
Liquid Biopsy to Detect Minimal Residual Disease: Methodology and Impact

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
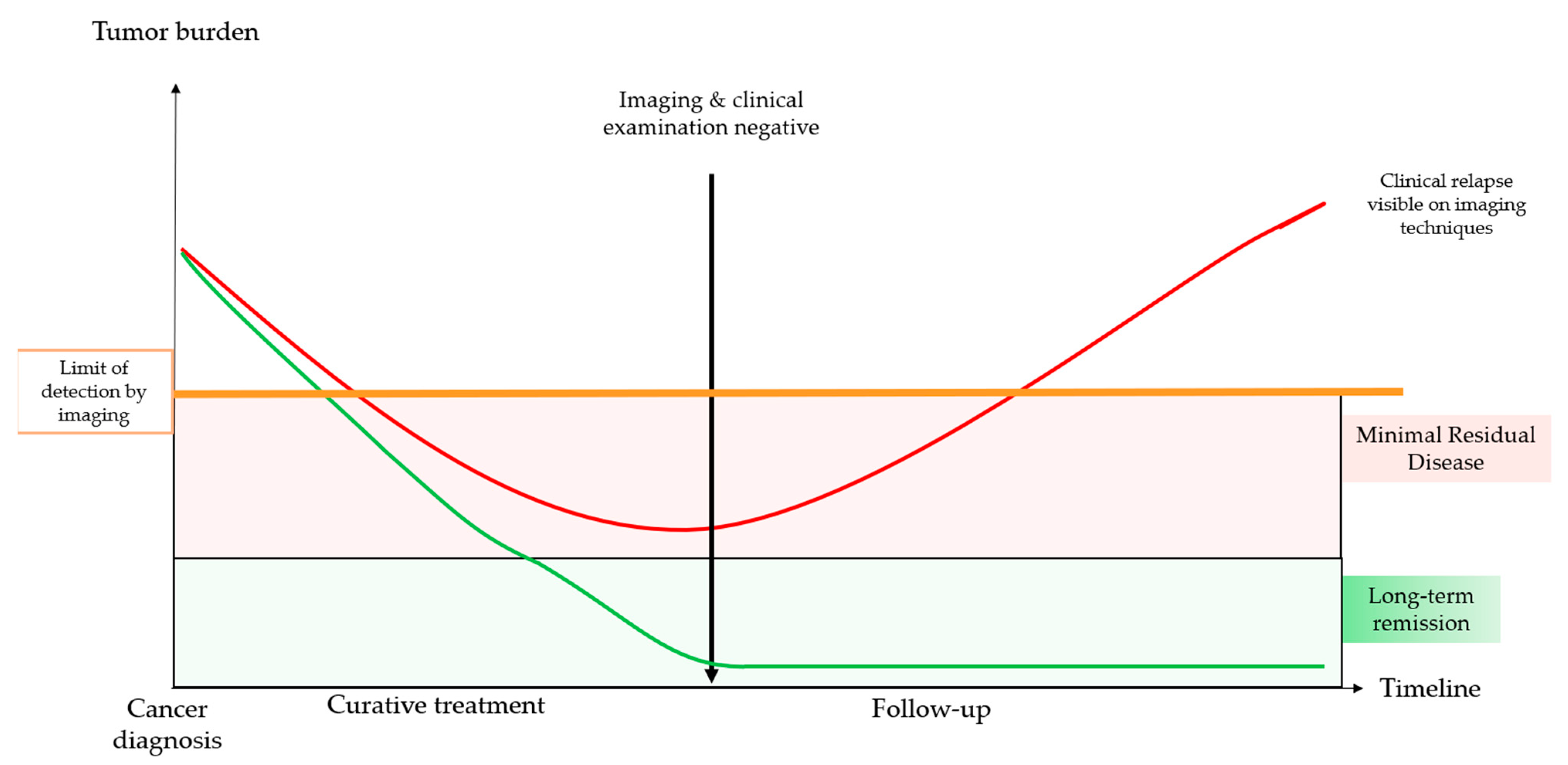
1.1. Minimal Residual Disease
1.2. Liquid Biopsy
1.2.1. Circulating Nucleic Acids
- ctDNA
- Mitochondrial ctDNA
- Methylation patterns
- Non-coding RNA
- Exosomes
1.2.2. Circulating Tumor Cells
1.2.3. Circulating Proteins
2. Methodology to Detect Minimal Residual Disease with ctDNA
2.1. Personalized Methods
2.1.1. Tumor-Customized Based Panels
2.1.2. Custom-Based PCR Assay
2.2. Non-Personalized Methods
2.3. Other Methods
3. Clinical Impact of Detecting MRD with ctDNA and Perspectives
3.1. Issues
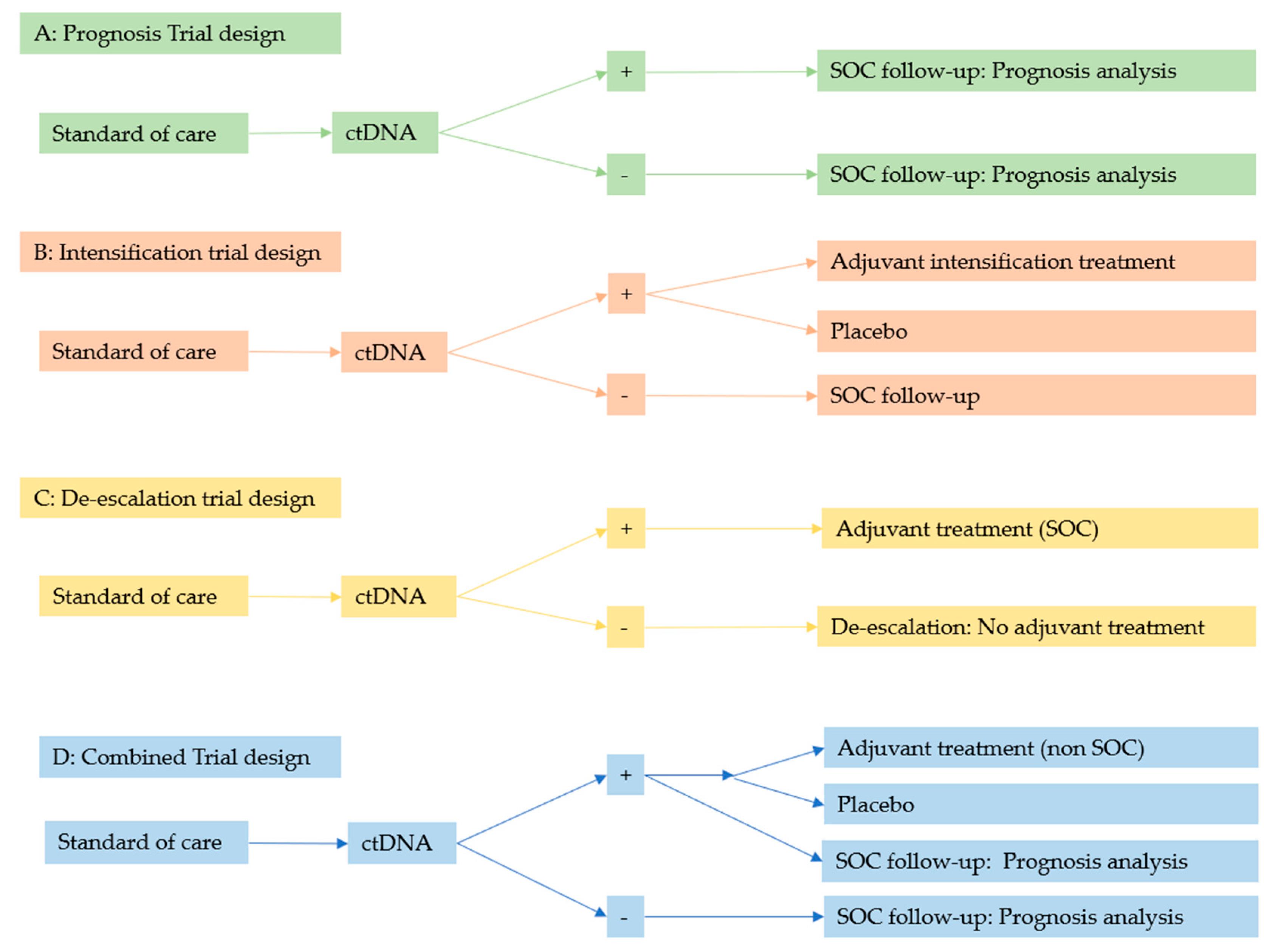
3.2. Current Prospective Trials
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- NIH. Minimal Residual Disease Definition. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/minimal-residual-disease (accessed on 20 March 2021).
- Szczepariski, T.; Orfão, A.; van der Valden, V.H.; Miguel, J.F.S.; van Dongen, J.J. Minimal residual disease in leukaemia patients. Lancet Oncol. 2001, 2, 409–417. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.C.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.B.; Siu, L.L.; et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 2019, 30, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, M.; Gibbs, P.; Tie, J. ctDNA and Adjuvant Therapy for Colorectal Cancer: Time to Re-Invent Our Treatment Paradigm. Cancers 2021, 13, 346. [Google Scholar] [CrossRef] [PubMed]
- Coakley, M.; Garcia-Murillas, I.; Turner, N.C. Molecular Residual Disease and Adjuvant Trial Design in Solid Tumors. Clin. Cancer Res. 2019, 25, 6026–6034. [Google Scholar] [CrossRef]
- Wills, B.; Gorse, E.; Lee, V. Role of liquid biopsies in colorectal cancer. Curr. Probl. Cancer 2018, 42, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.-I.; Chen, K.; Usmani, A.; Chua, C.; Harris, P.K.; Binkley, M.S.; Azad, T.D.; Dudley, J.C.; Chaudhuri, A.A. Detection of Solid Tumor Molecular Residual Disease (MRD) Using Circulating Tumor DNA (ctDNA). Mol. Diagn. Ther. 2019, 23, 311–331. [Google Scholar] [CrossRef]
- NIH. Liquid Biopsy Definition. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/liquid-biopsy (accessed on 21 March 2021).
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.; Lindeman, N.; Lockwood, C.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Nuclear acids in human blood plasma. Comptes Rendus Séances Société Biologie Ses Filiales 1948, 142, 241–243. [Google Scholar]
- Yan, Y.-Y.; Guo, Q.-R.; Wang, F.-H.; Adhikari, R.; Zhu, Z.-Y.; Zhang, H.-Y.; Zhou, W.-M.; Yu, H.; Li, J.-Q.; Zhang, J.-Y. Cell-Free DNA: Hope and Potential Application in Cancer. Front. Cell Dev. Biol. 2021, 9, 192. [Google Scholar] [CrossRef] [PubMed]
- Lui, Y.Y.; Chik, K.-W.; Chiu, R.W.; Ho, C.-Y.; Lam, C.W.; Lo, Y.D. Predominant Hematopoietic Origin of Cell-free DNA in Plasma and Serum after Sex-mismatched Bone Marrow Transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouliere, F.; Rosenfeld, N. Circulating tumor-derived DNA is shorter than somatic DNA in plasma. Proc. Natl. Acad. Sci. USA 2015, 112, 3178–3179. [Google Scholar] [CrossRef] [Green Version]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Stewart, C.M.; Kothari, P.; Mouliere, F.; Mair, R.; Somnay, S.; Benayed, R.; Zehir, A.; Weigelt, B.; Dawson, S.-J.; Arcila, M.E.; et al. The value of cell-free DNA for molecular pathology. J. Pathol. 2018, 244, 616–627. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinger, J.; Müller, D.C.; Müller, S.C.; Hauser, S.; Heukamp, L.; von Ruecker, A.; Bastian, P.J.; Walgenbach-Brunagel, G. Circulating mitochondrial DNA in serum: A universal diagnostic biomarker for patients with urological malignancies. Urol. Oncol. Semin. Orig. Investig. 2012, 30, 509–515. [Google Scholar] [CrossRef]
- Pagani, I.S.; Kok, C.H.; Saunders, V.A.; Van Der Hoek, M.B.; Heatley, S.L.; Schwarer, A.P.; Hahn, C.N.; Hughes, T.P.; White, D.L.; Ross, D.M. A Method for Next-Generation Sequencing of Paired Diagnostic and Remission Samples to Detect Mitochondrial DNA Mutations Associated with Leukemia. J. Mol. Diagn. 2017, 19, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Phillips, T. The role of methylation in gene expression. Nat. Educ. 2008, 1, 116. [Google Scholar]
- Ma, Y.; Chen, Y.; Petersen, I. Expression and promoter DNA methylation of MLH1 in colorectal cancer and lung cancer. Pathol. Res. Pr. 2017, 213, 333–338. [Google Scholar] [CrossRef]
- Jin, N.; George, T.L.; Otterson, G.A.; Verschraegen, C.; Wen, H.; Carbone, D.; Herman, J.; Bertino, E.M.; He, K. Advances in epigenetic therapeutics with focus on solid tumors. Clin. Epigenetics 2021, 13, 1–27. [Google Scholar] [CrossRef]
- Taieb, J.; Taly, V.; Vernerey, D.; Bourreau, C.; Bennouna, J.; Faroux, R.; Desrame, J.; Bouche, O.; Borg, C.; Egreteau, J.; et al. Analysis of circulating tumour DNA (ctDNA) from patients enrolled in the IDEA-FRANCE phase III trial: Prognostic and predictive value for adjuvant treatment duration. Ann. Oncol. 2019, 30, v867. [Google Scholar] [CrossRef]
- Takahashi, H.; Kagara, N.; Tanei, T.; Naoi, Y.; Shimoda, M.; Shimomura, A.; Shimazu, K.; Kim, S.J.; Noguchi, S. Correlation of methylated circulating tumor DNA with response to neoadjuvant chemotherapy in breast cancer patients. Clin. Breast Cancer 2017, 17, 61–69. [Google Scholar] [CrossRef]
- Barault, L.; Amatu, A.; Siravegna, G.; Ponzetti, A.; Moran, S.; Cassingena, A.; Mussolin, B.; Falcomatà, C.; Binder, A.M.; Cristiano, C.; et al. Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer. Gut 2017, 67, 1995–2005. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Koukaki, T.; Karamitrousis, E.; Nena, E.; Tsamardinos, I.; Kolios, G.; Lianidou, E.; et al. Circulating cell-free DNA in breast cancer: Size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 2019, 38, 3387–3401. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wei, W.; Ye, Z.; Zheng, J.; Xu, R.-H. Liquid Biopsy of Methylation Biomarkers in Cell-Free DNA. Trends Mol. Med. 2021, 27, 482–500. [Google Scholar] [CrossRef]
- Bhatti, G.K.; Khullar, N.; Sidhu, I.S.; Navik, U.S.; Reddy, A.P.; Reddy, P.H.; Bhatti, J.S. Emerging role of non-coding RNA in health and disease. Metab. Brain Dis. 2021, 36, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Rahat, B.; Ali, T.; Sapehia, D.; Mahajan, A.; Kaur, J. Circulating Cell-Free Nucleic Acids as Epigenetic Biomarkers in Precision Medicine. Front. Genet. 2020, 11, 844. [Google Scholar] [CrossRef]
- Rodriguez-Casanova, A.; Costa-Fraga, N.; Bao-Caamano, A.; López-López, R.; Muinelo-Romay, L.; Diaz-Lagares, A. Epigenetic Landscape of Liquid Biopsy in Colorectal Cancer. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Lafin, J.T.; Murray, M.J.; Coleman, N.; Frazier, A.L.; Amatruda, J.F.; Bagrodia, A. The Road Ahead for Circulating microRNAs in Diagnosis and Management of Testicular Germ Cell Tumors. Mol. Diagn. Ther. 2021, 25, 269–271. [Google Scholar] [CrossRef]
- Vacante, M.; Ciuni, R.; Basile, F.; Biondi, A. The Liquid Biopsy in the Management of Colorectal Cancer: An Overview. Biomedicines 2020, 8, 308. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Müller, V.; Milde-Langosch, K.; Trillsch, F.; Pantel, K.; Schwarzenbach, H. Diagnostic and prognostic relevance of circulating exosomal miR-373, miR-200a, miR-200b and miR-200c in patients with epithelial ovarian cancer. Oncotarget 2016, 7, 16923–16935. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Gao, W.; Chen, X.; Zhang, Y.; Wu, M.; Wang, S. A Pilot Study of Circulating MicroRNA-125b as a Diagnostic and Prognostic Biomarker for Epithelial Ovarian Cancer. Int. J. Gynecol. Cancer 2016, 27, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Necula, L.; Matei, L.; Dragu, D.; Neagu, A.I.; Mambet, C.; Nedeianu, S.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Recent advances in gastric cancer early diagnosis. World J. Gastroenterol. 2019, 25, 2029–2044. [Google Scholar] [CrossRef]
- Al-Khanbashi, M.; Caramuta, S.; Alajmi, A.M.; Al-Haddabi, I.; Al-Riyami, M.; Lui, W.-O.; Al-Moundhri, M.S. Tissue and Serum miRNA Profile in Locally Advanced Breast Cancer (LABC) in Response to Neo-Adjuvant Chemotherapy (NAC) Treatment. PLoS ONE 2016, 11, e0152032. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wang, Z.-F.; Lou, Q.-Y.; Rankine, A.N.; Zheng, W.-X.; Zhang, Z.-H.; Zhang, L.; Gu, H. Long non-coding RNAs in head and neck squamous cell carcinoma: Diagnostic biomarkers, targeted therapies, and prognostic roles. Eur. J. Pharmacol. 2021, 902, 174114. [Google Scholar] [CrossRef]
- Seborova, K.; Vaclavikova, R.; Rob, L.; Soucek, P.; Vodicka, P. Non-Coding RNAs as Biomarkers of Tumor Progression and Metastatic Spread in Epithelial Ovarian Cancer. Cancers 2021, 13, 1839. [Google Scholar] [CrossRef]
- Ramli, S.; Sim, M.S.; Guad, R.M.; Gopinath, S.C.B.; Subramaniyan, V.; Fuloria, S.; Fuloria, N.K.; Choy, K.W.; Rana, S.; Wu, Y.S. Long Noncoding RNA UCA1 in Gastrointestinal Cancers: Molecular Regulatory Roles and Patterns, Mechanisms, and Interactions. J. Oncol. 2021, 2021, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mortoglou, M.; Tabin, Z.K.; Arisan, E.D.; Kocher, H.M.; Uysal-Onganer, P. Non-coding RNAs in pancreatic ductal adenocarcinoma: New approaches for better diagnosis and therapy. Transl. Oncol. 2021, 14, 101090. [Google Scholar] [CrossRef]
- Wang, M.-Q.; Zhu, W.-J.; Gao, P. New insights into long non-coding RNAs in breast cancer: Biological functions and therapeutic prospects. Exp. Mol. Pathol. 2021, 120, 104640. [Google Scholar] [CrossRef]
- Tan, Q.; Zuo, J.; Qiu, S.; Yu, Y.; Zhou, H.; Li, N.; Wang, H.; Liang, C.; Yu, M.; Tu, J. Identification of circulating long non-coding RNA GAS5 as a potential biomarker for non-small cell lung cancer diagnosisnon-small cell lung cancer, long non-coding RNA, plasma, GAS5, biomarker. Int. J. Oncol. 2017, 50, 1729–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Xiang, Y.; Guo, X.; Zhang, Y.; Li, C.; Xie, W.; Wu, N.; Wu, L.; Cai, T.; Ma, X.; et al. Circulating Long Noncoding RNAs Act as Diagnostic Biomarkers in Non-Small Cell Lung Cancer. Front. Oncol. 2020, 10, 537120. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Guo, S.; Zhao, Y.; Wang, Y.; Piao, H.-Y.; Wu, Y.; Zhang, J. Circulating long non-coding RNA PCGEM1 as a novel biomarker for gastric cancer diagnosis. Pathol. Res. Pr. 2019, 215, 152569. [Google Scholar] [CrossRef]
- Arita, T.; Ichikawa, D.; Konishi, H.; Komatsu, S.; Shiozaki, A.; Shoda, K.; Kawaguchi, T.; Hirajima, S.; Nagata, H.; Kubota, T.; et al. Circulating long non-coding RNAs in plasma of patients with gastric cancer. Anticancer. Res. 2013, 33, 3185–3193. [Google Scholar]
- McAndrews, K.M.; Kalluri, R. Mechanisms associated with biogenesis of exosomes in cancer. Mol. Cancer 2019, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [Green Version]
- Bidarimath, M.; Lingegowda, H.; Miller, J.E.; Koti, M.; Tayade, C. Insights into Extracellular Vesicle/Exosome and miRNA Mediated Bi-Directional Communication During Porcine Pregnancy. Front. Vet. Sci. 2021, 8, 318. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Y.; Yin, G.; Xie, Q. Therapeutic prospects of MicroRNAs carried by mesenchymal stem cells-derived extracellular vesicles in autoimmune diseases. Life Sci. 2021, 277, 119458. [Google Scholar] [CrossRef]
- Benecke, L.; Coray, M.; Umbricht, S.; Chiang, D.; Figueiró, F.; Muller, L. Exosomes: Small EVs with Large Immunomodulatory Effect in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 3600. [Google Scholar] [CrossRef]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.; Enderle, D.; Noerholm, M.; Breakefield, X.; Skog, J. Exosome-based liquid biopsies in cancer: Opportunities and challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Chen, S.; He, H.; Wen, W.; Wang, H. The role and potential application of extracellular vesicles in liver cancer. Sci. China Life Sci. 2021, 64, 1281–1294. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Whiteside, T.L. Plasma-derived exosomes in acute myeloid leukemia for detection of minimal residual disease: Are we ready? Expert Rev. Mol. Diagn. 2016, 16, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Fu, M.; Liu, J.; Chong, W.; Fang, Z.; Du, F.; Liu, Y.; Shang, L.; Li, L. The role and application of small extracellular vesicles in gastric cancer. Mol. Cancer 2021, 20, 1–19. [Google Scholar] [CrossRef]
- Galvis, M.M.; Romero, C.S.; Bueno, T.O.; Teng, Y. Toward a New Era for the Management of Circulating Tumor Cells. In Reviews on New Drug Targets in Age-Related Disorders: Part II.; Guest, P.C., Ed.; Springer: Berlin/Heidelberg, Germany, 2021; pp. 125–134. [Google Scholar]
- Riethdorf, S.; Soave, A.; Rink, M. The current status and clinical value of circulating tumor cells and circulating cell-free tumor DNA in bladder cancer. Transl. Androl. Urol. 2017, 6, 1090–1110. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Orozco, J.I.J.; Hoon, D.S.B. Detection of Minimal Residual Disease and Its Clinical Applications in Melanoma and Breast Cancer Patients. In Biological Mechanisms of Minimal Residual Disease and Systemic Cancer; Aguirre-Ghiso, J.A., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 83–95. [Google Scholar]
- Tellez-Gabriel, M.; Knutsen, E.; Perander, M. Current Status of Circulating Tumor Cells, Circulating Tumor DNA, and Exosomes in Breast Cancer Liquid Biopsies. Int. J. Mol. Sci. 2020, 21, 9457. [Google Scholar] [CrossRef]
- Joosse, S.A.; Gorges, T.M.; Pantel, K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol. Med. 2014, 7, 1–11. [Google Scholar] [CrossRef]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.J.; Uhr, J.W.; Terstappen, L. Tumor Cells Circulate in the Peripheral Blood of All Major Carcinomas but not in Healthy Subjects or Patients with Nonmalignant Diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef] [Green Version]
- Andree, K.C.; van Dalum, G.; Terstappen, L.W. Challenges in circulating tumor cell detection by the CellSearch system. Mol. Oncol. 2016, 10, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Rack, B.; Schindlbeck, C.; Jückstock, J.; Andergassen, U.; Hepp, P.; Zwingers, T.; Friedl, T.W.P.; Lorenz, R.; Tesch, H.; Fasching, P.A.; et al. Circulating Tumor Cells Predict Survival in Early Average-to-High Risk Breast Cancer Patients. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Radovich, M.; Jiang, G.; Hancock, B.A.; Chitambar, C.; Nanda, R.; Falkson, C.; Lynce, F.C.; Gallagher, C.; Isaacs, C.; Blaya, M. Association of circulating tumor DNA and circulating tumor cells after neoadjuvant chemotherapy with disease recurrence in patients with triple-negative breast cancer: Preplanned secondary analysis of the BRE12-158 randomized clinical trial. JAMA Oncol. 2020, 6, 1410–1415. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.M.-Y.; Vardhanabhuti, V.; Ng, W.-T.; Lam, K.-O.; Ngan, R.K.-C.; Kwong, D.L.-W.; Lee, V.H.-F.; Lui, Y.-H.; Yau, C.-C.; Kwan, C.-K.; et al. Clinical utility of serial analysis of circulating tumour cells for detection of minimal residual disease of metastatic nasopharyngeal carcinoma. Br. J. Cancer 2020, 123, 114–125. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Lee, C.-L.; Wu, C.-F.; Fu, J.-Y.; Yang, C.-T.; Wen, C.-T.; Liu, Y.-H.; Liu, H.-P.; Hsieh, J.C.-H. Circulating Tumor Cells as a Tool of Minimal Residual Disease Can Predict Lung Cancer Recurrence: A longitudinal, Prospective Trial. Diagnostics 2020, 10, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoja, L.; Lorigan, P.; Zhou, C.; Lancashire, M.; Booth, J.; Cummings, J.; Califano, R.; Clack, G.; Hughes, A.; Dive, C. Biomarker Utility of Circulating Tumor Cells in Metastatic Cutaneous Melanoma. J. Investig. Dermatol. 2013, 133, 1582–1590. [Google Scholar] [CrossRef] [Green Version]
- Koyanagi, K.; O’Day, S.J.; Boasberg, P.; Atkins, M.B.; Wang, H.-J.; Gonzalez, R.; Lewis, K.; Thompson, J.A.; Anderson, C.M.; Lutzky, J.; et al. Serial Monitoring of Circulating Tumor Cells Predicts Outcome of Induction Biochemotherapy plus Maintenance Biotherapy for Metastatic Melanoma. Clin. Cancer Res. 2010, 16, 2402–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatiadis, M.; Xenidis, N.; Perraki, M.; Apostolaki, S.; Politaki, E.; Kafousi, M.; Stathopoulos, E.N.; Stathopoulou, A.; Lianidou, E.; Chlouverakis, G.; et al. Different Prognostic Value of Cytokeratin-19 mRNA–Positive Circulating Tumor Cells According to Estrogen Receptor and HER2 Status in Early-Stage Breast Cancer. J. Clin. Oncol. 2007, 25, 5194–5202. [Google Scholar] [CrossRef] [Green Version]
- Saloustros, E.; Perraki, M.; Apostolaki, S.; Kallergi, G.; Xyrafas, A.; Kalbakis, K.; Agelaki, S.; Kalykaki, A.; Georgoulias, V.; Mavroudis, D. Cytokeratin-19 mRNA-positive circulating tumor cells during follow-up of patients with operable breast cancer: Prognostic relevance for late relapse. Breast Cancer Res. 2011, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bidard, F.C.; Jacot, W.; Kiavue, N.; Dureau, S.; Kadi, A.; Brain, E.; Bachelot, T.; Bourgeois, H.; Gonçalves, A.; Pierga, J.Y.; et al. Efficacy of circulating tumor cell count–driven vs. clinician-driven first-line therapy choice in hormone receptor–positive, ERBB2-negative metastatic breast cancer: The STIC CTC randomized clinical trial. JAMA Oncol. 2021, 7, 34–41. [Google Scholar] [CrossRef]
- Society ELB. European Liquid Biopsy Society (ELBS). Available online: https://www.uke.de/english/departments-institutes/institutes/tumor-biology/european-liquid-biopsy-society-elbs/project/index.html (accessed on 18 May 2021).
- Henry, N.L.; Hayes, D.F. Cancer biomarkers. Mol. Oncol. 2012, 6, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, J.; Chahwan, C.; Said, K.A.; Vaudreuil, L.; Seddik, S.; Tillou, X. Evaluation of the residual prostate cancer rate on cystoprostatectomy specimen in patients treated with radiotherapy for prostate cancer. Int. Urol. Nephrol. 2019, 52, 279–285. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, I.H.; Kim, Y.-J.; Chung, Y.S.; Lee, J.-Y.; Nam, E.J.; Kim, S.; Kim, S.W.; Kim, Y.T. Evaluation of various kinetic parameters of CA-125 in patients with advanced-stage ovarian cancer undergoing neoadjuvant chemotherapy. PLoS ONE 2018, 13, e0203366. [Google Scholar] [CrossRef]
- Yoneoka, Y.; Ishikawa, M.; Uehara, T.; Shimizu, H.; Uno, M.; Murakami, T.; Kato, T. Treatment strategies for patients with advanced ovarian cancer undergoing neoadjuvant chemotherapy: Interval debulking surgery or additional chemotherapy? J. Gynecol. Oncol. 2019, 30, e81. [Google Scholar] [CrossRef]
- Salminen, L.; Nadeem, N.; Jain, S.; Grènman, S.; Carpén, O.; Hietanen, S.; Oksa, S.; Lamminmäki, U.; Pettersson, K.; Gidwani, K.; et al. A longitudinal analysis of CA125 glycoforms in the monitoring and follow up of high grade serous ovarian cancer. Gynecol. Oncol. 2019, 156, 689–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, D.K.; Panda, N.; Biswas, S.; Saha, M.L.; Majumder, A. Is immediate postoperative CA15. 3 assay a predictive marker of early postoperative recurrence of carcinoma breast? J. Indian Med Assoc. 2012, 110, 146–147. [Google Scholar]
- Valencia, C.A.; Pervaiz, M.A.; Husami, A.; Qian, Y.; Zhang, K. Sanger Sequencing Principles, History, and Landmarks. In Next Generation Sequencing Technologies in Medical Genetics; Springer: New York, NY, USA, 2013; pp. 3–11. [Google Scholar]
- Vestergaard, L.; Oliveira, D.; Høgdall, C.; Høgdall, E. Next Generation Sequencing Technology in the Clinic and Its Challenges. Cancers 2021, 13, 1751. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Petrackova, A.; Vasinek, M.; Sedlarikova, L.; Dyskova, T.; Schneiderova, P.; Novosad, T.; Papajik, T.; Kriegova, E. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front. Oncol. 2019, 9, 851. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.S.G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; Garrido, J.A.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Hilke, F.J.; Muyas, F.; Admard, J.; Kootz, B.; Nann, D.; Welz, S.; Rieß, O.; Zips, D.; Ossowski, S.; Schroeder, C.; et al. Dynamics of cell-free tumour DNA correlate with treatment response of head and neck cancer patients receiving radiochemotherapy. Radiother. Oncol. 2020, 151, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Magbanua, M.; Swigart, L.; Wu, H.-T.; Hirst, G.; Yau, C.; Wolf, D.; Tin, A.; Salari, R.; Shchegrova, S.; Pawar, H.; et al. Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response and survival. Ann. Oncol. 2020, 32, 229–239. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.R.; Contente-Cuomo, T.; Sammut, S.J.; Odenheimer-Bergman, A.; Ernst, B.; Perdigones, N.; Murtaza, M. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci. Transl. Med. 2019, 11, 504. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef]
- Christensen, E.; Birkenkamp-Demtröder, K.; Sethi, H.; Shchegrova, S.; Salari, R.; Nordentoft, I.K.; Wu, H.-T.; Knudsen, M.; Lamy, P.; Lindskrog, S.V.; et al. Early Detection of Metastatic Relapse and Monitoring of Therapeutic Efficacy by Ultra-Deep Sequencing of Plasma Cell-Free DNA in Patients with Urothelial Bladder Carcinoma. J. Clin. Oncol. 2019, 37, 1547–1557. [Google Scholar] [CrossRef]
- Tarazona, N.; Gimeno-Valiente, F.; Gambardella, V.; Zuniga, S.; Rentero-Garrido, P.; Huerta, M.; Roselló, S.; Martinez-Ciarpaglini, C.; Carbonell-Asins, J.; Carrasco, F.; et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann. Oncol. 2019, 30, 1804–1812. [Google Scholar] [CrossRef] [Green Version]
- McDuff, S.G.R.; Hardiman, K.M.; Ulintz, P.J.; Parikh, A.R.; Zheng, H.; Kim, D.W.; Lennerz, J.K.; Hazar-Rethinam, M.; Van Seventer, E.E.; Fetter, I.J.; et al. Circulating Tumor DNA Predicts Pathologic and Clinical Outcomes Following Neoadjuvant Chemoradiation and Surgery for Patients with Locally Advanced Rectal Cancer. JCO Precis. Oncol. 2021, 5, 123–132. [Google Scholar] [CrossRef]
- Shirasu, H.; Taniguchi, H.; Watanabe, J.; Kotaka, M.; Yamazaki, K.; Hirata, K.; Yoshino, T. O-11 monitoring molecular residual disease by circulating tumor DNA in resectable colorectal cancer: Molecular subgroup analyses of a prospective observational study GALAXY in CIRCULATE-Japan. Ann. Oncol. 2021, 32, S222–S223. [Google Scholar] [CrossRef]
- Abbosh, C.; Frankell, A.; Garnett, A.; Harrison, T.; Weichert, M.; Licon, A.; Veeriah, S.; Daber, B.; Moreau, M.; Chesh, A.; et al. Abstract CT023: Phylogenetic tracking and minimal residual disease detection using ctDNA in early-stage NSCLC: A lung TRACERx study. Cancer Res. 2020, 80, CT023. [Google Scholar] [CrossRef]
- Valpione, S.; Campana, L. Detection of circulating tumor DNA (ctDNA) by digital droplet polymerase chain reaction (dd-PCR) in liquid biopsies. Methods Enzymol. 2019, 629, 1–15. [Google Scholar] [CrossRef]
- Guerrini, F.; Paolicchi, M.; Ghio, F.; Ciabatti, E.; Grassi, S.; Salehzadeh, S.; Ercolano, G.; Metelli, M.R.; Del Re, M.; Iovino, L.; et al. The Droplet Digital PCR: A New Valid Molecular Approach for the Assessment of B-RAF V600E Mutation in Hairy Cell Leukemia. Front. Pharmacol. 2016, 7, 363. [Google Scholar] [CrossRef] [Green Version]
- Huerta, M.; Roselló, S.; Sabater, L.; Ferrer, A.; Tarazona, N.; Roda, D.; Gambardella, V.; Alfaro-Cervelló, C.; Garcés-Albir, M.; Cervantes, A.; et al. Circulating Tumor DNA Detection by Digital-Droplet PCR in Pancreatic Ductal Adenocarcinoma: A Systematic Review. Cancers 2021, 13, 994. [Google Scholar] [CrossRef] [PubMed]
- Consul, N.; Menias, C.O.; Lubner, M.G.; Katabathina, V.S.; Chahinian, R.A.; Mansour, J.; Elsayes, K.M. A Review of Viral-Related Malignancies and the Associated Imaging Findings. Am. J. Roentgenol. 2020, 214, W1–W10. [Google Scholar] [CrossRef]
- Mohamad, S.; Fauzi, F.H.; Hamzan, N.I.; Ab Rahman, N.; Suraiya, S. Detection of human papillomavirus in oropharyngeal squamous cell carcinoma. J. Zhejiang Univ. Sci. B 2020, 21, 961–976. [Google Scholar] [CrossRef]
- Goh, S.M.P.; Swaminathan, M.; Lai, J.U.-M.; Anwar, A.; Chan, S.H.; Cheong, I. Increasing the accuracy and scalability of the Immunofluorescence Assay for Epstein Barr Virus by inferring continuous titers from a single sample dilution. J. Immunol. Methods 2017, 440, 35–40. [Google Scholar] [CrossRef]
- Arbuthnot, P.; Kew, M. Hepatitis B virus and hepatocellular carcinoma. Int. J. Exp. Pathol. 2001, 82, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.-P.; Luo, Y.-S.; Chen, K.; Li, J.-Y.; Huo, L.-Q.; Shi, L.; Ou-Yang, Y.; Cao, X.-P. Circulating Epstein–Barr virus DNA level post induction chemotherapy contributes to prognostication in advanced-stage nasopharyngeal carcinoma. Eur. J. Cancer 2021, 151, 63–71. [Google Scholar] [CrossRef]
- Veyer, D.; Wack, M.; Mandavit, M.; Garrigou, S.; Hans, S.; Bonfils, P.; Tartour, E.; Bélec, L.; Wang-Renault, S.; Laurent-Puig, P.; et al. HPV circulating tumoral DNA quantification by droplet-based digital PCR: A promising predictive and prognostic biomarker for HPV-associated oropharyngeal cancers. Int. J. Cancer 2019, 147, 1222–1227. [Google Scholar] [CrossRef]
- Chera, B.S.; Kumar, S.; Shen, C.; Amdur, R.; Dagan, R.; Green, R.; Goldman, E.; Weiss, J.; Grilley-Olson, J.; Patel, S.; et al. Plasma Circulating Tumor HPV DNA for the Surveillance of Cancer Recurrence in HPV-Associated Oropharyngeal Cancer. J. Clin. Oncol. 2020, 38, 1050–1058. [Google Scholar] [CrossRef]
- Damerla, R.R.; Lee, N.Y.; You, D.; Soni, R.; Shah, R.; Reyngold, M.; Katabi, N.; Wu, V.; McBride, S.M.; Tsai, C.J.; et al. Detection of Early Human Papillomavirus–Associated Cancers by Liquid Biopsy. JCO Precis. Oncol. 2019, 3, 1–17. [Google Scholar] [CrossRef]
- Rungkamoltip, P.; Temisak, S.; Piboonprai, K.; Japrung, D.; Thangsunan, P.; Chanpanitkitchot, S.; Chaowawanit, W.; Chandeying, N.; Tangjitgamol, S.; Iempridee, T. Rapid and ultrasensitive detection of circulating human papillomavirus E7 cell-free DNA as a cervical cancer biomarker. Exp. Biol. Med. 2020, 246, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.; Li, W.; Ma, B.; Lam, W.; Chan, K.; Mo, F.; Ai, Q.; King, A.; Wong, C.; Guo, R.; et al. Integrating postradiotherapy plasma Epstein–Barr virus DNA and TNM stage for risk stratification of nasopharyngeal carcinoma to adjuvant therapy. Ann. Oncol. 2020, 31, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Cutts, R.J.; White, I.; Augustin, Y.; Garcia-Murillas, I.; Fenwick, K.; Bhide, S. Next generation sequencing assay for detection of circulating HPV DNA (cHPV-DNA) in patients undergoing radical (chemo) radiotherapy in anal squamous cell carcinoma (ASCC). Front. Oncol. 2020, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Lopez, E.M.; Tanner, A.M.; Du, E.; Patel, S.N.; Weiss, J.; Weissler, M.C.; Hackman, T.; Gupta, G.P.; Zevallos, J.; Elmore, S.; et al. Decline in circulating viral and human tumor markers after resection of head and neck carcinoma. Head Neck 2020, 43, 27–34. [Google Scholar] [CrossRef]
- Voelkerding, K.V.; Coonrod, E.M.; Durtschi, J.D.; Margraf, R.L. Next-Generation Sequencing: Principles for Clinical Application. In Molecular Pathology in Clinical Practice; Leonard, D.G.B., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 889–909. [Google Scholar]
- Bewicke-Copley, F.; Kumar, E.A.; Palladino, G.; Korfi, K.; Wang, J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 2019, 17, 1348–1359. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, S.C.; Kim, Y.J. A Universal Analysis Pipeline for Hybrid Capture-Based Targeted Sequencing Data with Unique Molecular Indexes. Genom. Inform. 2018, 16, e29. [Google Scholar] [CrossRef]
- Galot, R.; van Marcke, C.; Helaers, R.; Mendola, A.; Goebbels, R.O.; Caignet, X.; Ambroise, J.; Wittouck, K.; Vikkula, M.; Limaye, N.; et al. Liquid biopsy for mutational profiling of locoregional recurrent and/or metastatic head and neck squamous cell carcinoma. Oral Oncol. 2020, 104, 104631. [Google Scholar] [CrossRef]
- Jiang, J.; Ye, S.; Xu, Y.; Chang, L.; Hu, X.; Ru, G.; Guo, Y.; Yi, X.; Yang, L.; Huang, D. Circulating Tumor DNA as a Potential Marker to Detect Minimal Residual Disease and Predict Recurrence in Pancreatic Cancer. Front. Oncol. 2020, 10, 1220. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Diehn, M. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Newman, A.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.; Stehr, H.; Azad, T.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Azad, T.D.; Chaudhuri, A.A.; Fang, P.; Qiao, Y.; Esfahani, M.S.; Chabon, J.J.; Hamilton, E.G.; Yang, Y.D.; Lovejoy, A.; Newman, A.M.; et al. Circulating tumor DNA analysis for detection of minimal residual disease after chemoradiotherapy for localized esophageal cancer. Gastroenterology 2020, 158, 494–505. [Google Scholar] [CrossRef]
- Jia, R.; Zhao, C.-H.; Li, P.-S.; Liu, R.-R.; Zhang, Y.; Chen, H.-E.; Chang, L.-P.; Gong, Y.-H.; Guan, Y.-F.; Yi, X.; et al. Post-radiation circulating tumor DNA as a prognostic factor in locally advanced esophageal squamous cell carcinoma. Oncol. Lett. 2020, 21, 1. [Google Scholar] [CrossRef]
- Parikh, A.R.; van Seventer, E.E.; Siravegna, G.; Hartwig, A.V.; Jaimovich, A.; He, Y.; Kanter, K.; Fish, M.G.; Fosbenner, K.D.; Miao, B.; et al. Minimal Residual Disease Detection using a Plasma-Only Circulating Tumor DNA Assay in Colorectal Cancer Patients. Clin. Cancer Res. 2021, 27, 5586–5594. [Google Scholar] [CrossRef]
- Circulating Tumor DNA Panel Testing for Cancer (Liquid Biopsy). Available online: https://www.anthem.com/dam/medpolicies/abc/active/policies/mp_pw_d082650.html (accessed on 18 May 2021).
- Peng, M.; Huang, Q.; Yin, W.; Tan, S.; Chen, C.; Liu, W.; Tang, J.; Wang, X.; Zhang, B.; Zou, M.; et al. Circulating Tumor DNA as a Prognostic Biomarker in Localized Non-small Cell Lung Cancer. Front. Oncol. 2020, 10, 1875. [Google Scholar] [CrossRef]
- Yang, J.; Gong, Y.; Lam, V.K.; Shi, Y.; Guan, Y.; Zhang, Y.; Ji, L.; Chen, Y.; Zhao, Y.; Qian, F.; et al. Deep sequencing of circulating tumor DNA detects molecular residual disease and predicts recurrence in gastric cancer. Cell Death Dis. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Ohara, S.; Suda, K.; Sakai, K.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Fujino, T.; Koga, T.; Hamada, A.; et al. Prognostic implications of preoperative versus postoperative circulating tumor DNA in surgically resected lung cancer patients: A pilot study. Transl. Lung Cancer Res. 2020, 9, 1915–1923. [Google Scholar] [CrossRef] [PubMed]
- Kuang, P.-P.; Li, N.; Liu, Z.; Sun, T.-Y.; Wang, S.-Q.; Hu, J.; Ou, W.; Wang, S.-Y. Circulating Tumor DNA Analyses as a Potential Marker of Recurrence and Effectiveness of Adjuvant Chemotherapy for Resected Non-Small-Cell Lung Cancer. Front. Oncol. 2021, 10, 2892. [Google Scholar] [CrossRef] [PubMed]
- Mes, S.W.; Brink, A.; Sistermans, E.A.; Straver, R.; Oudejans, C.B.; Poell, J.B.; Leemans, C.R.; Brakenhoff, R.H. Comprehensive multiparameter genetic analysis improves circulating tumor DNA detection in head and neck cancer patients. Oral Oncol. 2020, 109, 104852. [Google Scholar] [CrossRef] [PubMed]
- de Aledo-Castillo, J.M.G.; Arcocha, A.; Victoria, I.; Martinez-Puchol, A.I.; Sánchez, C.; Jares, P.; Rodríguez, G.F.; Viñolas, N.; Reyes, R.; Reguart, N.; et al. Molecular characterization of advanced non-small cell lung cancer patients by cfDNA analysis: Experience from routine laboratory practice. J. Thorac. Dis. 2021, 13, 1658. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Şenbabaoğlu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Dutta, M.; Nakagawa, H.; Kato, H.; Maejima, K.; Sasagawa, S.; Nakano, K.; Sasaki-Oku, A.; Fujimoto, A.; Mateos, R.N.; Patil, A.; et al. Whole genome sequencing analysis identifies recurrent structural alterations in esophageal squamous cell carcinoma. PeerJ 2020, 8, e9294. [Google Scholar] [CrossRef]
- Chin, S.F.; Santonja, A.; Grzelak, M.; Ahn, S.; Sammut, S.J.; Clifford, H.; Rueda, O.M.; Pugh, M.; Goldgraben, M.A.; Bardwell, H.A.; et al. Shallow whole genome sequencing for robust copy number profiling of formalin-fixed paraffin-embedded breast cancers. Exp. Mol. Pathol. 2018, 104, 161–169. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, Z.; Su, Z.; Liu, Y.; Yang, T.; Cao, M.; Jiang, Y.; Tang, Y.; Chen, H.; Zhang, W.; et al. Novel Recurrent Altered Genes in Chinese Patients with Anaplastic Thyroid Cancer. J. Clin. Endocrinol. Metab. 2021, 106, 988–998. [Google Scholar] [CrossRef]
- Passaro, A.; Attili, I.; Rappa, A.; Vacirca, D.; Ranghiero, A.; Fumagalli, C.; Guarize, J.; Spaggiari, L.; de Marinis, F.; Barberis, M.; et al. Genomic Characterization of Concurrent Alterations in Non-Small Cell Lung Cancer (NSCLC) Harboring Actionable Mutations. Cancers 2021, 13, 2172. [Google Scholar] [CrossRef]
- Straver, R.; Sistermans, E.A.; Holstege, H.; Visser, A.; Oudejans, C.B.M.; Reinders, M.J.T. WISECONDOR: Detection of fetal aberrations from shallow sequencing maternal plasma based on a within-sample comparison scheme. Nucleic Acids Res. 2013, 42, e31. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chang, C.-W.; Spoerke, J.M.; Yoh, K.E.; Kapoor, V.; Baudo, C.D.; Aimi, J.; Yu, M.; Liang-Chu, M.M.; Suttmann, R.; et al. Low-pass Whole-genome Sequencing of Circulating Cell-free DNA Demonstrates Dynamic Changes in Genomic Copy Number in a Squamous Lung Cancer Clinical Cohort. Clin. Cancer Res. 2019, 25, 2254–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [Green Version]
- Soave, A.; Chun, F.K.-H.; Hillebrand, T.; Rink, M.; Weisbach, L.; Steinbach, B.; Fisch, M.; Pantel, K.; Schwarzenbach, H. Copy number variations of circulating, cell-free DNA in urothelial carcinoma of the bladder patients treated with radical cystectomy: A prospective study. Oncotarget 2017, 8, 56398–56407. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.A.; Page, K.; Blighe, K.; Hava, N.; Guttery, D.; Ward, B.; Brown, J.; Ruangpratheep, C.; Stebbing, J.; Payne, R.; et al. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res. 2011, 22, 220–231. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Korde, L.A.; Yurgelun, M.B. A Case-Based Approach to Understanding Complex Genetic Information in an Evolving Landscape. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, e328–e338. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Yang, Y.; Chen, X.; Jiang, H.; Wang, H.; Shen, M.; Yu, Y.; Liu, T.; Pan, B.; Wang, B.; et al. Chemotherapy-associated clonal hematopoiesis mutations should be taken seriously in plasma cell-free DNA KRAS/NRAS/BRAF genotyping for metastatic colorectal cancer. Clin. Biochem. 2021, 92, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Ococks, E.; Frankell, A.; Soler, N.M.; Grehan, N.; Northrop, A.; Coles, H.; Redmond, A.; Devonshire, G.; Weaver, J.; Hughes, C.; et al. Longitudinal tracking of 97 esophageal adenocarcinomas using liquid biopsy sampling. Ann. Oncol. 2020, 32, 522–532. [Google Scholar] [CrossRef]
- Chan, H.T.; Chin, Y.M.; Nakamura, Y.; Low, S.-K. Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers 2020, 12, 2277. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2018, 35, 1978–1980. [Google Scholar] [CrossRef]
- Henriksen, T.V.; Reinert, T.; Christensen, E.; Sethi, H.; Birkenkamp-Demtröder, K.; Gögenur, M.; Gögenur, I.; Zimmermann, B.G.; Dyrskjøt, L.; Andersen, C.L.; et al. The effect of surgical trauma on circulating free DNA levels in cancer patients—implications for studies of circulating tumor DNA. Mol. Oncol. 2020, 14, 1670–1679. [Google Scholar] [CrossRef]
- Chera, B.S.; Kumar, S.; Beaty, B.T.; Marron, D.; Jefferys, S.; Green, R.; Goldman, E.C.; Amdur, R.; Sheets, N.; Dagan, R.; et al. Rapid Clearance Profile of Plasma Circulating Tumor HPV Type 16 DNA during Chemoradiotherapy Correlates with Disease Control in HPV-Associated Oropharyngeal Cancer. Clin. Cancer Res. 2019, 25, 4682–4690. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Reference | Number of Patients Included (n) | Tumor Type and Indication | Methodology | Conclusions |
|---|---|---|---|---|
| Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patientswith urothelial bladder carcinoma [91] | 68 | Muscle invasive bladder cancer treated with neoadjuvant chemotherapy before cystectomy | Tumor sequencing: WES Plasma sequencing: 16 mutations/patient by multiplex PCR. | A total of 76% of ctDNA-positive patients post cystectomy had recurrence (median 96 days before). A total of 0% of ctDNA-negative had recurrence. |
| Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer [86] | 55 | Early breast cancer patients receiving neoadjuvant chemotherapy | Tumor sequencing: NGS on panel with 14 known breast cancer driver genes (26). Plasma sequencing: 1 (or more) mutation(s) was (were) followed using ddPCR. | ctDNA was detected in the single post-operative blood test in 19% (7 of 37) of patients. ctDNA detection was predictive of early relapse (median 6.5 months). |
| Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer [89] | 33 | Stage I to Stage III breast cancer | Tumor sequencing: WES Plasma sequencing: Using TARDIS (combinaison of NGS + PCR + UMIs): 6 to 115 mutations per patient. | Before treatment, ctDNA detected in 32 of 32 patients at tumor fractions of 0.002% to 1.06%. Plasma samples after completion of NAT were analyzed in 22 patients. ctDNA+ in 17 out of 22 patients, including 12 out of 13 patients with invasive or in situ residual disease and 5 out of 9 patients with pathological CR. In patients who achieved pathological CR, the median decrease in ctDNA was 96%, whereas in patients with residual disease observed at surgery, the median decrease was 77%. |
| Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer [92] | 94 | Resectable colon cancers with plasma available | Tumor sequencing: NGS on custom targeted panel of 29 genes. Plasma sequencing: personalized ddPCR assays for each somatic mutation identified in the tissue. | ctDNA was detected in 63.8% at baseline. ctDNA was detected at 6–8 weeks post-surgery, before starting adjuvant chemotherapy, in 20.3% (14 of 69) patients with plasma available at this time. In ctDNA-positive post-op: 57.1% (8 of 14 patients) experienced reccurence. The presence of ctDNA immediately after surgery was associated with poorer DFS. |
| Circulating tumor DNA analyses as markers of recurrence risk and benefit of adjuvant therapy for Stage III colon cancer [90] | 96 | Stage III colon cancer | Tumor sequencing: NGS on 15 genes recurrently mutated in colorectal cancer. Plasma sequencing: 1 mutation/patient with Safe-Seq (NGS + UMIs). |
A tumor-specific mutation was detected (ctDNA-positive finding) in the post-surgical plasma sample of 20 of 96 patients (21%). ctDNA was detectable in 15 of 88 (17%) post-chemotherapy samples. Post-surgical ctDNA was detectable in 10 of 24 patients (42%) with recurrence. |
| Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response andsurvival [88] | 84 | High-risk earlybreast cancer patients with NAT (I-SPY2 Trial) | Tumor sequencing: WES Plasma sequencing: 16 mutations/patient by multiplex PCR | After NAC, all patients who achieved pCR were ctDNA-negative (n = 17, 100%). For those who did not achieve pCR (n = 43), ctDNA-positive patients (14%) had significantly increased risk of metastatic recurrence (HR 10.4; 95% CI, 2.3–46.6). Patients who did not achieve pCR but were ctDNA negative (86%) had a similar outcome to those who achieved pCR. |
| Circulating Tumor DNA predicts pathologic and clinical outcomes following neoadjuvantchemoradiation and surgery for patients with locally advanced rectal cancer [93] | 29 | Locally advanced rectal cancer | Tumor sequencing: WES Plasma sequencing: personalized ddPCR assays for each somatic mutation identified in the tissue. | Patients with detectable postoperative ctDNA experienced poorer RFS (hazard ratio, 11.56; p = 0.007). All patients (4 out of 4) with detectable postoperative ctDNA recurred (positive predictive value = 100%), whereas only 2 out of 15 patients with undetectable ctDNA recurred (negative predictive value = 87%). |
| Galaxy Study: Preoperative ctDNA levels are detectable in the majority of patients with resectable colorectal cancer [94] | 808 | Resectable CRC | Tumor sequencing: WES Plasma sequencing: personalized ddPCR assays for each somatic mutation identified in the tissue. | Longitudinal ctDNA positivity at postoperative weeks 4, 12, and 24 was significantly associated with inferior disease-free survival (DFS) with a hazard ratio (HR) of 46.8. Sensitivity of relapse detection was 93.1%. Positivity at postoperative week 4 was significantly associated with inferior DFS with HR 19.5 overall, and HR 24.4 in pathologic Stage I–III, indicating it is a suitable time point for ctDNA-based adjuvant study. |
| Dynamics of cell-free tumour DNA correlate with treatment response of head and neck cancer patients receiving radiochemotherapy [87] | 20 | Non-resecable locally advanced head and neck squamous cell carcinoma | Tumor sequencing: NGS with 327 genes panel. Plasma sequencing: 127 driver mutations + E7 NGS panel | Baseline: ctDNA-positive: 17/20 patients Post RCT ctDNA-positive-: 2/16 patients Eight patients relapsed: 2ctDNA-positive Eight patients without relapse: 8ctDNA-negative PPV 100%, Sn 25% |
| Reference | Number of Patients (n) | Tumor Type and Indication | Methodology | Conclusions |
|---|---|---|---|---|
| Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling [118] | 40 54 healthy controls | Curative intent for Stage I–III lung cancer | Plasma sequencing: CAPP-Seq 128 genes most frequently mutated in lung cancer. | 94% of patients with MRD were ctDNA-positive in post-treatment plasma samples. Patients were ctDNA-positive before radiological relapse (72%) (5.2months). 53% of ctDNA-positive patients had actionable targets. |
| Circulating tumor DNA analysis for detection of minimal residual disease after chemoradiotherapy for localized esophagealcancer [119] | 45 | Stage IA to Stage IIIB esophageal cancers (adenocarinoma or squamous cell carinoma) | Plasma sequencing: CAPP-Seq Esophageal specific panel | Baseline ctDNA-positive: 27/45 (60%). Post CRT ctDNA-positive: 5/31 (16%). Patients with detectable ctDNA post-CRT also had significantly increased risk of disease progression (HR 18.7, p < 0.0001), distant metastasis (HR 32.1, p < 0.0001), and disease specific death (HR 23.1, p < 0.0001). |
| Post-radiation circulating tumor DNA as a prognostic factor in locally advanced esophageal squamous cell carcinoma [120] | 25 | Resectable esophageal squamous cell carcinoma | Plasma sequencing: NGS on a custom designed 180 genes panel | At baseline, 100% ctDNA-positive. Post radiotherapy: 14/24 (58%) ctDNA-positive 10/24 (42%) ctDNA-negative In the 14 ctDNA-positive patients, 11 patients had a documented follow-up: 90.9% (10/11) had documented disease recurrence. In the 10 ctDNA-negative patients, 8 patients had documented follow-up: 50% (4/8) had documented disease recurrence. Patients who were ctDNA-positive exhibited a marginally significant reduction in PFS (p = 0.047) and a significantly decreased OS (p = 0.005) compared to patients who were ctDNA-negative. |
| Minimal residual disease detection using a plasma-only circulating tumor DNA assay in colorectal cancer patients [121] | 84 | Resectable colorectal cancer | Plasma sequencing: Guardant Reveal™ test using NGS custom based panel for the detection of somatic and epigenitic abberations. | Fifteen patients had detectable ctDNA and all 15 recurred. Of 49 patients without detectable ctDNA at the landmark timepoint, 12 (24.5%) recurred. Landmark recurrence sensitivity and specificity were 55.6% and 100%. Integrating epigenomic signatures increased sensitivity by 25–36% versus genomic alterations alone. |
| Prognostic implications of preoperative versus postoperative circulating tumor DNA in surgically resected lung cancer patients: a pilot study [125] | 20 | Stage IIA–IIIA lung cancer | Plasma sequencing: CAPP-Seq on a commercial 197 genes panel (Roche Diagnostics). |
Eight patients (40%) were positive for preoperative ctDNA.
Four patients (20%) were positive for postoperative ctDNA, and this was significantly correlated with histological grade (3 vs. 1 or 2, p = 0.032). Postoperative positivity for ctDNA also predicted shorter recurrence-free survival (RFS). |
| Circulating tumor DNA as a prognostic biomarker in localized non-small cell lung cancer [123] | 77 | Resectable NSCLC | Plasma sequencing: NGS (cSMART assay) on a custom 127 gene panel | Postoperative ctDNA-positive patients also associated with a lower RFS (HR = 3.076, p = 0.0015) and OS (HR = 3.195, p = 0.0053). Disease recurrence occurred among 63.3% (19/30) of postoperative ctDNA-positive patients. Most of these patients 89.5% (17/19) had detectable ctDNA within 2 weeks after surgery. |
| Circulating tumor DNA as a potential marker to detect minimal residual disease and predict recurrence in pancreatic cancer [115] | 27 | Operable pancreatic cancer | Plasma sequencing: NGS on a large (1.017) gene panel | ctDNA was detected in 18 of 27 preoperative plasma samples, resulting in a detectable rate of 66.67%. Seven days after surgical resection, the status of ctDNA changed in 19 patients. Of these, one turned positive and 10 became completely negative. Patients who were ctDNA-positive postoperatively had a markedly reduced disease-free survival (DFS) compared to those who were ctDNA-negative. A positive postoperative ctDNA status was an independent prognostic factor for DFS. |
| Deep sequencing of circulating tumor DNA detects molecular residual disease and predicts recurrence in gastric cancer [124] | 46 | Stage I–III gastric cancer | Plasma sequencing: NGS with Enrich Rare Mutation Sequencing (ER-Seq) assay on a custom driver mutation panel | ctDNA was detected in 45% of treatment-naïve plasma samples. All patients with detectable ctDNA in the immediate post-operative period eventually experienced recurrence. Post-operative samples (collected prior to any adjuvant chemotherapy; 9–48 days after surgery) showed that ctDNA was detected in 18% (7 out of 38) of evaluable patients. ctDNA positivity after surgery was strongly associated with an increased risk of relapse (100% recurrence in the positive group vs. 32% in the negative group), worse DFS (p < 0.0001), and worse OS (p = 0.0007). |
| Circulating tumor DNA analyses as a potential marker of recurrence and effectiveness of adjuvant chemotherapy for resected non-small cell lung cancer [126] | 38 | Resectable NSCLC | Plasma sequencing: NGS on a custom 425 genes panel | Preoperative plasma samples, ctDNA+ in 19 (50%) patients ctDNA was detected post-chemotherapy in 8 out of 36 (22.2%) patients and was associated with an inferior RFS (HR, 8.76; p < 0.001). |
| Name of Trial | NCT | Tumor Type | Primary Endpoint | Type of Trial |
|---|---|---|---|---|
| circTeloDIAG: liquid biopsy in glioma tumor | NCT04931732 | Glioma | Sensitivity and specificity of the circTeloDIAG assay at the time of surgery | A: Prognosis trial design |
| Liquid biopsy in head and neck cancer | NCT099326468 | HNSCC | Compare liquid biopsy to PET-CT to evaluate MRD | A: Prognosis trial design |
| LIQUID | NCT049443406 | Gastric cancer | Evaluate the prognosis role of liquid biopsy in locally advanced gastric cancer | A: Prognosis trial design |
| NSCLC heterogeneity in early-stage patients and prediction of relapse using a personalized “liquid biopsy” | NCT03771404 | NSCLC | Correlate the liquid biopsy information to disease recurrence | A: Prognosis trial design |
| T-MENC | NCT03838588 | NSCLC | The concordance of the plasma ctDNA detection status with PFS and OS after radical resection or/and under adjuvant treatment | A: Prognosis trial design |
| PEGASUS trial | NCT04259944 | Colon cancer | Proving the feasibility of using liquid biopsy to guide post-surgical and post-adjuvant clinical management in MSS Stage III and Stage II T4N0 colon cancer | C: De-escalation trial design with several arms depending on de-escalation regime |
| HCCGenePanel | NCT04111029 | Hepatocarcinoma | Prove response to locoregional therapy | A: Prognosis trial design |
| Liquid biopsy in monitoring the neoadjuvant chemotherapy and operation in gastric cancer | NCT03957564 | Gastric cancer | Explore the clinical value of CTC, ctDNA, and cfDNA in neoadjuvant chemotherapy and operation of resectable or locally advanced gastric cancer | A: Prognosis trial design |
| PROJECTION | NCT04246203 | Pancreatic cancer | Prognostic role of circulating tumor DNA in resectable pancreatic cancer | A: Prognosis trial design |
| ctDNA Lung RCT | NCT049666663 | NSCLC | To evaluate whether the presence of circulating tumor DNA (ctDNA) in the blood can help to predict whether giving adjuvant treatment after surgery can decrease the risk of cancer recurrence. | B: Intensification trial design with several arms |
| Verification of predictive biomarkers for pancreatic cancer treatment using multicenter liquid biopsy | NCT04241367 | Pancreatic cancer | Verification of predictive biomarkers for pancreatic cancer treatment | A: Prognosis trial design |
| Cell-free tumor DNA in head and neck cancer patients | NCT03942380 | Head and neck cancer | Measure the percentage of recurrence in head and neck cancer patients through serial monitoring with liquid biopsy | A: Prognosis trial design |
| MARTINI | NCT04853420 | Solid malignancies | Minimal residual disease: a trial using liquid biopsies in solid malignancies | A: Prognosis trial design |
| WHENII | NCT03481101 | NSCLC | Evaluate early response to chemotherapy in NSCLC | A: Prognosis trial design |
| PECAN | NCT03540563 | HNSCC | ctDNA as a biomarker for treatment response | A: Prognosis trial design |
| Serial ctDNA monitoring during adjuvant capecitabine in early triple negative breast cancer | NCT04768426 | Triple negative breast cancer | Detection levels of ctDNA during adjuvant treatment | A: Prognosis trial design |
| LiBReCA | NCT03699410 | Rectal cancer | Investigate the value of liquid biopsies to predict tumor response after neoadjuvant chemo-radiotherapy in patients with locally advanced rectal cancer | A: Prognosis trial design |
| Monitoring efficacity of radiotherapy in lung cancer and esophageal cancer | NCT04014465 | Lung and esophageal cancer | Clinical value of efficacy evaluation and prognosis of ctDNA detecting technique in patients with radiotherapy | A: Prognosis trial design |
| MRD monitoring in lung cancer after resection | NCT04976296 | Lung cancer | MRD monitoring | A: Prognosis trial design |
| PRE-MERIDIAN | NCT04599309 | Locally advanced head and neck squamous cell carcinoma | Number of high-risk HNSCC with successful ctDNA detection after standard treatment | A: Prognosis trial design |
| TOMBOLA | NCT04138628 | Bladder cancer | Treatment of metastatic bladder cancer at the time of biochemical relapse following radical cystectomy | B: Intervention trial design |
| Adjuvant durvalumab in early-stage NSCLC patients with ctDNA MRD | NCT04585477 | NSCLC | Durvalumab as adjuvant treatment in ctDNA-positive patients | B: Intervention trial design |
| Study of ctDNA guided change of treatment for refractory MRD in colon adenocarcinoma | NCT04920032 | Colon adenocarcinoma | Adjuvant TAS-102 + iritotecan in ctDNA-positive colon cancer patients | B: Intervention trial design |
| Minimal residual disease assessment in patients with colorectal cancer: MIRDA-C study | NCT04739072 | Colorectal cancer | Improve the detection of MRD | A: Prognosis trial design |
| c-TRAK-TN | NCT03145961 | Early-stage triple negative breast cancer | A randomized trial using ctDNA mutation tracking to detect MRD and trigger patient intervention. | B: Intervention trial design |
| CITCCA | NCT04726800 | Colorectal cancer | ctDNA as a prognostic and predictive marker in colorectal cancer | A: Prognosis trial design |
| Clearance of ctDNA big ten cancer research consortium | NCT04367311 | NSCLC | Clearance of ctDNA under adjuvant treatment | A: Prognosis trial design |
| Personalized escalation of consolidation treatment following chemoradiotherapy and immunotherapy in Stage III NSCLC | NCT04585490 | NSCLC | Adjuvant therapy in ctDNA-postive patients | B: Intervention trial design |
| Measuring MRD in colorectal cancer after primary surgery and resection of metastases | NCT03189576 | Colorectal cancer | Measuring MRD | A: Prognosis trial design |
| IMPROVE-IT | NCT03748680 | Colorectal cancer | Implementing non-invasive ctDNA analysis to optimize the operative and post-operative treatment of colorectal cancer | B: Intervention trial design |
| DYNAMIC-III | ACTRN/12615000381583 | Colon cancer | Adjuvant therapy in ctDNA-positive patients | B: Intervention trial design |
| COBRA | NCT04068103 | Colon cancer | Adjuvant therapy in ctDNA-positive patients | B: Intervention trial design |
| IM-VIGOR 011 | NCT04660344 | Bladder cancer | Adjuvant therapy (atezolizumab) in ctDNA-positive patients | B: Intervention trial design |
| MERMAID-1 | NCT04385368 | NSCLC | Adjuvant therapy (durvalumab) in ctDNA-positive patients | B: Intervention trial design |
| BESPOKE Study of ctDNA Guided Therapy in Colorectal Cancer | NCT04264702 | Colon cancer | Adjuvant chemotherapy or observation (choice by treating clinician) in ctDNA positive patients | B: Intervention trial design |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honoré, N.; Galot, R.; van Marcke, C.; Limaye, N.; Machiels, J.-P. Liquid Biopsy to Detect Minimal Residual Disease: Methodology and Impact. Cancers 2021, 13, 5364. https://doi.org/10.3390/cancers13215364
Honoré N, Galot R, van Marcke C, Limaye N, Machiels J-P. Liquid Biopsy to Detect Minimal Residual Disease: Methodology and Impact. Cancers. 2021; 13(21):5364. https://doi.org/10.3390/cancers13215364
Chicago/Turabian StyleHonoré, Natasha, Rachel Galot, Cédric van Marcke, Nisha Limaye, and Jean-Pascal Machiels. 2021. "Liquid Biopsy to Detect Minimal Residual Disease: Methodology and Impact" Cancers 13, no. 21: 5364. https://doi.org/10.3390/cancers13215364
APA StyleHonoré, N., Galot, R., van Marcke, C., Limaye, N., & Machiels, J.-P. (2021). Liquid Biopsy to Detect Minimal Residual Disease: Methodology and Impact. Cancers, 13(21), 5364. https://doi.org/10.3390/cancers13215364