
Interplay between Cellular and Non-Cellular Components of the Tumour Microenvironment in Hepatocellular Carcinoma



Abstract
:Simple Summary
Abstract
1. Introduction
2. The Extracellular Matrix in the Tumour Microenvironment
2.1. The Liver ECM in Health
2.2. A Remodelled ECM in Liver Cancer
2.3. Cytokines Are Key Regulators of the Tumour Microenvironment
2.4. The Effect of the Remodelled ECM on the Tumour Immune Microenvironment
3. Models to Study the Cellular and Non-Cellular Components of the TME
3.1. Organoids
3.2. Spheroids
3.3. Organ-on-Chip
3.4. Precision Cut Tissue Slices
3.5. Bioengineered Cancer Models
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. GLOBOCAN Cancer Outlook. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/11-Liver-fact-sheet.pdf (accessed on 30 April 2021).
- WHO. Projections of Mortality and Causes of Death 2016 to 2060; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, A.B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SanGiovanni, A.; Prati, G.M.; Fasani, P.; Ronchi, G.; Romeo, R.; Manini, M.; Del Ninno, E.; Morabito, A.; Colombo, M. The natural history of compensated cirrhosis due to hepatitis C virus: A 17-year cohort study of 214 patients. Hepatology 2006, 43, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.J.; Von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Huang, X.; Zhang, G.; Hong, Z.; Bai, X.; Liang, T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Villanueva, A.; Marrero, J.A.; Schwartz, M.; Meyer, T.; Galle, P.R.; Lencioni, R.; Greten, T.F.; Kudo, M.; Mandrekar, S.J.; et al. Trial Design and Endpoints in Hepatocellular Carcinoma: AASLD Consensus Conference. Hepatology 2021, 73, 158–191. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Segal, N.H.; Jaeger, D.; Lee, K.-H.; Marshall, J.; Antonia, S.J.; Butler, M.; Sanborn, R.E.; Nemunaitis, J.J.; Carlson, C.A.; et al. Safety and clinical activity of durvalumab monotherapy in patients with hepatocellular carcinoma (HCC). J. Clin. Oncol. 2017, 35, 4071. [Google Scholar] [CrossRef]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.-I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.H.; Harding, J.J.; Merle, P.; et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.-Y.; Breder, V.V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Results of KEYNOTE-240: Phase 3 study of pembrolizumab (Pembro) vs best supportive care (BSC) for second line therapy in advanced hepatocellular carcinoma (HCC). J. Clin. Oncol. 2019, 37, 4004. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Ikeda, M.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. A phase Ib study of lenvatinib (LEN) plus pembrolizumab (PEMBRO) in unresectable hepatocellular carcinoma (uHCC). J. Clin. Oncol. 2020, 38, 4519. [Google Scholar] [CrossRef]
- Kelley, R.K.; Cheng, A.-L.; Braiteh, F.S.; Park, J.-W.; Benzaghou, F.; Milwee, S.; Borgman, A.; El-Khoueiry, A.B.; Kayali, Z.K.; Zhu, A.X.; et al. Phase 3 (COSMIC-312) study of cabozantinib (C) in combination with atezolizumab (A) versus sorafenib (S) in patients (pts) with advanced hepatocellular carcinoma (aHCC) who have not received previous systemic anticancer therapy. J. Clin. Oncol. 2019, 37, TPS4157. [Google Scholar] [CrossRef]
- Kelley, R.K.; Abou-Alfa, G.K.; Bendell, J.C.; Kim, T.-Y.; Borad, M.J.; Yong, W.-P.; Morse, M.; Kang, Y.-K.; Rebelatto, M.; Makowsky, M.; et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. J. Clin. Oncol. 2017, 35, 4073. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate. J. Clin. Oncol. 2019, 37, 4012. [Google Scholar] [CrossRef]
- Yin, Z.; Dong, C.; Jiang, K.; Xu, Z.; Li, R.; Guo, K.; Shao, S.; Wang, L. Heterogeneity of cancer-associated fibroblasts and roles in the progression, prognosis, and therapy of hepatocellular carcinoma. J. Hematol. Oncol. 2019, 12, 101. [Google Scholar] [CrossRef]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Ford, A.J.; Rajagopalan, P. Extracellular matrix remodeling in 3D: Implications in tissue homeostasis and disease progression. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1503. [Google Scholar] [CrossRef]
- Charras, G.; Sahai, E. Physical influences of the extracellular environment on cell migration. Nat. Rev. Mol. Cell Biol. 2014, 15, 813–824. [Google Scholar] [CrossRef]
- Hastings, J.F.; Skhinas, J.N.; Fey, D.; Croucher, D.R.; Cox, T.R. The extracellular matrix as a key regulator of intracellular signalling networks. Br. J. Pharmacol. 2019, 176, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.D. Extracellular matrix alters epithelial differentiation. Curr. Opin. Cell Biol. 1993, 5, 1029–1035. [Google Scholar] [CrossRef]
- Berger, C.; Bjørlykke, Y.; Hahn, L.; Mühlemann, M.; Kress, S.; Walles, H.; Luxenhofer, R.; Ræder, H.; Metzger, M.; Zdzieblo, D. Matrix decoded—A pancreatic extracellular matrix with organ specific cues guiding human iPSC differentiation. Biomaterials 2020, 244, 119766. [Google Scholar] [CrossRef]
- Ueno, T.; Sata, M.; Tanikawa, K. Chapter 6—Cells Responsible for Extracellular Matrix Production in the Liver. In Extracellular Matrix and the Liver; Oakazaki, I., Ninomiya, Y., Friedman, S.L., Tanikawa, K., Eds.; Academic Press: Cambridge, MA, USA, 2003; pp. 89–103. [Google Scholar]
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 518. [Google Scholar] [CrossRef] [Green Version]
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44-46, 147–156. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-P.; Chang, H.-L.; Bamodu, O.A.; Yadav, V.K.; Huang, T.-Y.; Wu, A.T.H.; Yeh, C.-T.; Tsai, S.-H.; Lee, W.-H. Collagen 1A1 (COL1A1) Is a Reliable Biomarker and Putative Therapeutic Target for Hepatocellular Carcinogenesis and Metastasis. Cancers 2019, 11, 786. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.-C.; Wang, C.J.; Liao, P.-C.; Yen, C.-J.; Shan, Y.-S. Hepatic stellate cells secretes type I collagen to trigger epithelial mesenchymal transition of hepatoma cells. Am. J. Cancer Res. 2014, 4, 751–763. [Google Scholar]
- Levental, K.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Mustonen, A.-M.; Salvén, A.; Kärjä, V.; Rilla, K.; Matilainen, J.; Nieminen, P. Hyaluronan histochemistry—A potential new tool to assess the progress of liver disease from simple steatosis to hepatocellular carcinoma. Glycobiology 2019, 29, 298–306. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tateishi, R.; Koike, K. Proteoglycans Are Attractive Biomarkers and Therapeutic Targets in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2018, 19, 3070. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Xie, X.; Wang, Z.; Hu, C.; Zheng, Q.; Wang, Y.; Chen, R.; Xue, T.; Chen, J.; Gao, D.; et al. Increasing matrix stiffness upregulates vascular endothelial growth factor expression in hepatocellular carcinoma cells mediated by integrin β1. Biochem. Biophys. Res. Commun. 2014, 444, 427–432. [Google Scholar] [CrossRef]
- You, Y.; Zheng, Q.; Dong, Y.; Wang, Y.; Zhang, L.; Xue, T.; Xie, X.; Hu, C.; Wang, Z.; Chen, R.; et al. Higher Matrix Stiffness Upregulates Osteopontin Expression in Hepatocellular Carcinoma Cells Mediated by Integrin β1/GSK3β/β-Catenin Signaling Pathway. PLoS ONE 2015, 10, e0134243. [Google Scholar] [CrossRef]
- Pan, H.-W.; Ou, Y.-H.; Peng, S.-Y.; Liu, S.-H.; Lai, P.-L.; Lee, P.-H.; Sheu, J.-C.; Chen, C.-L.; Hsu, H.-C. Overexpression of osteopontin is associated with intrahepatic metastasis, early recurrence, and poorer prognosis of surgically resected hepatocellular carcinoma. Cancer 2003, 98, 119–127. [Google Scholar] [CrossRef]
- Yuzhalin, A.E.; Lim, S.Y.; Kutikhin, A.G.; Gordon-Weeks, A.N. Dynamic matrisome: ECM remodeling factors licensing cancer progression and metastasis. Biochim. Biophys. Acta BBA Bioenerg. 2018, 1870, 207–228. [Google Scholar] [CrossRef]
- Ye, M.; Song, Y.; Pan, S.; Chu, M.; Wang, Z.-W.; Zhu, X. Evolving roles of lysyl oxidase family in tumorigenesis and cancer therapy. Pharmacol. Ther. 2020, 215, 107633. [Google Scholar] [CrossRef]
- Wong, C.C.-L.; Tse, A.P.-W.; Huang, Y.-P.; Zhu, Y.-T.; Chiu, D.K.-C.; Lai, R.K.-H.; Au, S.L.-K.; Kai, A.K.-L.; Lee, J.M.-F.; Wei, L.L.; et al. Lysyl oxidase-like 2 is critical to tumor microenvironment and metastatic niche formation in hepatocellular carcinoma. Hepatology 2014, 60, 1645–1658. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Scheau, C.; Badarau, I.A.; Costache, R.; Caruntu, C.; Mihai, G.L.; Didilescu, A.C.; Constantin, C.; Neagu, M. The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Anal. Cell. Pathol. 2019, 2019, 9423907. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.M.; Gaggar, A.; Blalock, J.E. MMP generated matrikines. Matrix Biol. 2015, 44-46, 122–129. [Google Scholar] [CrossRef]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.-B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef] [Green Version]
- Rivas, M.J.; Arii, S.; Furutani, M.; Harada, T.; Mizumoto, M.; Nishiyama, H.; Fujita, J.; Imamura, M. Expression of human macrophage metalloelastase gene in hepatocellular carcinoma: Correlation with angiostatin generation and its clinical significance. Hepatology 1998, 28, 986–993. [Google Scholar] [CrossRef]
- Gorrin-Rivas, M.J.; Arii, S.; Mori, A.; Takeda, Y.; Mizumoto, M.; Furutani, M.; Imamura, M. Implications of Human Macrophage Metalloelastase and Vascular Endothelial Growth Factor Gene Expression in Angiogenesis of Hepatocellular Carcinoma. Ann. Surg. 2000, 231, 67–73. [Google Scholar] [CrossRef]
- Korpi, J.T.; Kervinen, V.; Mäklin, H.; Väänänen, A.; Lahtinen, M.; Läärä, E.; Ristimäki, A.; Thomas, G.; Ylipalosaari, M.; Åström, P.; et al. Collagenase-2 (matrix metalloproteinase-8) plays a protective role in tongue cancer. Br. J. Cancer 2008, 98, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Acuff, H.B.; Carter, K.J.; Fingleton, B.; Gorden, D.L.; Matrisian, L.M. Matrix Metalloproteinase-9 from Bone Marrow–Derived Cells Contributes to Survival but not Growth of Tumor Cells in the Lung Microenvironment. Cancer Res. 2006, 66, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Cui, J.; Qin, Q.; Zhang, J.; Liu, L.; Deng, S.; Wu, C.; Yang, M.; Li, S.; Wang, C. Mechanical stiffness of liver tissues in relation to integrin β1 expression may influence the development of hepatic cirrhosis and hepatocellular carcinoma. J. Surg. Oncol. 2010, 102, 482–489. [Google Scholar] [CrossRef]
- Dong, Y.; Zheng, Q.; Wang, Z.; Lin, X.; You, Y.; Wu, S.; Wang, Y.; Hu, C.; Xie, X.; Chen, J.; et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J. Hematol. Oncol. 2019, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.V.; Sleiman, M.; Moriarty, T.; Herrick, W.G.; Peyton, S.R. Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials 2014, 35, 5749–5759. [Google Scholar] [CrossRef] [Green Version]
- Schrader, J.; Gordon-Walker, T.T.; Aucott, R.L.; Van Deemter, M.; Quaas, A.; Walsh, S.; Benten, D.; Forbes, S.J.; Wells, R.G.; Iredale, J.P. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011, 53, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Attieh, Y.; Barbazan, J.; et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Deng, Y.; Cheng, J.; Fu, B.; Liu, W.; Chen, G.; Zhang, Q.; Yang, Y. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene 2017, 36, 1090–1101. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Barry, A.; Baldeosingh, R.; Lamm, R.; Patel, K.; Zhang, K.; Dominguez, D.A.; Kirton, K.J.; Shah, A.P.; Dang, H. Hepatic Stellate Cells and Hepatocarcinogenesis. Front. Cell Dev. Biol. 2020, 8, 709. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gu, C.; Song, Q.; Zhu, M.; Xu, Y.; Xiao, M.; Zheng, W. Identifying cancer-associated fibroblasts as emerging targets for hepatocellular carcinoma. Cell Biosci. 2020, 10, 127. [Google Scholar] [CrossRef]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers 2020, 12, 641. [Google Scholar] [CrossRef] [Green Version]
- Cannon, J.G. Inflammatory Cytokines in Nonpathological States. Physiology 2000, 15, 298–303. [Google Scholar] [CrossRef]
- Lokau, J.; Schoeder, V.; Haybaeck, J.; Garbers, C. Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers 2019, 11, 1704. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Gingold, J.A.; Su, X. Immunomodulatory TGF-β Signaling in Hepatocellular Carcinoma. Trends Mol. Med. 2019, 25, 1010–1023. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, R.; Yano, H.; Iemura, A.; Ogasawara, S.; Haramaki, M.; Kojiro, M. Expression of vascular endothelial growth factor in human hepatocellular carcinoma. Hepatology 1998, 28, 68–77. [Google Scholar] [CrossRef]
- Poon, R.T.-P.; Ng, I.O.-L.; Lau, C.; Zhu, L.-X.; Yu, W.-C.; Lo, C.-M.; Fan, S.-T.; Wong, J. Serum Vascular Endothelial Growth Factor Predicts Venous Invasion in Hepatocellular Carcinoma: A Prospective Study. Ann. Surg. 2001, 233, 227–235. [Google Scholar] [CrossRef]
- Poon, R.T.-P.; Lau, C.; Yu, W.-C.; Fan, S.-T.; Wong, J. High serum levels of vascular endothelial growth factor predict poor response to transarterial chemoembolization in hepatocellular carcinoma: A prospective study. Oncol. Rep. 2004, 11, 1077–1084. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, D.; Wu, P.; Wang, Z.; Huang, J. Serum IL-10 Predicts Worse Outcome in Cancer Patients: A Meta-Analysis. PLoS ONE 2015, 10, e0139598. [Google Scholar] [CrossRef] [Green Version]
- Montanari, N.R.; Anugwom, C.M.; Boonstra, A.; Debes, J.D. The Role of Cytokines in the Different Stages of Hepatocellular Carcinoma. Cancers 2021, 13, 4876. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2019, 21, 157. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.; He, J.-C.; Yang, Y.; Wang, J.-M.; Qian, Y.-W.; Yang, T.; Jian-Ming, W. The Prognostic Value of Tumor-infiltrating Lymphocytes in Hepatocellular Carcinoma: A Systematic Review and Meta-analysis. Sci. Rep. 2017, 7, 7525. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; de Moura, M.C.; Putra, J.; Campreciós, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Huang, H.; Wang, Y.; Pan, C.; Yin, S.; Zhou, L.; Zheng, S. Tumor Immune Microenvironment Characterization in Hepatocellular Carcinoma Identifies Four Prognostic and Immunotherapeutically Relevant Subclasses. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef]
- McQuitty, C.E.; Williams, R.; Chokshi, S.; Urbani, L. Immunomodulatory Role of the Extracellular Matrix Within the Liver Disease Microenvironment. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Díaz, D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.G.; Gray, J.D.; Ohtsuka, K.; Yamagiwa, S.; Horwitz, D.A. Generation ex vivo of TGF-β-producing regulatory T cells from CD4+CD25− precursors. J. Immunol. 2002, 169, 4183–4189. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.J.; Lei, K.-J.; Li, J.; Marinos, N.J.; McGrady, G.; Wahl, S.M. Conversion of Peripheral CD4+CD25− Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-β Induction of Transcription Factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Nicolas-Boluda, A.; Donnadieu, E. Obstacles to T cell migration in the tumor microenvironment. Comp. Immunol. Microbiol. Infect. Dis. 2019, 63, 22–30. [Google Scholar] [CrossRef]
- Murdamoothoo, D.; Sun, Z.; Yilmaz, A.; Riegel, G.; Abou-Faycal, C.; Deligne, C.; Velazquez-Quesada, I.; Erne, W.; Nascimento, M.; Mörgelin, M.; et al. Tenascin-C immobilizes infiltrating T lymphocytes through CXCL12 promoting breast cancer progression. EMBO Mol. Med. 2021, 13, e13270. [Google Scholar] [CrossRef]
- Elahi-Gedwillo, K.Y.; Carlson, M.; Zettervall, J.; Provenzano, P.P. Antifibrotic Therapy Disrupts Stromal Barriers and Modulates the Immune Landscape in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, N.; Giese, N.A.; Giese, T.; Poschke, I.; Offringa, R.; Werner, J.; Ryschich, E. Prevailing Role of Contact Guidance in Intrastromal T-cell Trapping in Human Pancreatic Cancer. Clin. Cancer Res. 2014, 20, 3422–3433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.-L.; Carretta, M.; Kalvisa, A.; Siersbæk, M.S.; Simões, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.H.; Rodriguez, B.L.; Diao, L.; Chen, L.; Wang, J.; Byers, L.A.; Wei, Y.; Chapman, H.A.; Yamauchi, M.; Behrens, C.; et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8+ T cell exhaustion. Nat. Commun. 2020, 11, 4520. [Google Scholar] [CrossRef] [PubMed]
- Klement, J.; Poschel, D.; Lu, C.; Merting, A.; Yang, D.; Redd, P.; Liu, K. Osteopontin Blockade Immunotherapy Increases Cytotoxic T Lymphocyte Lytic Activity and Suppresses Colon Tumor Progression. Cancers 2021, 13, 1006. [Google Scholar] [CrossRef] [PubMed]
- Noda, Y.; Kishino, M.; Sato, S.; Hirose, K.; Sakai, M.; Fukuda, Y.; Murakami, S.; Toyosawa, S. Galectin-1 expression is associated with tumour immunity and prognosis in gingival squamous cell carcinoma. J. Clin. Pathol. 2016, 70, 126–133. [Google Scholar] [CrossRef]
- Nambiar, D.K.; Aguilera, T.; Cao, H.; Kwok, S.; Kong, C.; Bloomstein, J.; Wang, Z.; Rangan, V.S.; Jiang, D.; Von Eyben, R.; et al. Galectin-1–driven T cell exclusion in the tumor endothelium promotes immunotherapy resistance. J. Clin. Investig. 2019, 129, 5553–5567. [Google Scholar] [CrossRef] [Green Version]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Ochi, A.; Heindel, D.W.; Lee, K.B.; Zambirinis, C.P.; Pandian, G.S.B.; Savadkar, S.; et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat. Med. 2017, 23, 556–567. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Dirkx, A.E.; oude Egbrink, M.G.; Kuijpers, M.J.; van der Niet, S.T.; Heijnen, V.V.; Bouma-ter Steege, J.C.; Wagstaff, J.; Griffioen, A.W. Tumor Angiogenesis Modulates Leukocyte-Vessel Wall Interactions in vivo by Reducing Endothelial Adhesion Molecule Expression. Cancer Res. 2003, 63, 2322. [Google Scholar]
- Tromp, S.C.; Egbrink, M.G.A.O.; Dings, R.; Van Velzen, S.; Slaaf, D.W.; Hillen, H.F.P.; Tangelder, G.J.; Reneman, R.S.; Griffioen, A.W. Tumor angiogenesis factors reduce leukocyte adhesion in vivo. Int. Immunol. 2000, 12, 671–676. [Google Scholar] [CrossRef]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Dirkx, A.E.M.; Egbrink, M.G.A.O.; Castermans, K.; van der Schaft, D.W.J.; Thijssen, V.L.J.L.; Dings, R.P.M.; Kwee, L.; Mayo, K.H.; Wagstaff, J.; Steege, J.C.A.B.; et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006, 20, 621–630. [Google Scholar] [CrossRef] [Green Version]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours—implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Blanco, C.; Fondevila, F.; Palomo, A.G.; González-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Arrowsmith, J.; Miller, P. Phase II and Phase III attrition rates 2011–2012. Nat. Rev. Drug Discov. 2013, 12, 569. [Google Scholar] [CrossRef]
- Costa, E.C.; Moreira, A.F.; Diogo, D.M.D.M.; Gaspar, V.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: An overview on the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Porter, R.J.; Murray, G.I.; McLean, M.H. Current concepts in tumour-derived organoids. Br. J. Cancer 2020, 123, 1209–1218. [Google Scholar] [CrossRef]
- Lv, D.; Hu, Z.; Lu, L.; Lu, H.; Xu, X. Three-dimensional cell culture: A powerful tool in tumor research and drug discovery (Review). Oncol. Lett. 2017, 14, 6999–7010. [Google Scholar] [CrossRef] [Green Version]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef]
- nbsp; Petersen, O.W.; Rønnov-Jessen, L.; Howlett, A.R.; Bissell, M.J. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA 1992, 89, 9064. [Google Scholar] [CrossRef] [Green Version]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [Green Version]
- nbsp; Fischbach, C.; Kong, H.J.; Hsiong, S.X.; Evangelista, M.B.; Yuen, W.; Mooney, D.J. Cancer cell angiogenic capability is regulated by 3D culture and integrin engagement. Proc. Natl. Acad. Sci. USA 2009, 106, 399–404. [Google Scholar] [CrossRef] [Green Version]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [Green Version]
- Lagies, S.; Schlimpert, M.; Neumann, S.; Wäldin, A.; Kammerer, B.; Borner, C.; Peintner, L. Cells grown in three-dimensional spheroids mirror in vivo metabolic response of epithelial cells. Commun. Biol. 2020, 3, 246. [Google Scholar] [CrossRef]
- Melissaridou, S.; Wiechec, E.; Magan, M.; Jain, M.V.; Chung, M.K.; Farnebo, L.; Roberg, K. The effect of 2D and 3D cell cultures on treatment response, EMT profile and stem cell features in head and neck cancer. Cancer Cell Int. 2019, 19, 16. [Google Scholar] [CrossRef] [Green Version]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT–mTOR–S6K signaling and drug responses. J. Cell Sci. 2017, 130, 203–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Sci. 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; Van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer–derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Finnberg, N.K.; Gokare, P.; Lev, A.; Grivennikov, S.I.; MacFarlane, A.W., IV; Campbell, K.S.; Winters, R.M.; Kaputa, K.; Farma, J.M.; Abbas, E.-S.A.; et al. Application of 3D tumoroid systems to define immune and cytotoxic therapeutic responses based on tumoroid and tissue slice culture molecular signatures. Oncotarget 2017, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, K.; Mochizuki, W.; Matsumoto, Y.; Matsumoto, T.; Fukuda, M.; Mizutani, T.; Watanabe, M.; Nakamura, T. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J. Gastroenterol. 2016, 51, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Pasch, C.A.; Favreau, P.F.; Yueh, A.E.; Babiarz, C.P.; Gillette, A.A.; Sharick, J.T.; Karim, M.R.; Nickel, K.P.; DeZeeuw, A.K.; Sprackling, C.M.; et al. Patient-Derived Cancer Organoid Cultures to Predict Sensitivity to Chemotherapy and Radiation. Clin. Cancer Res. 2019, 25, 5376–5387. [Google Scholar] [CrossRef]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; Van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; Van De Haar, J.; et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019, 11, eaay2574. [Google Scholar] [CrossRef]
- Bruun, J.; Kryeziu, K.; Eide, P.W.; Moosavi, S.H.; Eilertsen, I.A.; Langerud, J.; Røsok, B.I.; Totland, M.Z.; Brunsell, T.H.; Pellinen, T.; et al. Patient-Derived Organoids from Multiple Colorectal Cancer Liver Metastases Reveal Moderate Intra-patient Pharmacotranscriptomic Heterogeneity. Clin. Cancer Res. 2020, 26, 4107–4119. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, P.; Wang, L.; Li, M.; Ge, Z.; Noordam, L.; Lieshout, R.; Verstegen, M.M.; Ma, B.; Su, J.; et al. Cancer-Associated Fibroblasts Provide a Stromal Niche for Liver Cancer Organoids That Confers Trophic Effects and Therapy Resistance. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 407–431. [Google Scholar] [CrossRef]
- Białkowska, K.; Komorowski, P.; Bryszewska, M.; Miłowska, K. Spheroids as a Type of Three-Dimensional Cell Cultures—Examples of Methods of Preparation and the Most Important Application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef]
- Amann, T.; Bataille, F.; Spruss, T.; Mühlbauer, M.; Gäbele, E.; Schölmerich, J.; Kiefer, P.; Bosserhoff, A.-K.; Hellerbrand, C. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009, 100, 646–653. [Google Scholar] [CrossRef]
- Jung, H.R.; Kang, H.M.; Hong-Ryul, J.; Kim, D.-S.; Noh, K.H.; Kim, E.-S.; Lee, H.-J.; Chung, K.-S.; Cho, H.-S.; Kim, N.-S.; et al. Cell Spheroids with Enhanced Aggressiveness to Mimic Human Liver Cancer In Vitro and In Vivo. Sci. Rep. 2017, 7, 10499. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Kim, S.-H.; Kim, K.M.; Choi, E.K.; Kim, J.; Seo, H.R. Activated hepatic stellate cells play pivotal roles in hepatocellular carcinoma cell chemoresistance and migration in multicellular tumor spheroids. Sci. Rep. 2016, 6, 36750. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Kim, J.-S.; Kim, S.-H.; Park, Y.K.; Yu, E.; Kim, K.-H.; Seo, E.-J.; Oh, H.-B.; Lee, H.C.; Kim, K.M.; et al. Patient-derived multicellular tumor spheroids towards optimized treatment for patients with hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 109. [Google Scholar] [CrossRef]
- Ingber, D.E. Developmentally inspired human ‘organs on chips’. Development 2018, 145, dev156125. [Google Scholar] [CrossRef] [Green Version]
- Beckwitt, C.; Clark, A.; Wheeler, S.; Taylor, D.L.; Stolz, D.B.; Griffith, L.; Wells, A. Liver ‘organ on a chip’. Exp. Cell Res. 2018, 363, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Benam, K.H.; Villenave, R.; Lucchesi, C.; Varone, A.; Hubeau, C.; Lee, H.-H.; Alves, S.E.; Salmon, M.; Ferrante, T.C.; Weaver, J.C.; et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat. Methods 2016, 13, 151–157. [Google Scholar] [CrossRef]
- Stucki, A.O.; Stucki, J.D.; Hall, S.R.R.; Felder, M.; Mermoud, Y.; Schmid, R.A.; Geiser, T.; Guenat, O.T. A lung-on-a-chip array with an integrated bio-inspired respiration mechanism. Lab Chip 2015, 15, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Luo, Z.; Lee, J.; Kim, H.-J.; Lee, K.; Tebon, P.; Feng, Y.; Dokmeci, M.R.; Sengupta, S.; Khademhosseini, A. Organ-on-a-Chip for Cancer and Immune Organs Modeling. Adv. Healthc. Mater. 2019, 8, e1801363. [Google Scholar] [CrossRef]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Rehman, S.; Virumbrales-Munoz, M.; McMinn, P.H.; Geiger, P.; Fitzgerald, C.; Heaster, T.; Skala, M.C.; Beebe, D.J. Microfluidic tumor-on-a-chip model to evaluate the role of tumor environmental stress on NK cell exhaustion. Sci. Adv. 2021, 7, eabc2331. [Google Scholar] [CrossRef]
- Aref, A.R.; Campisi, M.; Ivanova, E.; Portell, A.; Larios, D.; Piel, B.P.; Mathur, N.; Zhou, C.; Coakley, R.V.; Bartels, A.; et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 2018, 18, 3129–3143. [Google Scholar] [CrossRef] [Green Version]
- Businaro, L.; De Ninno, A.; Schiavoni, G.; Lucarini, V.; Ciasca, G.; Gerardino, A.; Belardelli, F.; Gabriele, L.; Mattei, F. Cross talk between cancer and immune cells: Exploring complex dynamics in a microfluidic environment. Lab Chip 2013, 13, 229–239. [Google Scholar] [CrossRef]
- Misra, S.; Moro, C.F.; Del Chiaro, M.; Pouso, S.; Sebestyén, A.; Löhr, M.; Björnstedt, M.; Verbeke, C.S. Ex vivo organotypic culture system of precision-cut slices of human pancreatic ductal adenocarcinoma. Sci. Rep. 2019, 9, 2133. [Google Scholar] [CrossRef] [PubMed]
- Roelants, C.; Pillet, C.; Franquet, Q.; Sarrazin, C.; Peilleron, N.; Giacosa, S.; Guyon, L.; Fontanell, A.; Fiard, G.; Long, J.-A.; et al. Ex-Vivo Treatment of Tumor Tissue Slices as a Predictive Preclinical Method to Evaluate Targeted Therapies for Patients with Renal Carcinoma. Cancers 2020, 12, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivakumar, R.; Chan, M.; Shin, J.S.; Nishida-Aoki, N.; Kenerson, H.L.; Elemento, O.; Beltran, H.; Yeung, R.; Gujral, T.S. Organotypic tumor slice cultures provide a versatile platform for immuno-oncology and drug discovery. OncoImmunology 2019, 8, e1670019. [Google Scholar] [CrossRef] [PubMed]
- Kishan, A.T.N.; Nicole, S.V.; Humberto, S.; van Deurzen Carolien, H.M.; den Bakker Michael, A.; Jan, H.J.H.; Roland, K.; Vreeswijk Maaike, P.G.; Agnes, J.; van Gent, D.C. Tumor slice culture system to assess drug response of primary breast cancer. BMC Cancer 2016, 16, 78. [Google Scholar] [CrossRef] [Green Version]
- Kenerson, H.L.; Sullivan, K.M.; Seo, Y.D.; Stadeli, K.M.; Ussakli, C.; Yan, X.; Lausted, C.; Pillarisetty, V.G.; Park, J.O.; Riehle, K.J.; et al. Tumor slice culture as a biologic surrogate of human cancer. Ann. Transl. Med. 2020, 8, 114. [Google Scholar] [CrossRef]
- Roife, D.; Dai, B.; Kang, Y.; Perez, M.V.R.; Pratt, M.; Li, X.; Fleming, J.B. Ex Vivo Testing of Patient-Derived Xenografts Mirrors the Clinical Outcome of Patients with Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2016, 22, 6021–6030. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.; Armeanu, S.; Smirnow, I.; Kupka, S.; Wagner, S.; Wehrmann, M.; Rots, M.G.; Groothuis, G.M.M.; Weiss, T.S.; Königsrainer, A.; et al. Human precision-cut liver tumor slices as a tumor patient-individual predictive test system for oncolytic measles vaccine viruses. Int. J. Oncol. 2009, 34, 1247–1256. [Google Scholar]
- Doornebal, E.; Harris, N.; Cooksley, H.; Pizanias, M.; Miquel, R.; Zen, Y.; Zamalloa, A.; Preziosi, M.; Heaton, N.; Prachalias, A.; et al. Development of personalised human immunocompetent ex vivo models of primary and secondary liver cancers using precision cut tissue slice technology. J. Hepatol. 2020, 73, S635–S636. [Google Scholar] [CrossRef]
- Cassim, S.; Raymond, V.-A.; Lacoste, B.; Lapierre, P.; Bilodeau, M. Metabolite profiling identifies a signature of tumorigenicity in hepatocellular carcinoma. Oncotarget 2018, 9, 26868–26883. [Google Scholar] [CrossRef] [Green Version]
- Palma, E.; Doornebal, E.J.; Chokshi, S. Precision-cut liver slices: A versatile tool to advance liver research. Hepatol. Int. 2019, 13, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Graaf, I.A.M.D.; Olinga, P.; De Jager, M.H.; Merema, M.T.; de Kanter, R.; Van De Kerkhof, E.G.; Groothuis, G.M.M. Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat. Protoc. 2010, 5, 1540–1551. [Google Scholar] [CrossRef]
- Palma, E.; Riva, A.; Moreno, C.; Odena, G.; Mudan, S.; Manyakin, N.; Miquel, R.; Degré, D.; Trepo, E.; Sancho-Bru, P.; et al. Perturbations in Mitochondrial Dynamics Are Closely Involved in the Progression of Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2020, 44, 856–865. [Google Scholar] [CrossRef]
- Sadasivan, S.K.; Nethra, S.; Khan, K.M.; Vasamsetti, B.; Kumar, N.R.; Haridas, V.; Reddy, M.B.; Somesh, B.P.; Oommen, A.; Rao, R.P. Developing an in vitro screening assay platform for evaluation of antifibrotic drugs using precision-cut liver slices. Fibrogenes. Tissue Repair 2015, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Roberto, J.B.; Knupp, A.; Kenerson, H.L.; Truong, C.D.; Yuen, S.Y.; Brempelis, K.J.; Tuefferd, M.; Chen, A.; Horton, H.; et al. Precision-cut human liver slice cultures as an immunological platform. J. Immunol. Methods 2018, 455, 71–79. [Google Scholar] [CrossRef]
- Stärkel, P.; Schnabl, B.; Leclercq, S.; Komuta, M.; Bataller, R.; Argemi, J.; Palma, E.; Chokshi, S.; Hellerbrand, C.; Maccioni, L.; et al. Deficient IL-6/Stat3 Signaling, High TLR7, and Type I Interferons in Early Human Alcoholic Liver Disease: A Triad for Liver Damage and Fibrosis. Hepatol. Commun. 2019, 3, 867–882. [Google Scholar] [CrossRef] [Green Version]
- Voabil, P.; de Bruijn, M.; Roelofsen, L.M.; Hendriks, S.H.; Brokamp, S.; Braber, M.V.D.; Broeks, A.; Sanders, J.; Herzig, P.; Zippelius, A.; et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat. Med. 2021, 27, 1250–1261. [Google Scholar] [CrossRef]
- Davies, E.J.; Dong, M.; Gutekunst, M.; Närhi, K.; Van Zoggel, H.J.A.A.; Blom, S.; Nagaraj, A.; Metsalu, T.; Oswald, E.; Erkens-Schulze, S.; et al. Capturing complex tumour biology in vitro: Histological and molecular characterisation of precision cut slices. Sci. Rep. 2015, 5, 17187. [Google Scholar] [CrossRef]
- Paish, H.L.; Reed, L.H.; Brown, H.; Bryan, M.C.; Govaere, O.; Leslie, J.; Barksby, B.S.; Macia, M.G.; Watson, A.; Xu, X.; et al. A Bioreactor Technology for Modeling Fibrosis in Human and Rodent Precision-Cut Liver Slices. Hepatology 2019, 70, 1377–1391. [Google Scholar] [CrossRef] [Green Version]
- Moysidou, C.-M.; Barberio, C.; Owens, R.M. Advances in Engineering Human Tissue Models. Front. Bioeng. Biotechnol. 2021, 8. [Google Scholar] [CrossRef]
- Crapo, P.M.; Gilbert, T.W.; Badylak, S.F. An overview of tissue and whole organ decellularization processes. Biomaterials 2011, 32, 3233–3243. [Google Scholar] [CrossRef] [Green Version]
- Giobbe, G.G.; Crowley, C.; Luni, C.; Campinoti, S.; Khedr, M.; Kretzschmar, K.; De Santis, M.M.; Zambaiti, E.; Michielin, F.; Meran, L.; et al. Extracellular matrix hydrogel derived from decellularized tissues enables endodermal organoid culture. Nat. Commun. 2019, 10, 5658. [Google Scholar] [CrossRef] [Green Version]
- Ott, H.C.; Matthiesen, T.S.; Goh, S.-K.; Black, L.D.; Kren, S.; Netoff, T.I.; Taylor, D.A. Perfusion-decellularized matrix: Using nature’s platform to engineer a bioartificial heart. Nat. Med. 2008, 14, 213–221. [Google Scholar] [CrossRef]
- Uygun, B.; Soto-Gutierrez, A.; Yagi, H.; Izamis, M.-L.; Guzzardi, M.; Shulman, C.; Milwid, J.; Kobayashi, N.; Tilles, A.; Berthiaume, F.; et al. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat. Med. 2010, 16, 814–820. [Google Scholar] [CrossRef]
- Petersen, T.H.; Calle, E.A.; Zhao, L.; Lee, E.J.; Gui, L.; Raredon, M.B.; Gavrilov, K.; Yi, T.; Zhuang, Z.W.; Breuer, C.; et al. Tissue-Engineered Lungs for in Vivo Implantation. Science 2010, 329, 538–541. [Google Scholar] [CrossRef] [Green Version]
- Mazza, G.; Telese, A.; Al-Akkad, W.; Frenguelli, L.; Levi, A.; Marrali, M.; Longato, L.; Thanapirom, K.; Vilia, M.G.; Lombardi, B.; et al. Cirrhotic Human Liver Extracellular Matrix 3D Scaffolds Promote Smad-Dependent TGF-β1 Epithelial Mesenchymal Transition. Cells 2019, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Huleihel, L.; Dziki, J.L.; Bartolacci, J.G.; Rausch, T.; Scarritt, M.E.; Cramer, M.C.; Vorobyov, T.; LoPresti, S.T.; Swineheart, I.T.; White, L.; et al. Macrophage phenotype in response to ECM bioscaffolds. Semin. Immunol. 2017, 29, 2–13. [Google Scholar] [CrossRef]
- Piccoli, M.; D’Angelo, E.; Crotti, S.; Sensi, F.; Urbani, L.; Maghin, E.; Burns, A.; De Coppi, P.; Fassan, M.; Rugge, M.; et al. Decellularized colorectal cancer matrix as bioactive microenvironment for in vitro 3D cancer research. J. Cell. Physiol. 2018, 233, 5937–5948. [Google Scholar] [CrossRef]
- D’Angelo, E.; Natarajan, D.; Sensi, F.; Ajayi, O.; Fassan, M.; Mammano, E.; Pilati, P.; Pavan, P.; Bresolin, S.; Preziosi, M.; et al. Patient-Derived Scaffolds of Colorectal Cancer Metastases as an Organotypic 3D Model of the Liver Metastatic Microenvironment. Cancers 2020, 12, 364. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, D.; Wu, G.; Wu, J.; Lu, S.; Lo, J.; He, Y.; Zhao, C.; Zhao, X.; Zhang, H.; et al. Metastasis-on-a-chip mimicking the progression of kidney cancer in the liver for predicting treatment efficacy. Theranostics 2020, 10, 300–311. [Google Scholar] [CrossRef]
- Lu, S.; Cuzzucoli, F.; Jiang, J.; Liang, L.-G.; Wang, Y.; Kong, M.; Zhao, X.; Cui, W.; Li, J.; Wang, S. Development of a biomimetic liver tumor-on-a-chip model based on decellularized liver matrix for toxicity testing. Lab Chip 2018, 18, 3379–3392. [Google Scholar] [CrossRef]
- Micek, H.M.; Visetsouk, M.R.; Masters, K.S.; Kreeger, P.K. Engineering the Extracellular Matrix to Model the Evolving Tumor Microenvironment. iScience 2020, 23, 101742. [Google Scholar] [CrossRef] [PubMed]
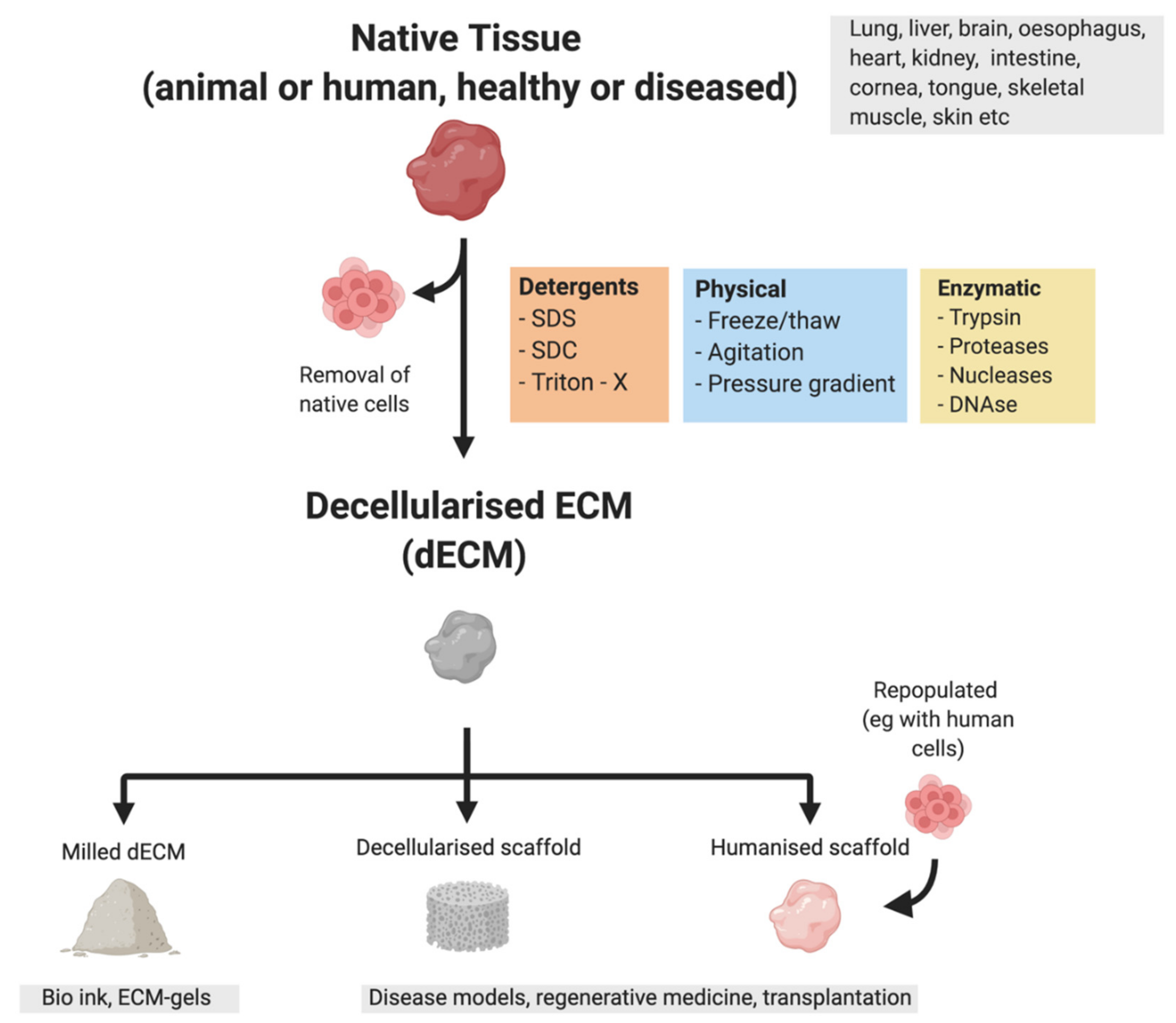

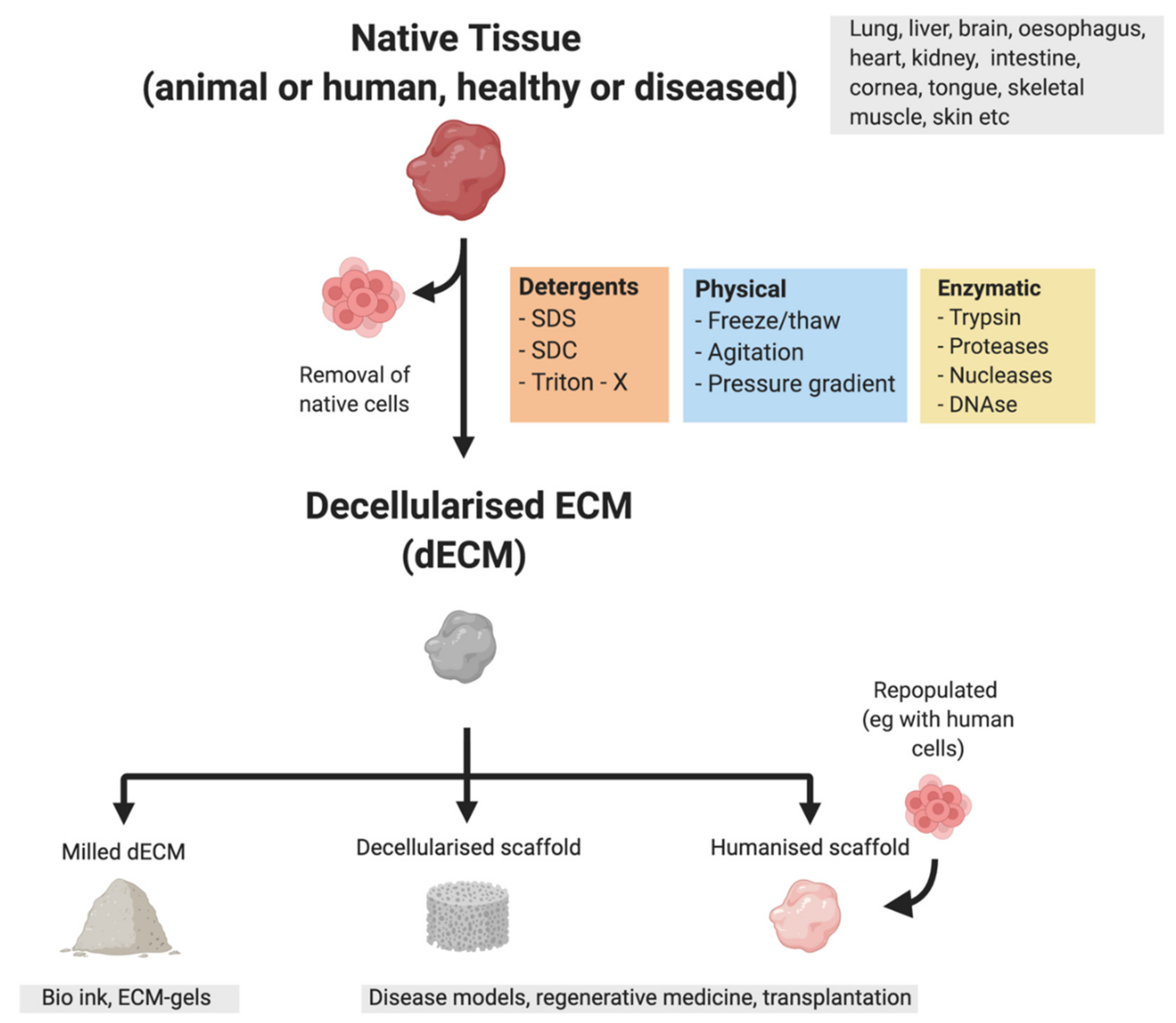{kind=link}
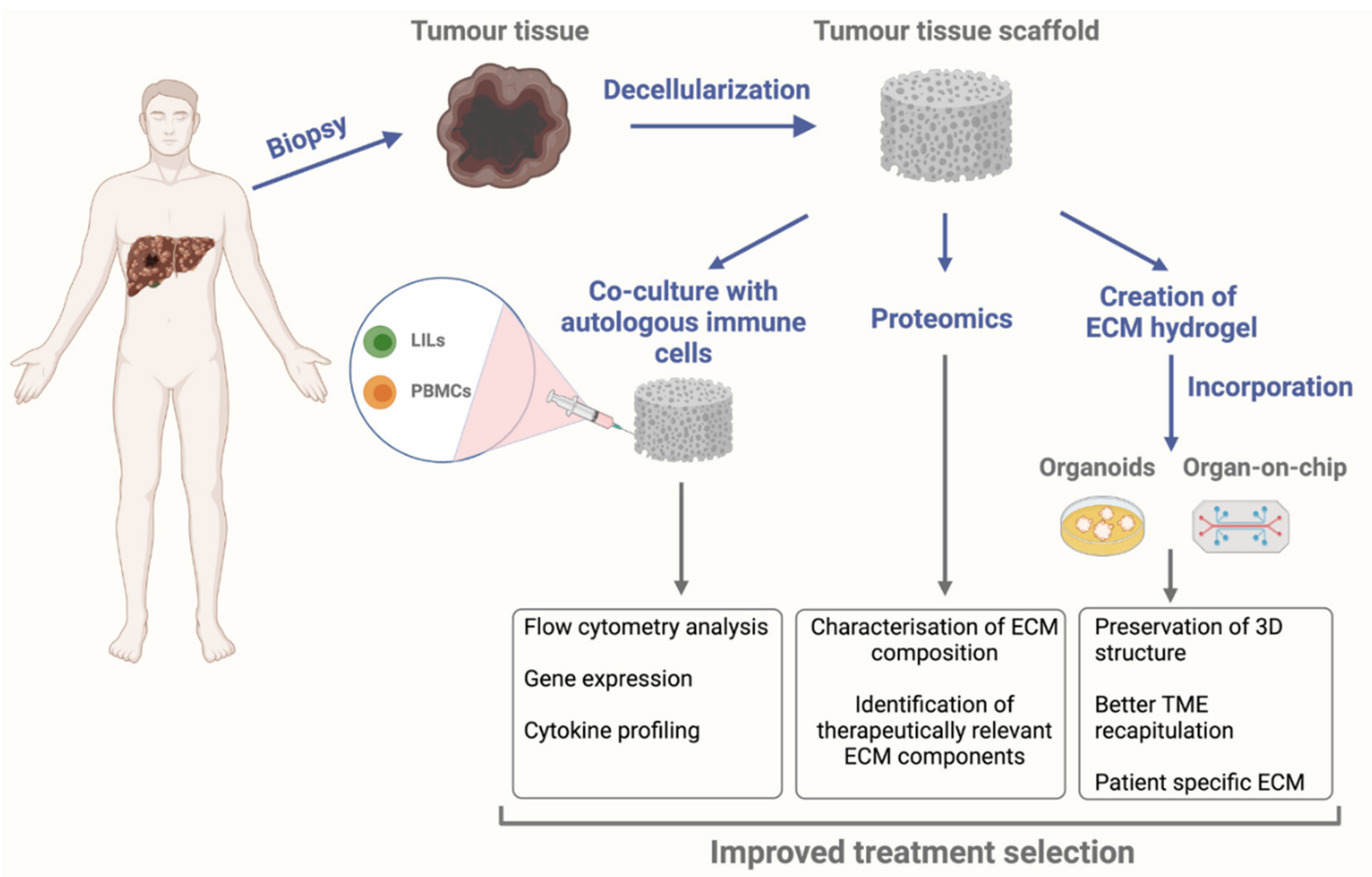
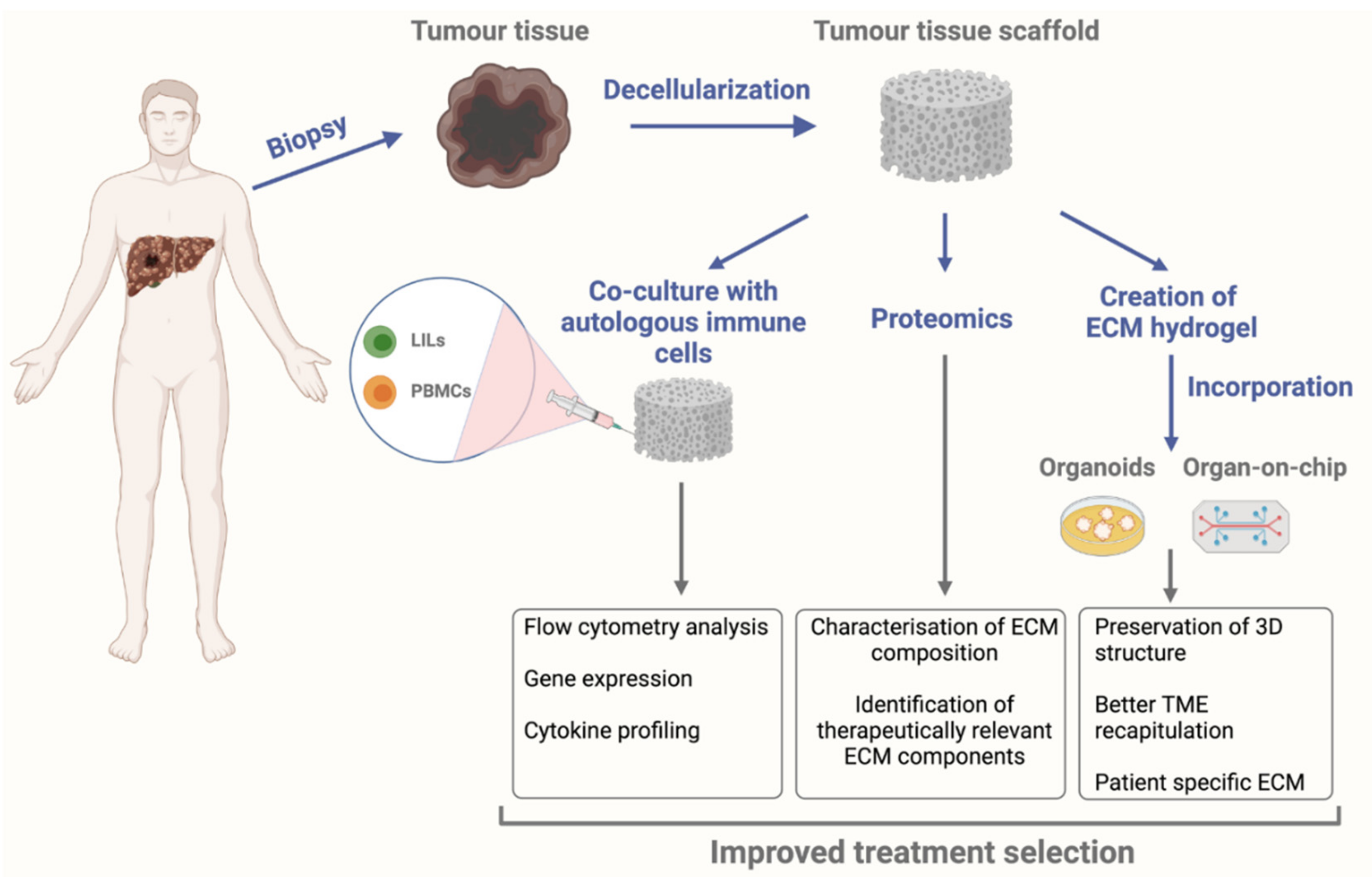{kind=link}
| Increased ECM Deposition | ECM Degradation | Altered Post-Translational Modifications |
|---|---|---|
| ↑Fibronectin, collagens I, III, IV | ↑ECM degradation ↑Creation of bioactive molecules (matrikines) ↑Release of growth factors ↑Angiogenesis ↑ Metastasis | ↑LOX activity ↑ECM stiffness ↑Mechanosignalling ↑Interstitial fluid pressure ↓ Impeded immune cell activity |
| Variation | 2D Cell Lines | Spheroids | Organoids | Organ on Chip | Bioengineered Models | PCTS |
|---|---|---|---|---|---|---|
| Advantages | -Easy to use -Affordable -Highly reproducible -Long culture times | -Can be patient specific -Can be multicellular and be used in co-cultures -High reproducibility | -Patient specific -Various cell lineages -May contain tissue-specific ECM -Ability to co-culture | -Patient specific -Vascularisation -Ability for co-culture -Controlled environment | -Patient specific -Inclusion of native microenvironment -Ability for co-culture | -Preservation of in vivo microenvironment -Fully immunocompetent -Preservation of patient-specific characteristics |
| Disadvantages | -Lack of microenvironment -No cell heterogeneity -No patient specificity | -Poor structural organization -Forms a necrotic core -Limited oxygen and nutrients supply | -High inter patient variability -High cost -Lack of vascularization -Difficult to achieve maturation | -Low throughput -Difficulty with longevity -Technically challenging to use -Lack of technical robustness | -High inter patient variability -Lack of standardised protocols -Costly -Difficulty with obtaining tissue | -Short culture time -High inter patient variability -Difficulty with obtaining tissue |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sükei, T.; Palma, E.; Urbani, L. Interplay between Cellular and Non-Cellular Components of the Tumour Microenvironment in Hepatocellular Carcinoma. Cancers 2021, 13, 5586. https://doi.org/10.3390/cancers13215586
Sükei T, Palma E, Urbani L. Interplay between Cellular and Non-Cellular Components of the Tumour Microenvironment in Hepatocellular Carcinoma. Cancers. 2021; 13(21):5586. https://doi.org/10.3390/cancers13215586
Chicago/Turabian StyleSükei, Tamás, Elena Palma, and Luca Urbani. 2021. "Interplay between Cellular and Non-Cellular Components of the Tumour Microenvironment in Hepatocellular Carcinoma" Cancers 13, no. 21: 5586. https://doi.org/10.3390/cancers13215586
APA StyleSükei, T., Palma, E., & Urbani, L. (2021). Interplay between Cellular and Non-Cellular Components of the Tumour Microenvironment in Hepatocellular Carcinoma. Cancers, 13(21), 5586. https://doi.org/10.3390/cancers13215586