mg/m2 D1 to D10 B, AZA 75 mg/m2 D1 to D7 | ||||||
| IDH inhibitor (ivosidenib) | Ib/II; NCT03471260 | >18 years old IDH1mut MDS, ND secondary AML, or RR AML | Cohort 1 IVO + VEN 400 mg D1 to D14 Cohort 2 IVO + VEN 800 mg Cohort 3 IVO + VEN 400 mg + AZA | IVO; 500 mg daily from D15, +/− AZA; 75mg/m2 D1 to D7 every 28 days. | 92% (ORR), 84% cCR (CR + CRi + CRh) | NR (median OS). 68% (1-year OS) | [53] |
| FLT3 inhibitors | Ib; NCT03625505 | RR FLT3mut AML | 400 mg D1 to D28 | GILT 150 mg D1-D28 | 83.8% (modified cCR = CR + CRi + CRh + MLFS), | 5.1 months (median EFS) | [54] |
| II; NCT03404193 | ND patients with AML >60 years RR patients >18 years | DEC10-VEN | GILT, SORA and MIDO at recommended doses | 92% (cCR, ND AML), 62% (cCR, RR AML) | NR (median OS in ND AML) 6.8 months (median OS in RR AML) | [42,55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garciaz, S.; Saillard, C.; Hicheri, Y.; Hospital, M.-A.; Vey, N. Venetoclax in Acute Myeloid Leukemia: Molecular Basis, Evidences for Preclinical and Clinical Efficacy and Strategies to Target Resistance. Cancers 2021, 13, 5608. https://doi.org/10.3390/cancers13225608
Garciaz S, Saillard C, Hicheri Y, Hospital M-A, Vey N. Venetoclax in Acute Myeloid Leukemia: Molecular Basis, Evidences for Preclinical and Clinical Efficacy and Strategies to Target Resistance. Cancers. 2021; 13(22):5608. https://doi.org/10.3390/cancers13225608
Chicago/Turabian StyleGarciaz, Sylvain, Colombe Saillard, Yosr Hicheri, Marie-Anne Hospital, and Norbert Vey. 2021. "Venetoclax in Acute Myeloid Leukemia: Molecular Basis, Evidences for Preclinical and Clinical Efficacy and Strategies to Target Resistance" Cancers 13, no. 22: 5608. https://doi.org/10.3390/cancers13225608
APA StyleGarciaz, S., Saillard, C., Hicheri, Y., Hospital, M.-A., & Vey, N. (2021). Venetoclax in Acute Myeloid Leukemia: Molecular Basis, Evidences for Preclinical and Clinical Efficacy and Strategies to Target Resistance. Cancers, 13(22), 5608. https://doi.org/10.3390/cancers13225608