
Intratumor Heterogeneity in Hepatocellular Carcinoma: Challenges and Opportunities


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
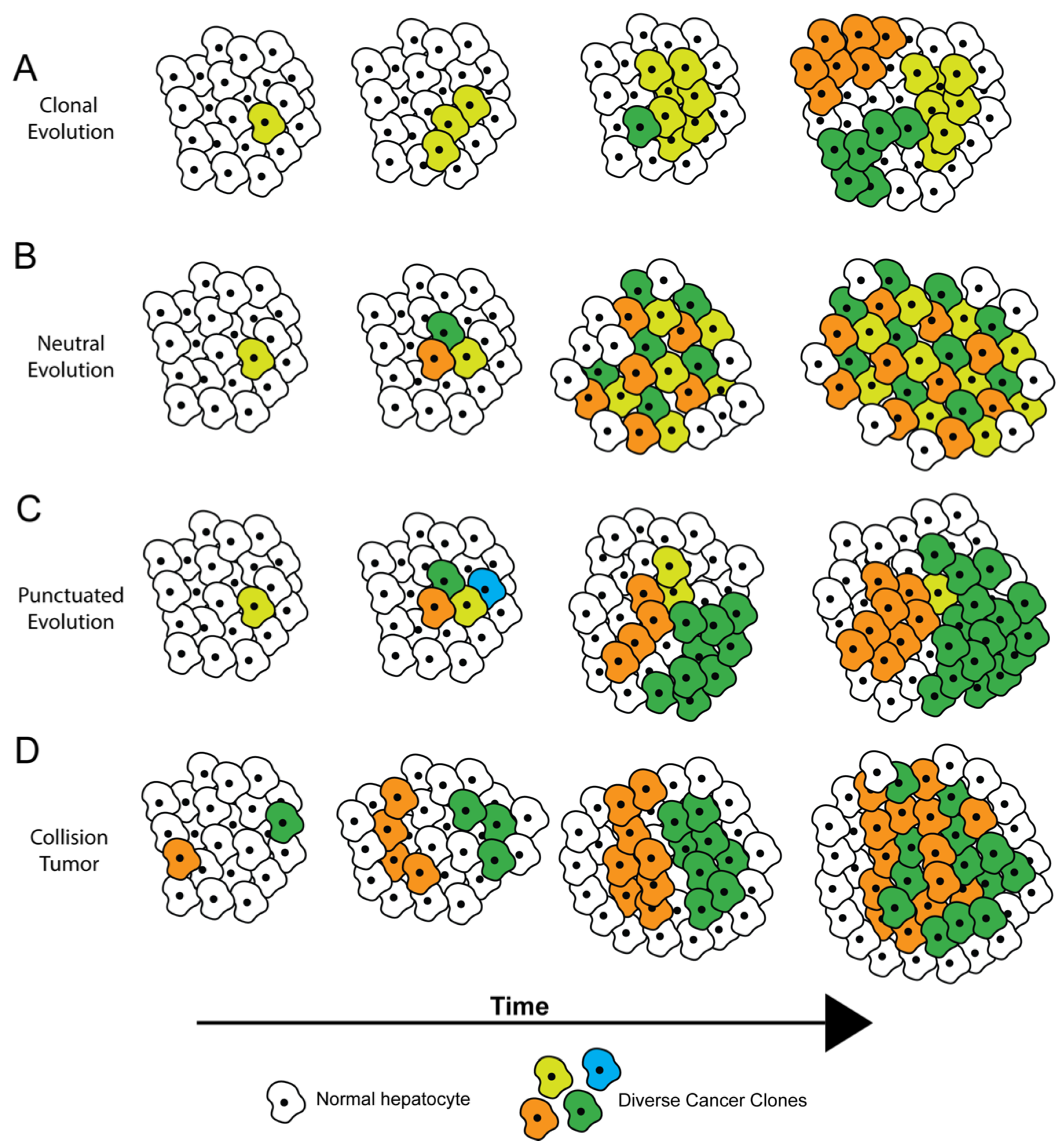
2. How Tumors Become Heterogeneous
3. Subtypes of Genomic ITH
4. Phenotypic/Functional ITH
5. Tumor Microenvironment ITH
6. Clonal Cooperation
7. ITH of TERT and Wnt/β-Catenin Signaling
7.1. Wnt/β-Catenin Pathway ITH
7.2. TERT ITH
8. Clinical Consequences of ITH for HCC
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Tabassum, D.P.; Polyak, K. Tumorigenesis: It Takes a Village. Nat. Rev. Cancer 2015, 15, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.J.; von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour Evolution in Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, D.B.; Bayramoğlu, Z.; Ünay, G.; Ayık, E.; Başsorgun, C.İ.; Elpek, G.Ö. Incidental Collision Tumor of Hepatocellular Carcinoma and Neuroendocrine Carcinoma. J. Clin. Transl. Hepatol. 2018, 6, 339–344. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward Understanding and Exploiting Tumor Heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving Genetic Heterogeneity in Cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- Wagner, D.E.; Klein, A.M. Lineage Tracing Meets Single-Cell Omics: Opportunities and Challenges. Nat. Rev. Genet. 2020, 21, 410–427. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Sottoriva, A.; Graham, T.A. Measuring Clonal Evolution in Cancer with Genomics. Annu. Rev. Genom. Hum. Genet. 2019, 20, 309–329. [Google Scholar] [CrossRef]
- Xu, L.X.; He, M.H.; Dai, Z.H.; Yu, J.; Wang, J.G.; Li, X.C.; Jiang, B.B.; Ke, Z.F.; Su, T.H.; Peng, Z.W.; et al. Genomic and Transcriptional Heterogeneity of Multifocal Hepatocellular Carcinoma. Ann. Oncol. 2019, 30, 990–997. [Google Scholar] [CrossRef]
- Sirivatanauksorn, Y.; Sirivatanauksorn, V.; Bhattacharya, S.; Davidson, B.R.; Dhillon, A.P.; Kakkar, A.K.; Williamson, R.C.N.; Lemoine, N.R. Evolution of Genetic Abnormalities in Hepatocellular Carcinomas Demonstrated by DNA Fingerprinting. J. Pathol. 1999, 189, 344–350. [Google Scholar] [CrossRef]
- Xue, R.; Li, R.; Guo, H.; Guo, L.; Su, Z.; Ni, X.; Qi, L.; Zhang, T.; Li, Q.; Zhang, Z.; et al. Variable Intra-Tumor Genomic Heterogeneity of Multiple Lesions in Patients with Hepatocellular Carcinoma. Gastroenterology 2016, 150, 998–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrecilla, S.; Sia, D.; Harrington, A.N.; Zhang, Z.; Cabellos, L.; Cornella, H.; Moeini, A.; Camprecios, G.; Leow, W.-Q.; Fiel, M.I.; et al. Trunk Mutational Events Present Minimal Intra- and Inter-Tumoral Heterogeneity in Hepatocellular Carcinoma. J. Hepatol. 2017, 67, 1222–1231. [Google Scholar] [CrossRef] [Green Version]
- Uchi, R.; Takahashi, Y.; Niida, A.; Shimamura, T.; Hirata, H.; Sugimachi, K.; Sawada, G.; Iwaya, T.; Kurashige, J.; Shinden, Y.; et al. Integrated Multiregional Analysis Proposing a New Model of Colorectal Cancer Evolution. PLoS Genet. 2016, 12, e1005778. [Google Scholar] [CrossRef]
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and Temporal Diversity in Genomic Instability Processes Defines Lung Cancer Evolution. Science 2014, 346, 251. [Google Scholar] [CrossRef] [Green Version]
- Harbst, K.; Lauss, M.; Cirenajwis, H.; Isaksson, K.; Rosengren, F.; Törngren, T.; Kvist, A.; Johansson, M.C.; Vallon-Christersson, J.; Baldetorp, B.; et al. Multiregion Whole-Exome Sequencing Uncovers the Genetic Evolution and Mutational Heterogeneity of Early-Stage Metastatic Melanoma. Cancer Res. 2016, 76, 4765–4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.P.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic Architecture and Evolution of Clear Cell Renal Cell Carcinomas Defined by Multiregion Sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Van Loo, P.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Subclonal Diversification of Primary Breast Cancer Revealed by Multiregion Sequencing. Nat. Med. 2015, 21, 751–759. [Google Scholar] [CrossRef]
- Bashashati, A.; Ha, G.; Tone, A.; Ding, J.; Prentice, L.M.; Roth, A.; Rosner, J.; Shumansky, K.; Kalloger, S.; Senz, J.; et al. Distinct Evolutionary Trajectories of Primary High-Grade Serous Ovarian Cancers Revealed through Spatial Mutational Profiling. J. Pathol. 2013, 231, 21–34. [Google Scholar] [CrossRef]
- Colombino, M.; Sperlongano, P.; Izzo, F.; Tatangelo, F.; Botti, G.; Lombardi, A.; Accardo, M.; Tarantino, L.; Sordelli, I.; Agresti, M.; et al. BRAF and PIK3CA Genes Are Somatically Mutated in Hepatocellular Carcinoma among Patients from South Italy. Cell Death Dis. 2012, 3, e259. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Lee, H.W.; Park, W.S.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. PIK3CA Gene Is Frequently Mutated in Breast Carcinomas and Hepatocellular Carcinomas. Oncogene 2005, 24, 1477–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janku, F.; Kaseb, A.O.; Tsimberidou, A.M.; Wolff, R.A.; Kurzrock, R. Identification of Novel Therapeutic Targets in the PI3K/AKT/MTOR Pathway in Hepatocellular Carcinoma Using Targeted next Generation Sequencing. Oncotarget 2014, 5, 3012–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Liu, W.; Engelmann, J.C.; Nuhn, P.; Gurel, M.; Haffner, M.C.; Esopi, D.; Irizarry, R.A.; Getzenberg, R.H.; Nelson, W.G.; et al. DNA Methylation Alterations Exhibit Intraindividual Stability and Interindividual Heterogeneity in Prostate Cancer Metastases. Sci. Transl. Med. 2013, 5, 169ra10. [Google Scholar] [CrossRef] [Green Version]
- Mazor, T.; Pankov, A.; Johnson, B.E.; Hong, C.; Hamilton, E.G.; Bell, R.J.A.; Smirnov, I.V.; Reis, G.F.; Phillips, J.J.; Barnes, M.J.; et al. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell 2015, 28, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.-C.; Mayakonda, A.; Dinh, H.Q.; Huang, P.; Lin, L.; Liu, X.; Ding, L.; Wang, J.; Berman, B.P.; Song, E.-W.; et al. Genomic and Epigenomic Heterogeneity of Hepatocellular Carcinoma. Cancer Res. 2017, 77, 2255–2265. [Google Scholar] [CrossRef] [Green Version]
- Mroz, E.A.; Tward, A.D.; Pickering, C.R.; Myers, J.N.; Ferris, R.L.; Rocco, J.W. High Intratumor Genetic Heterogeneity Is Related to Worse Outcome in Patients with Head and Neck Squamous Cell Carcinoma. Cancer 2013, 119, 3034–3042. [Google Scholar] [CrossRef] [PubMed]
- Mroz, E.A.; Tward, A.D.; Tward, A.M.; Hammon, R.J.; Ren, Y.; Rocco, J.W. Intra-Tumor Genetic Heterogeneity and Mortality in Head and Neck Cancer: Analysis of Data from the Cancer Genome Atlas. PLoS Med. 2015, 12, e1001786. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.M.; Budhu, A.; Woo, H.G.; Chaisaingmongkol, J.; Dang, H.; Forgues, M.; Harris, C.C.; Zhang, G.; Auslander, N.; Ruppin, E.; et al. Functional Genomic Complexity Defines Intratumor Heterogeneity and Tumor Aggressiveness in Liver Cancer. Sci. Rep. 2019, 9, 16930. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Celià-Terrassa, T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J. Clin. Med. 2019, 8, 1542. [Google Scholar] [CrossRef] [Green Version]
- Ho, D.W.-H.; Tsui, Y.-M.; Sze, K.M.-F.; Chan, L.-K.; Cheung, T.-T.; Lee, E.; Sham, P.-C.; Tsui, S.K.-W.; Lee, T.K.-W.; Ng, I.O.-L. Single-Cell Transcriptomics Reveals the Landscape of Intra-Tumoral Heterogeneity and Stemness-Related Subpopulations in Liver Cancer. Cancer Lett. 2019, 459, 176–185. [Google Scholar] [CrossRef]
- Zheng, H.; Pomyen, Y.; Hernandez, M.O.; Li, C.; Livak, F.; Tang, W.; Dang, H.; Greten, T.F.; Davis, J.L.; Zhao, Y.; et al. Single-Cell Analysis Reveals Cancer Stem Cell Heterogeneity in Hepatocellular Carcinoma. Hepatology 2018, 68, 127–140. [Google Scholar] [CrossRef]
- Veryaskina, Y.A.; Titov, S.E.; Kometova, V.V.; Rodionov, V.V.; Zhimulev, I.F. Intratumoral Heterogeneity of Expression of 16 MiRNA in Luminal Cancer of the Mammary Gland. Non-Coding RNA 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D.; et al. Single-Cell Analyses Reveal Increased Intratumoral Heterogeneity after the Onset of Therapy Resistance in Small-Cell Lung Cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Yang, Z.; Zhou, L.; Zhu, Q.; Xie, H.; Zhang, F.; Wu, L.; Chen, L.; Zheng, S. Long Non-Coding RNA MALAT-1 Overexpression Predicts Tumor Recurrence of Hepatocellular Carcinoma after Liver Transplantation. Med. Oncol. 2012, 29, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic Stellate Cells and Extracellular Matrix in Hepatocellular Carcinoma: More Complicated than Ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef] [Green Version]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of Immune Microenvironment in Hepatocellular Carcinoma and Its Additional Impact on Histological and Molecular Classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; D’Avola, D.; et al. Intratumoral Heterogeneity and Clonal Evolution in Liver Cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef]
- Calderaro, J.; Petitprez, F.; Becht, E.; Laurent, A.; Hirsch, T.Z.; Rousseau, B.; Luciani, A.; Amaddeo, G.; Derman, J.; Charpy, C.; et al. Intra-Tumoral Tertiary Lymphoid Structures Are Associated with a Low Risk of Early Recurrence of Hepatocellular Carcinoma. J. Hepatol. 2019, 70, 58–65. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Hsu, C.-L.; Jeng, Y.-M.; Ho, M.-C.; Ho, C.-M.; Yeh, C.-P.; Yeh, C.-Y.; Hsu, M.-C.; Hu, R.-H.; Cheng, A.-L. Reliability of a Single-Region Sample to Evaluate Tumor Immune Microenvironment in Hepatocellular Carcinoma. J. Hepatol. 2020, 72, 489–497. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The Role of PD-L1 Expression as a Predictive Biomarker: An Analysis of All US Food and Drug Administration (FDA) Approvals of Immune Checkpoint Inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Sun, W.; Kim, R.; He, A.R.; Abada, P.B.; Mynderse, M.; Finn, R.S. The Role of Angiogenesis in Hepatocellular Carcinoma. Clin. Cancer Res. 2019, 25, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wu, J.; Bai, X.; Liang, T. Evaluation of Intra-Tumoral Vascularization in Hepatocellular Carcinomas. Front. Med. 2020, 7, 584250. [Google Scholar] [CrossRef]
- Jang, H.-J.; Lim, H.K.; Lee, W.J.; Kim, S.H.; Kim, M.J.; Choi, D.; Lee, S.J.; Lim, J.H. Focal Hepatic Lesions: Evaluation with Contrast-Enhanced Gray-Scale Harmonic US. Korean J. Radiol. 2003, 4, 91. [Google Scholar] [CrossRef] [PubMed]
- Hayano, K.; Yoshida, H.; Zhu, A.X.; Sahani, D.V. Fractal Analysis of Contrast-Enhanced CT Images to Predict Survival of Patients with Hepatocellular Carcinoma Treated with Sunitinib. Dig. Dis. Sci. 2014, 59, 1996–2003. [Google Scholar] [CrossRef]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y.R. Hepatic Stellate Cells in Liver Development, Regeneration, and Cancer. J. Clin. Invest. 2013, 123, 1902–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukowati, C.H.C.; Anfuso, B.; Crocé, L.S.; Tiribelli, C. The Role of Multipotent Cancer Associated Fibroblasts in Hepatocarcinogenesis. BMC Cancer 2015, 15, 188. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-Associated Fibroblasts: Overview, Progress, Challenges, and Directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Dong, C.; Jiang, K.; Xu, Z.; Li, R.; Guo, K.; Shao, S.; Wang, L. Heterogeneity of Cancer-Associated Fibroblasts and Roles in the Progression, Prognosis, and Therapy of Hepatocellular Carcinoma. J. Hematol. Oncol. 2019, 12, 101. [Google Scholar] [CrossRef]
- Takamura, H.; Nakanuma, S.; Hayashi, H.; Tajima, H.; Kakinoki, K.; Sakai, S.; Makino, I.; Nakagawara, H.; Miyashita, T.; Okamoto, K.; et al. Evaluation of Eligibility Criteria in Living Donor Liver Transplantation for Hepatocellular Carcinoma by α-SMA-Positive Cancer-Associated Fibroblasts. Oncol. Rep. 2013, 30, 1561–1574. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.; Yuan, J.; Chen, M.; Sun, Z.; Liu, L.; Cheng, G.; Ying, H.; Yang, S.; Chen, M. The Heterogenic Tumor Microenvironment of Hepatocellular Carcinoma and Prognostic Analysis Based on Tumor Neo-Vessels, Macrophages and α-SMA. Oncol. Lett. 2018, 15, 4805–4812. [Google Scholar] [CrossRef]
- Kim, G.J.; Rhee, H.; Yoo, J.E.; Ko, J.E.; Lee, J.S.; Kim, H.; Choi, J.S.; Park, Y.N. Increased Expression of CCN2, Epithelial Membrane Antigen, and Fibroblast Activation Protein in Hepatocellular Carcinoma with Fibrous Stroma Showing Aggressive Behavior. PLoS ONE 2014, 9, e105094. [Google Scholar] [CrossRef]
- Vinci, M.; Burford, A.; Molinari, V.; Kessler, K.; Popov, S.; Clarke, M.; Taylor, K.R.; Pemberton, H.N.; Lord, C.J.; Gutteridge, A.; et al. Functional Diversity and Cooperativity between Subclonal Populations of Pediatric Glioblastoma and Diffuse Intrinsic Pontine Glioma Cells. Nat. Med. 2018, 24, 1204–1215. [Google Scholar] [CrossRef]
- Cleary, A.S.; Leonard, T.L.; Gestl, S.A.; Gunther, E.J. Tumour Cell Heterogeneity Maintained by Cooperating Subclones in Wnt-Driven Mammary Cancers. Nature 2014, 508, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Thirumalai, D. Share, but Unequally: A Plausible Mechanism for Emergence and Maintenance of Intratumour Heterogeneity. J. R. Soc. Interface 2019, 16, 20180820. [Google Scholar] [CrossRef] [Green Version]
- Schulze, K.; Nault, J.-C.; Villanueva, A. Genetic Profiling of Hepatocellular Carcinoma Using Next-Generation Sequencing. J. Hepatol. 2016, 65, 1031–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouzé, E.; Imbeaud, S.; Pilati, C.; Nault, J.-C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-Phenotype Correlation of CTNNB1 Mutations Reveals Different β-Catenin Activity Associated with Liver Tumor Progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Wang, Y.-P.; Wang, J.; Fu, P.-Y.; Zhang, X.; Cao, Y.; Fan, J.; Yang, X.-R.; Zhou, J. Limited Bias Effect of Intratumoral Heterogeneity on Genetic Profiling of Hepatocellular Carcinoma. J. Gastrointest. Oncol. 2020, 11, 112–120. [Google Scholar] [CrossRef]
- Xue, Y.; San Luis, B.; Lane, D.P. Intratumour Heterogeneity of P53 Expression; Causes and Consequences. J. Pathol. 2019, 249, 274–285. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt Signalling in Stem Cells and Cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.O.; Monga, S.P. Wnt/β-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 351–378. [Google Scholar] [CrossRef] [Green Version]
- Krutsenko, Y.; Singhi, A.D.; Monga, S.P. β-Catenin Activation in Hepatocellular Cancer: Implications in Biology and Therapy. Cancers 2021, 13, 1830. [Google Scholar] [CrossRef]
- Friemel, J.; Rechsteiner, M.; Frick, L.; Böhm, F.; Struckmann, K.; Egger, M.; Moch, H.; Heikenwalder, M.; Weber, A. Intratumor Heterogeneity in Hepatocellular Carcinoma. Clin. Cancer Res. 2015, 21, 1951–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evason, K.J.; Francisco, M.T.; Juric, V.; Balakrishnan, S.; del Lopez Pazmino, M.P.; Gordan, J.D.; Kakar, S.; Spitsbergen, J.; Goga, A.; Stainier, D.Y.R. Identification of Chemical Inhibitors of β-Catenin-Driven Liver Tumorigenesis in Zebrafish. PLOS Genet. 2015, 11, e1005305. [Google Scholar] [CrossRef]
- Kalasekar, S.M.; Kotiyal, S.; Conley, C.; Phan, C.; Young, A.; Evason, K.J. Heterogeneous Beta-Catenin Activation Is Sufficient to Cause Hepatocellular Carcinoma in Zebrafish. Biol. Open 2019, 8, bio.047829. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable Beta-Catenin Expression in Colorectal Cancers Indicates Tumor Progression Driven by the Tumor Environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Jung, A.; Hermann, K.; Günther, K.; Hohenberger, W.; Kirchner, T. Nuclear Overexpression of the Oncoprotein β-Catenin in Colorectal Cancer Is Localized Predominantly at the Invasion Front. Pathol. Res. Pract. 1998, 194, 701–704. [Google Scholar] [CrossRef]
- Torre, C.; Perret, C.; Colnot, S. Transcription Dynamics in a Physiological Process: β-Catenin Signaling Directs Liver Metabolic Zonation. Int. J. Biochem. Cell Biol. 2011, 43, 271–278. [Google Scholar] [CrossRef]
- Leibing, T.; Géraud, C.; Augustin, I.; Boutros, M.; Augustin, H.G.; Okun, J.G.; Langhans, C.-D.; Zierow, J.; Wohlfeil, S.A.; Olsavszky, V.; et al. Angiocrine Wnt Signaling Controls Liver Growth and Metabolic Maturation in Mice. Hepatol. Baltim. Md. 2018, 68, 707–722. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/β-Catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; De Castro Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-Specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Autexier, C.; Lue, N.F. The Structure and Function of Telomerase Reverse Transcriptase. Annu. Rev. Biochem. 2006, 75, 493–517. [Google Scholar] [CrossRef] [PubMed]
- Dogan, F.; Forsyth, N.R. Telomerase Regulation: A Role for Epigenetics. Cancers 2021, 13, 1213. [Google Scholar] [CrossRef]
- Ségal-Bendirdjian, E.; Geli, V. Non-Canonical Roles of Telomerase: Unraveling the Imbroglio. Front. Cell Dev. Biol. 2019, 7, 332. [Google Scholar] [CrossRef] [PubMed]
- Romaniuk, A.; Paszel-Jaworska, A.; Totoń, E.; Lisiak, N.; Hołysz, H.; Królak, A.; Grodecka-Gazdecka, S.; Rubiś, B. The Non-Canonical Functions of Telomerase: To Turn off or Not to Turn off. Mol. Biol. Rep. 2019, 46, 1401–1411. [Google Scholar] [CrossRef] [Green Version]
- Nagore, E.; Heidenreich, B.; Rachakonda, S.; Garcia-Casado, Z.; Requena, C.; Soriano, V.; Frank, C.; Traves, V.; Quecedo, E.; Sanjuan-Gimenez, J.; et al. TERT Promoter Mutations in Melanoma Survival: TERT Promoter Mutations in Melanoma Survival. Int. J. Cancer 2016, 139, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Scott, G.A.; Laughlin, T.S.; Rothberg, P.G. Mutations of the TERT Promoter Are Common in Basal Cell Carcinoma and Squamous Cell Carcinoma. Mod. Pathol. 2014, 27, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.-S.; Wang, Z.; He, X.-J.; Diplas, B.H.; Yang, R.; Killela, P.J.; Meng, Q.; Ye, Z.-Y.; Wang, W.; Jiang, X.-T.; et al. Recurrent TERT Promoter Mutations Identified in a Large-Scale Study of Multiple Tumour Types Are Associated with Increased TERT Expression and Telomerase Activation. Eur. J. Cancer 2015, 51, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Rachakonda, P.S.; Hosen, I.; de Verdier, P.J.; Fallah, M.; Heidenreich, B.; Ryk, C.; Wiklund, N.P.; Steineck, G.; Schadendorf, D.; Hemminki, K.; et al. TERT Promoter Mutations in Bladder Cancer Affect Patient Survival and Disease Recurrence through Modification by a Common Polymorphism. Proc. Natl. Acad. Sci. USA 2013, 110, 17426–17431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Xing, M. TERT Promoter Mutations in Thyroid Cancer. Endocr. Relat. Cancer 2016, 23, R143–R155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwa, W.T.; Effendi, K.; Yamazaki, K.; Kubota, N.; Hatano, M.; Ueno, A.; Masugi, Y.; Sakamoto, M. Telomerase Reverse Transcriptase (TERT) Promoter Mutation Correlated with Intratumoral Heterogeneity in Hepatocellular Carcinoma. Pathol. Int. 2020, 70, 624–632. [Google Scholar] [CrossRef]
- Abedalthagafi, M.S.; Bi, W.L.; Merrill, P.H.; Gibson, W.J.; Rose, M.F.; Du, Z.; Francis, J.M.; Du, R.; Dunn, I.F.; Ligon, A.H.; et al. ARID1A and TERT Promoter Mutations in Dedifferentiated Meningioma. Cancer Genet. 2015, 208, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Juratli, T.A.; Thiede, C.; Koerner, M.V.A.; Tummala, S.S.; Daubner, D.; Shankar, G.M.; Williams, E.A.; Martinez-Lage, M.; Soucek, S.; Robel, K.; et al. Intratumoral Heterogeneity and TERT Promoter Mutations in Progressive/Higher-Grade Meningiomas. Oncotarget 2017, 8, 109228–109237. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Park, T.I.; Jang, S.Y.; Park, S.Y.; Park, W.-J.; Jung, S.-J.; Lee, J.-H. Clinicopathological Characteristics of TERT Promoter Mutation and Telomere Length in Hepatocellular Carcinoma. Medicine 2017, 96, e5766. [Google Scholar] [CrossRef]
- Huang, A.; Zhang, X.; Zhou, S.-L.; Cao, Y.; Huang, X.-W.; Fan, J.; Yang, X.-R.; Zhou, J. Detecting Circulating Tumor DNA in Hepatocellular Carcinoma Patients Using Droplet Digital PCR Is Feasible and Reflects Intratumoral Heterogeneity. J. Cancer 2016, 7, 1907–1914. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Hu, G.; Zhang, Y.; Ouyang, K.; Xie, F.; Fang, H.; Yang, X.; Liu, K.; Wang, Z.; Tang, X.; Liu, J.; et al. In vivo Acquired Sorafenib-Resistant Patient-Derived Tumor Model Displays Alternative Angiogenic Pathways, Multi-Drug Resistance and Chromosome Instability. Oncol. Lett. 2018, 16, 3439–3446. [Google Scholar] [CrossRef] [Green Version]
- Firtina Karagonlar, Z.; Koc, D.; Iscan, E.; Erdal, E.; Atabey, N. Elevated Hepatocyte Growth Factor Expression as an Autocrine C-Met Activation Mechanism in Acquired Resistance to Sorafenib in Hepatocellular Carcinoma Cells. Cancer Sci. 2016, 107, 407–416. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalasekar, S.M.; VanSant-Webb, C.H.; Evason, K.J. Intratumor Heterogeneity in Hepatocellular Carcinoma: Challenges and Opportunities. Cancers 2021, 13, 5524. https://doi.org/10.3390/cancers13215524
Kalasekar SM, VanSant-Webb CH, Evason KJ. Intratumor Heterogeneity in Hepatocellular Carcinoma: Challenges and Opportunities. Cancers. 2021; 13(21):5524. https://doi.org/10.3390/cancers13215524
Chicago/Turabian StyleKalasekar, Sharanya Maanasi, Chad H. VanSant-Webb, and Kimberley J. Evason. 2021. "Intratumor Heterogeneity in Hepatocellular Carcinoma: Challenges and Opportunities" Cancers 13, no. 21: 5524. https://doi.org/10.3390/cancers13215524
APA StyleKalasekar, S. M., VanSant-Webb, C. H., & Evason, K. J. (2021). Intratumor Heterogeneity in Hepatocellular Carcinoma: Challenges and Opportunities. Cancers, 13(21), 5524. https://doi.org/10.3390/cancers13215524