
The Growing Relevance of Immunoregulation in Pediatric Brain Tumors

Abstract
:Simple Summary
Abstract
1. Introduction
2. The (Postnatal) Brain as a Unique Tumor Site and CNS Immune Surveillance
2.1. Microglia as Brain-Resident Innate Immune Cell Populations
2.2. Brain-Infiltrating Immune Cell Populations
3. Targeting Immune Cells in Pediatric Brain Tumors
3.1. Immunophenotyping Studies of the Tumor Microenvironment in Pediatric Brain Tumors

{kind=link}
| Entity | Immunological Profile/Immune Population | Associated Molecular Features | Prognosis | Sample Cohort | Study |
|---|---|---|---|---|---|
| pLGG, pHGG | Hot (IS-I): more pLGGs, no DIPGs. | BRAF mutation (69.6%) | MS 1: 29.8y/>18y | 384 from CBTTC/111 from ICGC | [61] |
| Altered (IS-II): transitional stage. | SVIL mutation (55.5%) | MS 1: 19.2y/13.3y | |||
| Cold (IS-III): large fractions of pHGGs, DIPGs. | CACNA1A mutation (74.2%) | MS 1: 14.5y/1.99y | |||
| Monocytic lineage expression ↑ | Improved OS 2 | 113 | [62] | ||
| pHGG | Monocytes ↑ | MAPK mutation | 143 | [21] | |
| NK cells ↑ | G34/WT-C | Poor OS 2: 3.0 | |||
| B cells ↓ | WT-A | Poor OS 2: 4.3 | |||
| CD8 T cells ↑ | hypermutator tumors (MMRD 4; POLE/POLD1 mutations); BRAFV600E or NF1 | 113 | [67] | ||
| Gr.4-MB | Monocytes ↑ | 408 | [21] | ||
| SHH-MB 3 | Tregs ↑ | Poor OS 2: HR 1.7 | |||
| Gr.3-MB | CD8 T cells ↑; B cells ↑; Tregs ↓ | MYC amplification | Poor OS 2: HR 3.3 | ||
| SHH-MB | AIF1 expression (MAC/MG 5) ↑ | Improved OS 2 | 172 | [66] | |
| B cells ↑ | Improved OS 2 | 35 | [21] | ||
| ATRT | CD68+ MAC/MG 5 ↑ | Poor OS 2: HR 11.9 | 34 | [18] | |
| CD4/8 T cells ↑ | PBRM1 ↑ | Improved OS 2 | 33 | [68] | |
| CD163+ macrophages ↑ | PBRM1 ↑ | Poor OS 2 |
3.2. Impact of Epigenetic Dysregulation and Aberrant Signaling Pathways in Tumors on Immune Cells
3.3. Immunomodulation by Chemotherapy, Radiotherapy and Targeted Anticancer Agents
3.4. Immunotherapeutic Strategies
3.4.1. Immune Checkpoint Inhibition (ICI)
3.4.2. Targets for CAR-T Cell Therapy and Innate Immune Cells
| Entity | Targets | Therapy Approach | Study |
|---|---|---|---|
| GBM, MB, EPN | EGFRvIII | CAR T cells | [112] |
| MB, EPN | IL13Rα2 | CAR T cells | [109,113] |
| MB, EPN | HER2 | CAR T cells | [109,114,115] |
| MB, EPN | EPHA2 | CAR T cells | [109] |
| SHH-MB | CD1d | Vα24-invariant (type-I) NKT cells | [116] |
| MB | PRAME | CAR T cells (SLL TCR T cells with inducible caspase-9 gene) | [117] |
| MB | No target | allogeneic cord blood-derived NK cells expressing a dominant negative TGF-β receptor | [118] |
| SHH-MB | CSF1R | CSF1R inhibitor (PLX5622) for TAM depletion | [119] |
| ATRT, MB, ETMR, EPN, HGG | B7-H3 | CAR T cells | [106,107,108] |
| ATRT, MB, ETMR, EPN, HGG | GD2 | CAR T cells | [106,120] |
| Gr.3-MB, ATRT, PNET, GBM, DMG | CD47 | α-CD47 against tumor cells | [111] |
| ATRT | PD-1 | α-PD-1 against PD-1+ immune cells | [15] |
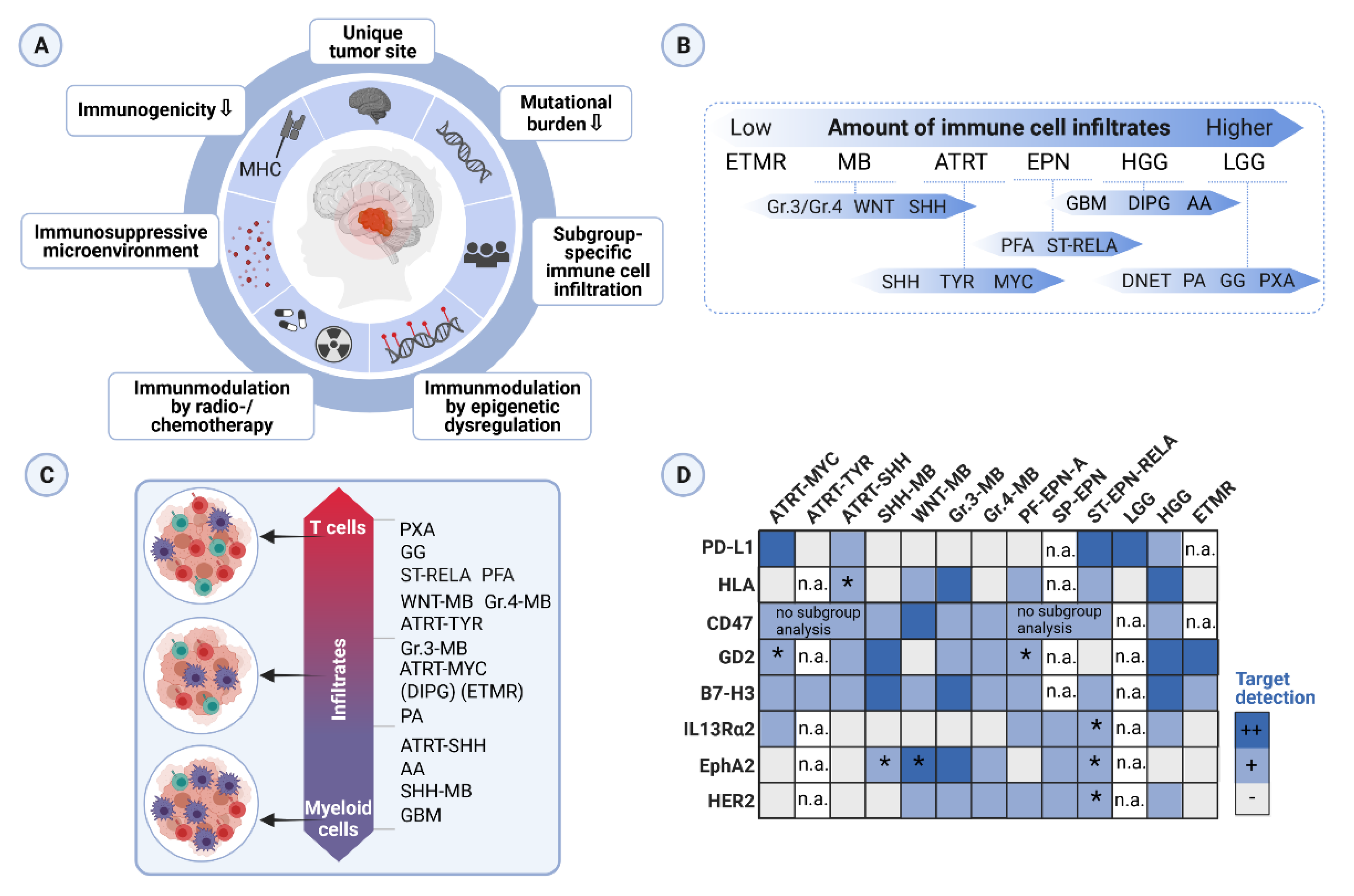
3.5. Challenges in Clinical Translation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Johnson, K.J.; Cullen, J.; Barnholtz-Sloan, J.S.; Ostrom, Q.T.; Langer, C.E.; Turner, M.C.; McKean-Cowdin, R.; Fisher, J.L.; Lupo, P.J.; Partap, S.; et al. Childhood Brain Tumor Epidemiology: A Brain Tumor Epidemiology Consortium Review. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2716–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2010, 29, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I.; et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Cacciotti, C.; Fleming, A.; Ramaswamy, V. Advances in the molecular classification of pediatric brain tumors: A guide to the galaxy. J. Pathol. 2020, 251, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Fangusaro, J.; Bandopadhayay, P. Advances in the classification and treatment of pediatric brain tumors. Curr. Opin. Pediatr. 2021, 33, 26–32. [Google Scholar] [CrossRef]
- Rutkowski, S.; Bode, U.; Deinlein, F.; Ottensmeier, H.; Warmuth-Metz, M.; Soerensen, N.; Graf, N.; Emser, A.; Pietsch, T.; Wolff, J.E.A.; et al. Treatment of Early Childhood Medulloblastoma by Postoperative Chemotherapy Alone. N. Engl. J. Med. 2005, 352, 978–986. [Google Scholar] [CrossRef] [Green Version]
- Vinchon, M.; Baroncini, M.; Leblond, P.; Delestret, I. Morbidity and tumor-related mortality among adult survivors of pediatric brain tumors: A review. Child’s Nerv. Syst. 2011, 27, 697–704. [Google Scholar] [CrossRef]
- Lieberman, N.A.P.; Vitanza, N.A.; Crane, C.A. Immunotherapy for brain tumors: Understanding early successes and limitations. Expert Rev. Neurother. 2018, 18, 251–259. [Google Scholar] [CrossRef]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.R.; Ramkissoon, S.H.; Ross, J.; Weintraub, L. Tumor mutational burden and driver mutations: Characterizing the genomic landscape of pediatric brain tumors. Pediatr. Blood Cancer 2020, 67, e28338. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Leruste, A.; Tosello, J.; Ramos, R.N.; Tauziède-Espariat, A.; Brohard, S.; Han, Z.Y.; Beccaria, K.; Andrianteranagna, M.; Caudana, P.; Nikolic, J.; et al. Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell 2019, 36, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.J.E.; Johann, P.D.; Milne, K.; Zapatka, M.; Buellesbach, A.; Ishaque, N.; Iskar, M.; Erkek, S.; Wei, L.; Tessier-Cloutier, B.; et al. Identification and Analyses of Extra-Cranial and Cranial Rhabdoid Tumor Molecular Subgroups Reveal Tumors with Cytotoxic T Cell Infiltration. Cell Rep. 2019, 29, 2338.e7–2354.e7. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.H.; Vasquez, J.; Kaushal, A.; MacDonald, T.J.; Velázquez Vega, J.E.; Schniederjan, M.; Dhodapkar, K. Subtype and grade-dependent spatial heterogeneity of T-cell infiltration in pediatric glioma. J. Immunother. Cancer 2020, 8, 6–11. [Google Scholar] [CrossRef]
- Melcher, V.; Graf, M.; Interlandi, M.; Moreno, N.; de Faria, F.W.; Kim, S.N.; Kastrati, D.; Korbanka, S.; Alfert, A.; Gerß, J.; et al. Macrophage-tumor cell interaction promotes ATRT progression and chemoresistance. Acta Neuropathol. 2020, 139, 913–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, D.; Knoll, R.; Gentles, A.J.; Vokuhl, C.; Buness, A.; Gütgemann, I. Prognostic Gene Expression, Stemness and Immune Microenvironment in Pediatric Tumors. Cancers 2021, 13, 854. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, C.; Luo, H.; Cao, J.; Ma, C.; Cheng, L.; Yang, Y. Prognostic implications of immune-related eight-gene signature in pediatric brain tumors. Braz. J. Med. Biol. Res. 2021, 54, 1–12. [Google Scholar] [CrossRef]
- Grabovska, Y.; Mackay, A.; O’Hare, P.; Crosier, S.; Finetti, M.; Schwalbe, E.C.; Pickles, J.C.; Fairchild, A.R.; Avery, A.; Cockle, J.; et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat. Commun. 2020, 11, 4324. [Google Scholar] [CrossRef]
- Vermeulen, J.F.; Van Hecke, W.; Adriaansen, E.J.M.; Jansen, M.K.; Bouma, R.G.; Villacorta Hidalgo, J.; Fisch, P.; Broekhuizen, R.; Spliet, W.G.M.; Kool, M.; et al. Prognostic relevance of tumor-infiltrating lymphocytes and immune checkpoints in pediatric medulloblastoma. Oncoimmunology 2018, 7, e1398877. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanabe, S.; Yamashita, T. The role of immune cells in brain development and neurodevelopmental diseases. Int. Immunol. 2018, 30, 437–444. [Google Scholar] [CrossRef]
- Morimoto, K.; Nakajima, K. Role of the Immune System in the Development of the Central Nervous System. Front. Neurosci. 2019, 13, 916. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, T.; Saddawi-Konefka, R.; Koebel, W.V.; Arthur, C.; White, J.M.; Uppaluri, R.; Andrews, D.M.; Ngiow, S.F.; Teng, M.W.L.; Smyth, M.J.; et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 2012, 209, 1869–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, A.K.; Hollander, G.A.; McMichael, A. Evolution of the immune system in humans from infancy to old age. Proc. R. Soc. 2015, 282, 20143085. [Google Scholar] [CrossRef]
- Rieber, N.; Gille, C.; Köstlin, N.; Schäfer, I.; Spring, B.; Ost, M.; Spieles, H.; Kugel, H.A.; Pfeiffer, M.; Heininger, V.; et al. Neutrophilic myeloid-derived suppressor cells in cord blood modulate innate and adaptive immune responses. Clin. Exp. Immunol. 2013, 174, 45–52. [Google Scholar] [CrossRef]
- McGovern, N.; Shin, A.; Low, G.; Low, D.; Duan, K.; Yao, L.J.; Msallam, R.; Low, I.; Shadan, N.B.; Sumatoh, H.R.; et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature 2017, 546, 662–666. [Google Scholar] [CrossRef]
- Olin, A.; Henckel, E.; Chen, Y.; Lakshmikanth, T.; Pou, C.; Mikes, J.; Gustafsson, A.; Bernhardsson, A.K.; Zhang, C.; Bohlin, K.; et al. Stereotypic Immune System Development in Newborn Children. Cell 2018, 174, 1277–1292. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, T.R.; Kampmann, B.; Mazmanian, S.K.; Marchant, A.; Levy, O. Protecting the Newborn and Young Infant from Infectious Diseases: Lessons from Immune Ontogeny. Immunity 2017, 46, 350–363. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef]
- Vitkovic, L.; Konsman, J.P.; Bockaert, J.; Dantzer, R.; Homburger, V.; Jacque, C. Cytokine signals propagate through the brain. Mol. Psychiatry 2000, 5, 604–615. [Google Scholar] [CrossRef] [Green Version]
- Warren, K.E. Beyond the blood: Brain barrier: The importance of central nervous system (CNS) pharmacokinetics for the treatment of CNS tumors, including diffuse intrinsic pontine glioma. Front. Oncol. 2018, 8, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef] [Green Version]
- Ginn, K.F.; Gajjar, A. Atypical teratoid rhabdoid tumor: Current therapy and future directions. Front. Oncol. 2012, 2, 114. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.T.; Strother, D.R.; Judkins, A.R.; Burger, P.C.; Pollack, I.F.; Krailo, M.D.; Buxton, A.B.; Williams-Hughes, C.; Fouladi, M.; Mahajan, A.; et al. Efficacy of High-Dose Chemotherapy and 3-D Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report from the Children’s Oncology Group Trial ACNS0333. J. Clin. Oncol. 2020, 38, 117. [Google Scholar] [CrossRef] [PubMed]
- Meel, M.H.; Guillén Navarro, M.; De Gooijer, M.C.; Metselaar, D.S.; Waranecki, P.; Breur, M.; Lagerweij, T.; Wedekind, L.E.; Koster, J.; Van De Wetering, M.D.; et al. Mek/melk inhibition and blood-brain barrier deficiencies in atypical teratoid/rhabdoid tumors. Neuro-Oncology 2020, 22, 58–69. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; Van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Jessa, S.; Blanchet-Cohen, A.; Krug, B.; Vladoiu, M.; Coutelier, M.; Faury, D.; Poreau, B.; De Jay, N.; Hébert, S.; Monlong, J.; et al. Stalled developmental programs at the root of pediatric brain tumors. Nat. Genet. 2019, 51, 1702–1713. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223. [Google Scholar] [CrossRef] [Green Version]
- Svalina, M.N.; Kikuchi, K.; Abraham, J.; Lal, S.; Davare, M.A.; Settelmeyer, T.P.; Young, M.C.; Peckham, J.L.; Cho, Y.J.; Michalek, J.E.; et al. IGF1R as a Key Target in High Risk, Metastatic Medulloblastoma. Sci. Rep. 2016, 6, 27012. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Ventura, P.B.; Jiang, Y.; Rodriguez, F.J.; Wang, L.; Perry, J.S.A.; Yang, Y.; Wahl, K.; Crittenden, R.B.; Bennett, M.L.; et al. Astrocytic trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell 2020, 180, 502–520. [Google Scholar] [CrossRef] [PubMed]
- Guadagno, E.; Presta, I.; Maisano, D.; Donato, A.; Pirrone, C.K.; Cardillo, G.; Corrado, S.D.; Mignogna, C.; Mancuso, T.; Donato, G.; et al. Role of macrophages in brain tumor growth and progression. Int. J. Mol. Sci. 2018, 19, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohme, M.; Neidert, M.C. Tumor-Specific T Cell Activation in Malignant Brain Tumors. Front. Immunol. 2020, 11, 205. [Google Scholar] [CrossRef]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.T.; Gonzalez, M.V.; Gaonkar, K.S.; Rathi, K.S.; Young, P.; Arif, S.; Zhai, L.; Alam, Z.; Devalaraja, S.; To, T.K.J.; et al. Macrophages in SHH subgroup medulloblastoma display dynamic heterogeneity that varies with treatment modality. Cell Rep. 2021, 34, 108917. [Google Scholar] [CrossRef] [PubMed]
- Crotty, E.E.; Smith, S.M.C.; Brasel, K.; Pakiam, F.; Girard, E.J.; Connor, Y.D.; Zindy, F.; Mhyre, A.J.; Roussel, M.F.; Olson, J.M. Medulloblastoma recurrence and metastatic spread are independent of colony—Stimulating factor 1 receptor signaling and macrophage survival. J. Neurooncol. 2021, 153, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Furness, A.; Joshi, K.; Ghorani, E.; Ford, K.; Ward, M.J.; King, E.V.; Lechner, M.; Marafioti, T.; Quezada, S.A.; et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat. Commun. 2018, 9, 3220. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, J.; Bizet, M.; Desmedt, C.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Koch, A.; Larsimont, D.; Salgado, R.; Van Den Eynden, G.; et al. DNA methylation-based immune response signature improves patient diagnosis in multiple cancers. J. Clin. Investig. 2017, 127, 3090–3102. [Google Scholar] [CrossRef] [Green Version]
- Safaei, S.; Mohme, M.; Niesen, J.; Schüller, U.; Bockmayr, M. DIMEimmune: Robust estimation of infiltrating lymphocytes in CNS tumors from DNA methylation profiles. Oncoimmunology 2021, 10, e1932365. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, X.; Gao, L.; Wang, Y.; Guo, Y.; Xing, B.; Ma, W. Classification of pediatric gliomas based on immunological profiling: Implications for immunotherapy strategies. Mol. Ther. Oncolytics 2021, 20, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Bockmayr, M.; Klauschen, F.; Maire, C.L.; Rutkowski, S.; Westphal, M.; Lamszus, K.; Scheuller, U.; Mohme, M. Immunologic Profiling of Mutational and Transcriptional Subgroups in Pediatric and Adult High-Grade Gliomas. Cancer Immunol. Res. 2019, 7, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Petralia, F.; Tignor, N.; Reva, B.; Koptyra, M.; Chowdhury, S.; Rykunov, D.; Krek, A.; Ma, W.; Zhu, Y.; Ji, J.; et al. Integrated Proteogenomic Characterization across Major Histological Types of Pediatric Brain Cancer. Cell 2020, 183, 1962.e31–1985.e31. [Google Scholar] [CrossRef]
- Albert, T.K.; Interlandi, M.; Sill, M.; Graf, M.; Moreno, N.; Menck, K.; Rohlmann, A.; Melcher, V.; Korbanka, S.; Meyer zu Hörste, G.; et al. An extracellular vesicle-related gene expression signature identifies high-risk patients in medulloblastoma. Neuro-Oncology 2021, 23, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Plant, A.S.; Koyama, S.; Sinai, C.; Solomon, I.H.; Griffin, G.K.; Ligon, K.L.; Bandopadhayay, P.; Betensky, R.; Emerson, R.; Dranoff, G.; et al. Immunophenotyping of pediatric brain tumors: Correlating immune infiltrate with histology, mutational load, and survival and assessing clonal T cell response. J. Neurooncol. 2018, 137, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Maximov, V.; Chen, Z.; Wei, Y.; Robinson, M.H.; Herting, C.J.; Shanmugam, N.S.; Rudneva, V.A.; Goldsmith, K.C.; MacDonald, T.J.; Northcott, P.A.; et al. Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nat. Commun. 2019, 10, 2410. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-Q.; Wilson, B.A.; Yong, V.W.; Pugh, J.; Mehta, V. Immune cell infiltrates in atypical teratoid/rhabdoid tumors. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 2012, 39, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcu, A.; Schlosser, A.; Keupp, A.; Trautwein, N.; Johann, P.; Wölfl, M.; Lager, J.; Monoranu, C.M.; Walz, J.S.; Henkel, L.M.; et al. Natural and cryptic peptides dominate the immunopeptidome of atypical teratoid rhabdoid tumors. J. Immunother. Cancer 2021, 9, e003404. [Google Scholar] [CrossRef] [PubMed]
- Griesinger, A.M.; Birks, D.K.; Donson, A.M.; Amani, V.; Hoffman, L.M.; Waziri, A.; Wang, M.; Handler, M.H.; Foreman, N.K. Characterization of Distinct Immunophenotypes across Pediatric Brain Tumor Types. J. Immunol. 2013, 191, 4880–4888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, W.Y.; Elghetany, M.T.; Shen, J.; Man, T.K.; Li, X.; Chintagumpala, M.; Su, J.M.F.; Dauser, R.; Whitehead, W.; Adesina, A.M.; et al. Therapeutic implications of CD1d expression and tumor-infiltrating macrophages in pediatric medulloblastomas. J. Neurooncol. 2014, 120, 293–301. [Google Scholar] [CrossRef]
- Margol, A.S.; Robison, N.J.; Gnanachandran, J.; Hung, L.T.; Kennedy, R.J.; Vali, M.; Dhall, G.; Finlay, J.L.; Erdreich-Epstein, A.; Krieger, M.D.; et al. Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin. Cancer Res. 2015, 21, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, C.D.; Flores, C.; Yang, C.; Pinheiro, E.M.; Yearley, J.H.; Sayour, E.J.; Pei, Y.; Moore, C.; McLendon, R.E.; Huang, J.; et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin. Cancer Res. 2016, 22, 582–595. [Google Scholar] [CrossRef] [Green Version]
- Aoki, T.; Hino, M.; Koh, K.; Kyushiki, M.; Kishimoto, H.; Arakawa, Y.; Hanada, R.; Kawashima, H.; Kurihara, J.; Shimojo, N.; et al. Low Frequency of Programmed Death Ligand 1 Expression in Pediatric Cancers. Pediatr. Blood Cancer 2016, 63, 1461–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockmayr, M.; Mohme, M.; Klauschen, F.; Winkler, B.; Budczies, J.; Rutkowski, S.; Schüller, U. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 2018, 7, e1462430. [Google Scholar] [CrossRef] [Green Version]
- Diao, S.; Gu, C.; Zhang, H.; Yu, C. Immune cell infiltration and cytokine secretion analysis reveal a non-inflammatory microenvironment of medulloblastoma. Oncol. Lett. 2020, 20, 397. [Google Scholar] [CrossRef] [PubMed]
- Witt, D.A.; Donson, A.M.; Amani, V.; Moreira, D.C.; Sanford, B.; Hoffman, L.M.; Handler, M.H.; Levy, J.M.M.; Jones, K.L.; Nellan, A.; et al. Specific expression of PD-L1 in RELA-fusion supratentorial ependymoma: Implications for PD-1-targeted therapy. Pediatr. Blood Cancer 2018, 65, e26960. [Google Scholar] [CrossRef]
- Martin, A.M.; Bell, W.R.; Yuan, M.; Harris, L.; Poore, B.; Arnold, A.; Engle, E.L.; Asnaghi, L.; Lim, M.; Raabe, E.H.; et al. PD-L1 expression in pediatric low-grade gliomas is independent of BRAF V600E mutational status. J. Neuropathol. Exp. Neurol. 2020, 79, 74–85. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Molinari, V.; Jones, D.T.W.; Izquierdo, E.; Brouwer-Visser, J.; Giangaspero, F.; Haberler, C.; Pietsch, T.; Jacques, T.S.; et al. Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 2018, 33, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panwalkar, P.; Pratt, D.; Chung, C.; Dang, D.; Le, P.; Martinez, D.; Bayliss, J.M.; Smith, K.S.; Adam, M.; Potter, S.; et al. SWI/SNF complex heterogeneity is related to polyphenotypic differentiation, prognosis, and immune response in rhabdoid tumors. Neuro-Oncology 2020, 22, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Alfert, A.; Moreno, N.; Kerl, K. The BAF complex in development and disease. Epigenet. Chromatin 2019, 12, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Pfister, S.M.; Jones, D.T.W. Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies. Lancet Oncol. 2015, 16, e293–e302. [Google Scholar] [CrossRef]
- Pan, D.; Kobayashi, A.; Jiang, P.; De Andrade, L.F.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biegel, J.A.; Zhou, J.Y.; Rorke, L.B.; Stenstrom, C.; Wainwright, L.M.; Fogelgren, B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999, 59, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselblatt, M.; Nagel, I.; Oyen, F.; Bartelheim, K.; Russell, R.B.; Schüller, U.; Junckerstorff, R.; Rosenblum, M.; Alassiri, A.H.; Rossi, S.; et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 2014, 128, 453–456. [Google Scholar] [CrossRef]
- Price, G.; Bouras, A.; Hambardzumyan, D.; Hadjipanayis, C.G. Current knowledge on the immune microenvironment and emerging immunotherapies in diffuse midline glioma. EBioMedicine 2021, 69, 103453. [Google Scholar] [CrossRef]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Hasselblatt, M.; Pfister, S.M.; Kool, M. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/b-catenin pathway activation correlates with immune exclusion across human cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [Green Version]
- Grund-Gröschke, S.; Stockmaier, G.; Aberger, F. Hedgehog/GLI signaling in tumor immunity—New therapeutic opportunities and clinical implications. Cell Commun. Signal. 2019, 17, 172. [Google Scholar] [CrossRef] [Green Version]
- Mehlman, C.; Kamga, P.T.; Costantini, A.; Julié, C.; Dumenil, C.; Dumoulin, J.; Ouaknine, J.; Giraud, V.; Chinet, T.; Emile, J.F.; et al. Baseline hedgehog pathway activation and increase of plasma Wnt1 protein are associated with resistance to immune checkpoint inhibitors in advanced non-small-cell lung cancer. Cancers 2021, 13, 1107. [Google Scholar] [CrossRef]
- Sevenich, L. Turning “Cold” into “Hot” tumors—Opportunities and challenges for radio-immunotherapy against primary and metastatic brain cancers. Front. Oncol. 2019, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Karachi, A.; Dastmalchi, F.; Mitchell, D.A.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro-Oncology 2018, 20, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Petroni, G.; Buqué, A.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Immunomodulation by targeted anticancer agents. Cancer Cell 2021, 39, 310–345. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, Z.; Zhou, J.; Huang, J.; Zhou, L.; Luo, J.; Wan, Y.Y.; Long, H.; Zhu, B. EZH2 inhibitor GSK126 suppresses antitumor immunity by driving production of myeloid-derived suppressor cells. Cancer Res. 2019, 79, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Alimova, I.; Birks, D.K.; Harris, P.S.; Knipstein, J.A.; Venkataraman, S.; Marquez, V.E.; Foreman, N.K.; Vibhakar, R. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro-Oncology 2013, 15, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Scott, M.P.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, S.N.; Bourdeaut, F.; Laetsch, T.W.; Fouladi, M.; Macy, M.E.; Makin, G.W.J.; Shukla, N.N.; Wetmore, C.; Margol, A.S.; Casanova, M.; et al. Phase I study of tazemetostat, an enhancer of zeste homolog-2 inhibitor, in pediatric pts with relapsed/refractory integrase interactor 1-negative tumors. J. Clin. Oncol. 2020, 38, 10525. [Google Scholar] [CrossRef]
- Jones, R.L.; Blay, J.; Agulnik, M.; Chugh, R.; Mir, O.; Italiano, A.; Thomas, D.; Gupta, A.; Jahan, T.; Cote, G.; et al. A phase 2, multicenter study of the EZH2 inhibitor tazemetostat in adults (rhabdoid tumor cohort) (NCT02601950). Ann. Oncol. 2018, 29, viii580–viii581. [Google Scholar] [CrossRef]
- Gounder, M.; Schöffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.W.; Jahan, T.; et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: An international, open-label, phase 2 basket study. Lancet Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef]
- Keane, L.; Cheray, M.; Saidi, D.; Kirby, C.; Friess, L.; Gonzalez-rodriguez, P.; Gerdes, M.E.; Grabert, K.; McColl, B.W. Inhibition of microglial EZH2 leads to anti-tumoral effects in pediatric diffuse midline gliomas. Neuro-Oncology 2021, 3, vdab096. [Google Scholar] [CrossRef]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [Green Version]
- Knox, T.; Sahakian, E.; Banik, D.; Hadley, M.; Palmer, E.; Noonepalle, S.; Kim, J.; Powers, J.; Gracia-Hernandez, M.; Oliveira, V.; et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci. Rep. 2019, 9, 6136. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, E.K.; Markert, J.M.; Gillespie, G.Y.; Friedman, G.K. Checkpoint proteins in pediatric brain and extracranial solid tumors: Opportunities for immunotherapy. Clin. Cancer Res. 2017, 23, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef] [Green Version]
- Haydar, D.; Houke, H.; Chiang, J.; Yi, Z.; Odé, Z.; Caldwell, K.; Zhu, X.; Mercer, K.S.; Stripay, J.L.; Shaw, T.I.; et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro-Oncology 2021, 23, 999–1011. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020, 26, 712–719. [Google Scholar] [CrossRef]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Taslim, C.; Tullius, B.P.; Cripe, T.P. Therapeutic modulation of the CD47-SIRPα axis in the pediatric tumor microenvironment: Working up an appetite. Cancer Drug Resist. 2020, 3, 550–562. [Google Scholar] [CrossRef]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, e001341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravanpay, A.C.; Gust, J.; Johnson, A.J.; Rolczynski, L.S.; Chang, C.A.; Hoglund, V.J.; Mukherjee, R.; Nicholas, A.; Orentas, R.J.; Jensen, M.C. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotraget 2019, 10, 7080–7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stastny, M.J.; Brown, C.E.; Ruel, C.; Jensen, M.C. Medulloblastomas Expressing IL13Ra2 are Targets for IL13-zetakine+ Cytolytic T Cells. J. Pediatr. Hematol. Oncol. 2007, 29, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Ratnayake, M.; Savoldo, B.; Perlaky, L.; Dotti, G.; Wels, W.S.; Bhattacharjee, M.B.; Gilbertson, R.J.; Shine, H.D.; Weiss, H.L.; et al. Regression of Experimental Medulloblastoma following Transfer of HER2-Specific T Cells. Cancer Res. 2007, 67, 5957–5964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nellan, A.; Rota, C.; Majzner, R.; Lester-McCully, C.M.; Griesinger, A.M.; Mulcahy Levy, J.M.; Foreman, N.K.; Warren, K.E.; Lee, D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 2018, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Song, L.; Brawley, V.S.; Robison, N.; Wei, J.; Gao, X.; Tian, G.; Margol, A.; Ahmed, N.; Asgharzadeh, S.; et al. Medulloblastoma expresses CD1d and can be targeted for immunotherapy with NKT cells. Clin. Immunol. 2013, 149, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, D.; Miele, E.; De Angelis, B.; Guercio, M.; Boffa, I.; Sinibaldi, M.; Po, A.; Caruana, I.; Abballe, L.; Carai, A.; et al. Adoptive immunotherapy using PRAME-specific T cells in medulloblastoma. Cancer Res. 2018, 78, 3337–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, A.B.; Yadavilli, S.; Saunders, D.; Van Pelt, S.; Chorvinsky, E.; Burga, R.A.; Albihani, S.; Hanley, P.J.; Xu, Z.; Pei, Y.; et al. Medulloblastoma rendered susceptible to NK-cell attack by TGFβ neutralization. J. Transl. Med. 2019, 17, 321. [Google Scholar] [CrossRef]
- Tan, I.; Arifa, R.D.N.; Rallapalli, H.; Kana, V.; Lao, Z.; Sanghrajka, R.M.; Bayin, N.S.; Tanne, A.; Wojcinski, A.; Korshunov, A.; et al. CSF1R Inhibition Depletes Tumor-Associated Macrophages and Attenuates Tumor Progression in a Mouse Sonic Hedgehog- Medulloblastoma Model. Oncogene 2021, 40, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef]
- van Gool, S.W.; Holm, S.; Rachor, J.; Adamson, L.; Technau, A.; Maass, E.; Frühwald, M.C.; Schlegel, P.G.; Eyrich, M. Immunotherapy in atypical teratoid-rhabdoid tumors: Data from a survey of the HGG-Immuno Group. Cytotherapy 2016, 18, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Caruso, D.A.; Orme, L.M.; Neale, A.M.; Radcliff, F.J.; Armor, G.M.; Maixner, W.; Downie, P.; Hassall, T.E.; Tang, M.L.K.; Ashley, D.M. Results of a phase 1 study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro-Oncology 2004, 6, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Ardon, H.; De Vleeschouwer, S.; Claes, L.; Van Calenbergh, F.; Kramm, C.M.; Rutkowski, S.; Wolff, J.E.A.; Van Gool, S.W. Adjuvant Dendritic Cell-Based Tumour Vaccination for Children With Malignant Brain Tumours. Pediatr. Blood Cancer 2010, 54, 519–525. [Google Scholar] [CrossRef]
- Garcia-Moure, M.; Gonzalez-Huarriz, M.; Labiano, S.; Guruceaga, E.; Bandres, E.; Zalacain, M.; Marrodan, L.; de Andrea, C.; Villalba, M.; Martinez-Velez, N.; et al. Delta-24-RGD, an oncolytic adenovirus, increases survival and promotes proinflammatory immune landscape remodeling in models of AT/RT and CNS-PNET. Clin. Cancer Res. 2021, 27, 1807–1820. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
| Entity | Findings | Sample Origin | Techniques |
|---|---|---|---|
| ATRT | ▪ Immune cell infiltration with CD68+ microglia/macrophages and CD4+/CD8+ T cells [69] | H | IHC |
| ▪ Immune cell infiltration with myeloid and T cells; ATRT-MYC highly infiltrated; clonally expanded T cells [15] | H, M | IHC, FACS, RNAseq, sc-RNAseq | |
| ▪ Subgroup-specific immune cell infiltration; CD68+ cells as negative prognostic factor [18] | H, M | IHC, scRNA-seq | |
| ▪ CD8+ T cell infiltration higher in ATRT-MYC than ATRT-TYR/-SHH; PD-L1 and PD-1 expression [16] | H | RNAseq, IHC | |
| ▪ Analysis of naturally & cryptic presented HLA-class-I and class-II ligands [70] | H | MS | |
| MB | ▪ Immunosuppressed myeloid cells; T cells are less frequent than in PA and EPN [71] | H | FACS, RNAseq |
| ▪ CD163 expression is most enriched in SHH-MB compared to other MB subtypes; CD1d expression in a subset of infantile MB [72] | H | RNAseq, IHC | |
| ▪ Increased expression of inflammation-related genes; greatest number of CD163+ TAMs in SHH-MB across MB subtypes [73] | H | RNAseq, IHC | |
| ▪ CD4+, CD8+ T cells, MDSCs, DCs and TAMs in SHH-MB > Gr.3-MB; CD8+ PD-1+ T cells in Gr.3-MB > SHH-MB; Gr.3-MB respond to PD-1 blockade, MB-SHH not [74] | M | In vivo, FACS, IHC | |
| ▪ No PD-L1 expression in four MBs [75] | H | IHC | |
| ▪ No PD-L1 expression in 26 MBs; no correlation of TILs with overall survival; expression of granzyme inhibitor SERPINB1 was associated with better survival [22] | H | IHC | |
| ▪ Subgroup-specific immune microenvironment [76] | H | RNAseq | |
| ▪ Antitumoral role of TAMs in SHH-MB [66] | H, M | RNAseq, in/ex vivo, FACS | |
| ▪ Increased expression of immune-related genes in SHH-MB compared to other MB subtypes; CD8+ T cells and neutrophils enriched in G4-MB; no PD-L1 expression in 19 MBs [77] | H | RNAseq, IHC | |
| ▪ TAMs in SHH-MB are of microglial origin and monocyte-derived; radiation therapy, but not targeted therapy, recruited immunosuppressive monocyte-derived macrophages that reduced T cells and neutrophils [53] | H, M | RNAseq, scRNAseq, in vivo, IHC, RNA in situ hybridization, FACS | |
| EPN | ▪ Higher numbers of T cells and myeloid cells compared to MB and GBM [71] | H | FACS, RNAseq |
| ▪ High expression of PD-L1 in ST-EPN-RELA with PD-1+ CD4+ and CD8+ T cells [78] | H | RNAseq, WB, IHC, FACS | |
| LGG | ▪ T cell infiltrates higher in LGG compared to HGG; Within LGG greater T-cell density in PXA and GG; CD3+ T cell infiltration correlates inversely with SOX2 expression [17] | H | Multiplex IHC, Single-cell mass cytometry |
| ▪ PD-L1 expression independent of BRAFV600E mutational status [79] | H | IHC, in vitro | |
| ▪ Highest CD8+ T-cell density in PXA and hypermutator LGGs. Histon mutant tumors immune “cold” [67] | H | RNAseq, IHC | |
| HGG | ▪ T cells and myeloid cells are less frequent than in PA and EPN [71] | H | FACS, RNAseq |
| ▪ Immunologic profiling of pediatric and adult HGGs [62] | H | RNAseq | |
| ▪ Immunologic profiling of pediatric LGG and HGG [61] | H | RNAseq |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melcher, V.; Kerl, K. The Growing Relevance of Immunoregulation in Pediatric Brain Tumors. Cancers 2021, 13, 5601. https://doi.org/10.3390/cancers13225601
Melcher V, Kerl K. The Growing Relevance of Immunoregulation in Pediatric Brain Tumors. Cancers. 2021; 13(22):5601. https://doi.org/10.3390/cancers13225601
Chicago/Turabian StyleMelcher, Viktoria, and Kornelius Kerl. 2021. "The Growing Relevance of Immunoregulation in Pediatric Brain Tumors" Cancers 13, no. 22: 5601. https://doi.org/10.3390/cancers13225601
APA StyleMelcher, V., & Kerl, K. (2021). The Growing Relevance of Immunoregulation in Pediatric Brain Tumors. Cancers, 13(22), 5601. https://doi.org/10.3390/cancers13225601