
Disruption of Tumor Suppressors HNF4α/HNF1α Causes Tumorigenesis in Liver


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. The Roles of HNF4α and HNF1α in Liver Cancer Development
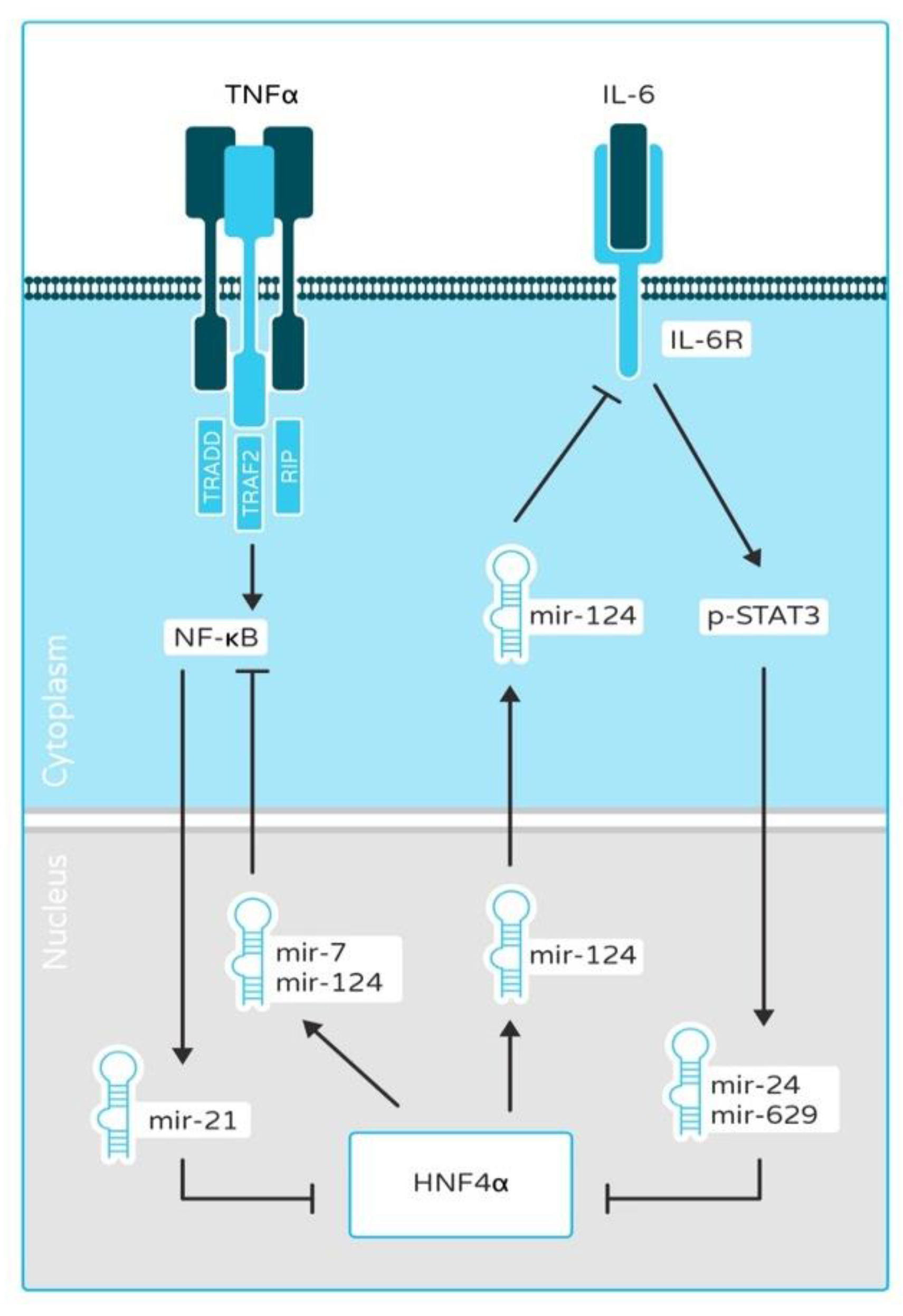
2.1. HNF4α and HNF1α as a Link between Inflammation and Cancer
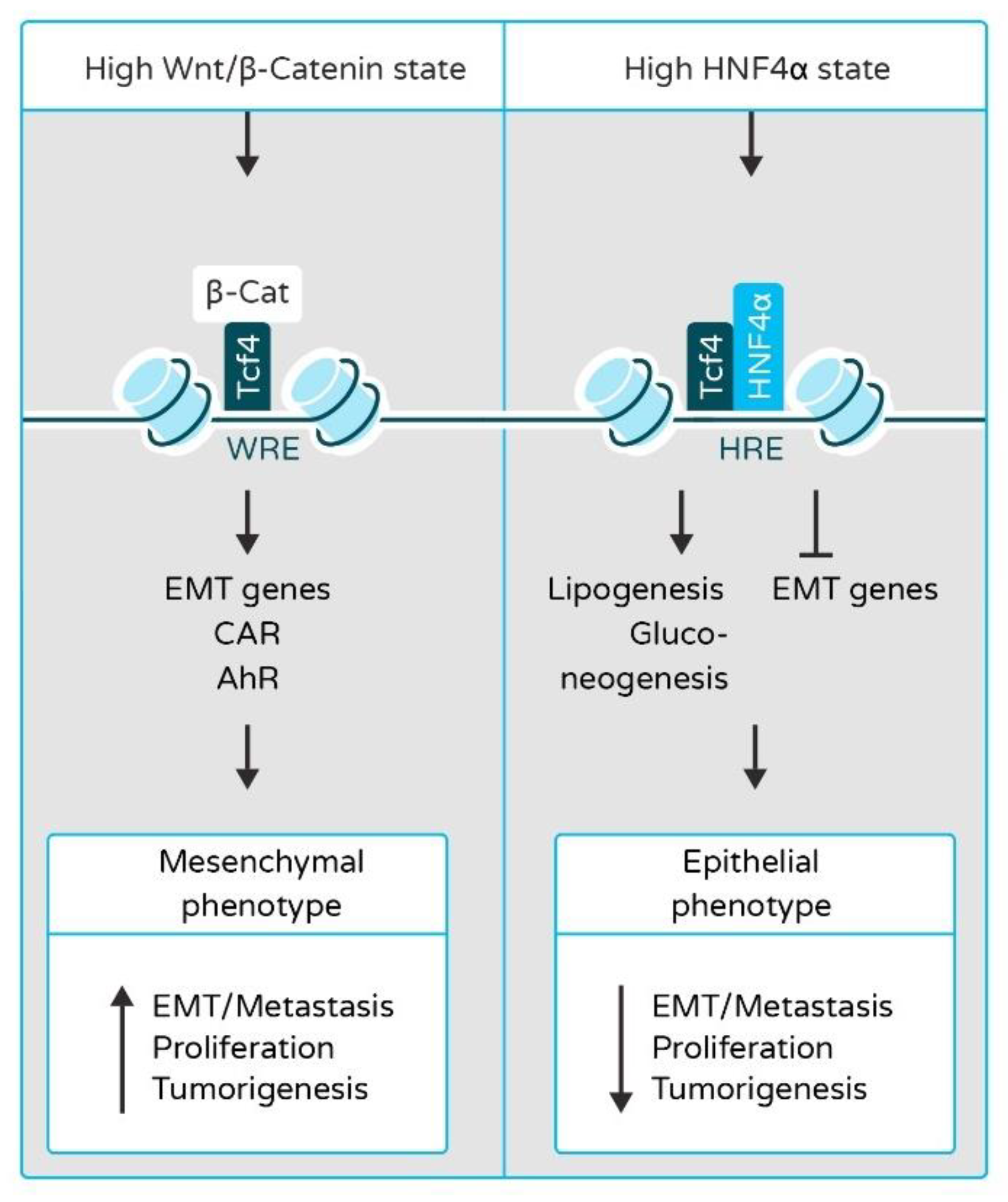
2.2. HNF4α and HNF1α in Epithelial-to-Mesenchymal Transition (EMT) and Liver Cancer Metastasis
2.3. HNF4α and HNF1α in Metabolism and Liver Cancer
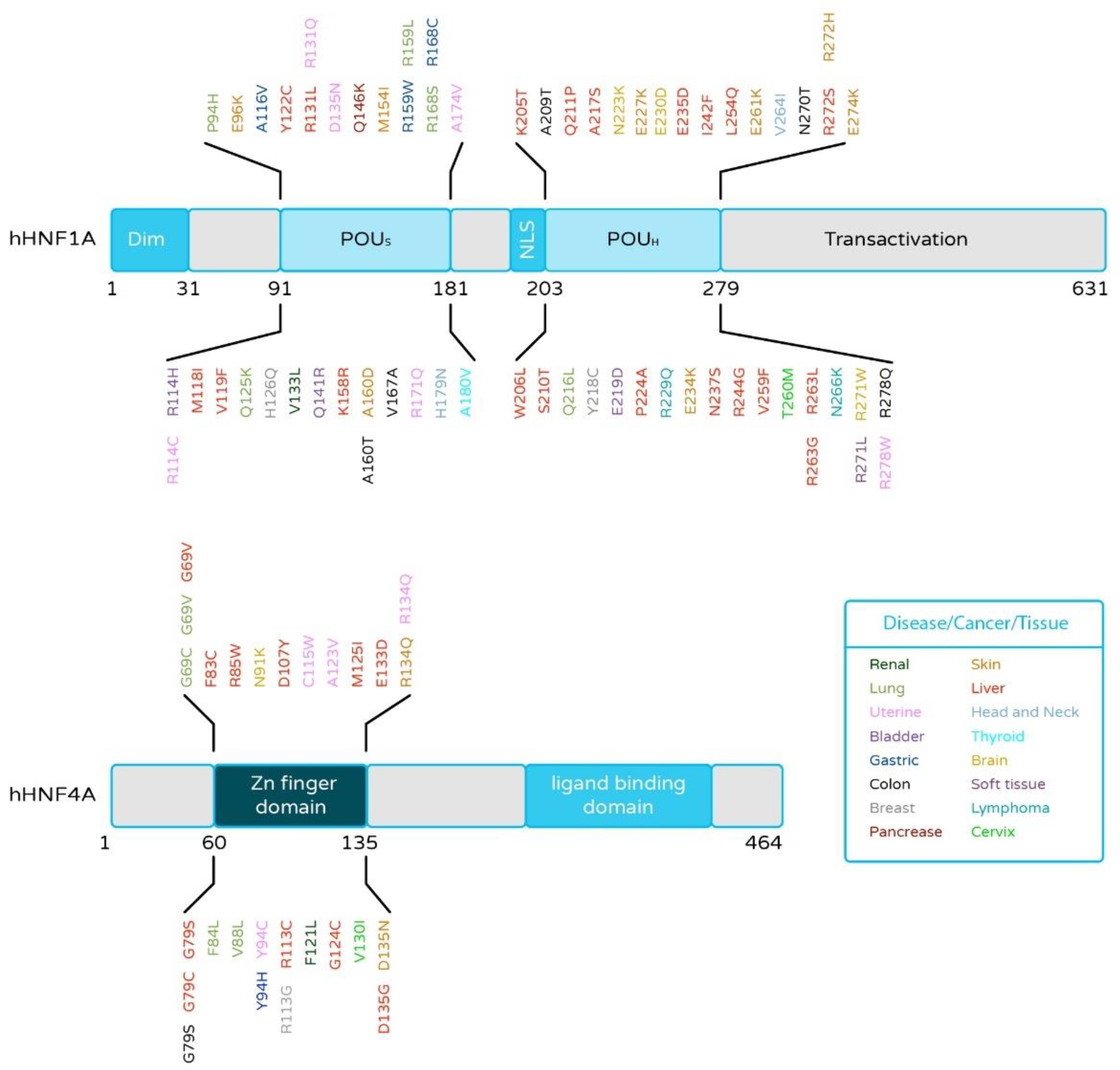
2.4. HNF4α and HNF1α Mutations in Liver Cancer
2.5. Phenotypic Consequences of Downregulated Expression of HNF4α and HNF1α in Liver Cancer
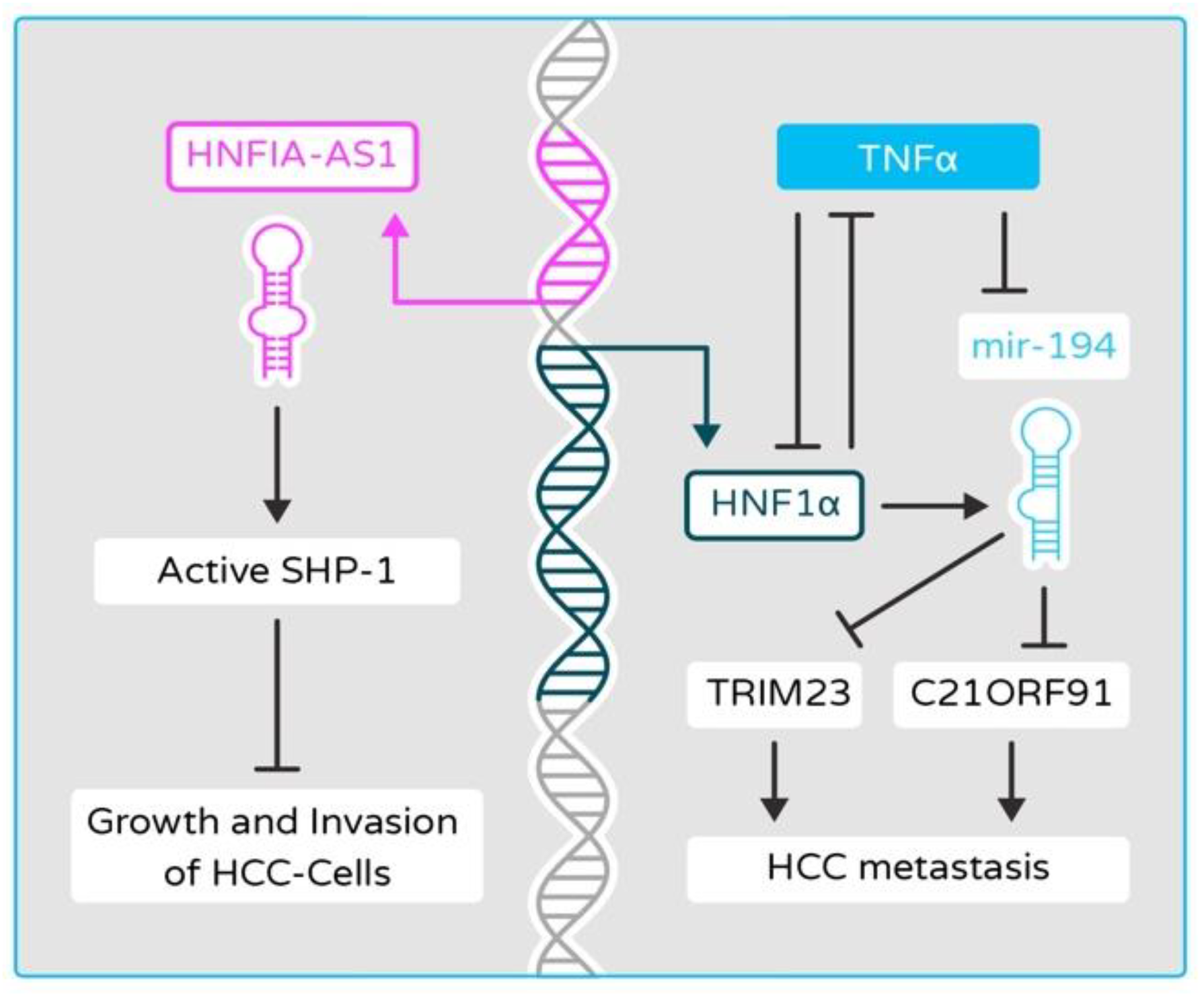
2.6. Disruptions of Epigenetic and miRNA Controls in HNF4α and HNF1α Networks Contribute to Cancer Development in Liver Cancer
2.7. Function of HNF4α and HNF1α Is Disrupted by HBV and HCV Infection
2.8. HNF4α in Colorectal Cancer (CRC): Friend or Foe?
3. HNF4α as a Therapeutic Target in Cancer
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Walesky, C.; Apte, U. Role of Hepatocyte Nuclear Factor 4 Alpha (HNF4α) in Cell Proliferation and Cancer. Gene Expr. 2015, 16, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.H.; Ng, N.H.J.; Loo, L.S.W.; Jasmen, J.B.; Teo, A.K.K. The Molecular Functions of Hepatocyte Nuclear Factors—In and beyond the Liver. J. Hepatol. 2018, 68, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Oda, N.; Kaisaki, P.J.; Menzel, S.; Furuta, H.; Vaxillaire, M.; Southam, L.; Cox, R.D.; Lathrop, G.M.; Boriraj, V.V.; et al. Mutations in the Hepatocyte Nuclear Factor-1alpha Gene in Maturity-Onset Diabetes of the Young (MODY3). Nature 1996, 384, 455–458. [Google Scholar] [CrossRef]
- Horikawa, Y.; Iwasaki, N.; Hara, M.; Furuta, H.; Hinokio, Y.; Cockburn, B.N.; Lindner, T.; Yamagata, K.; Ogata, M.; Tomonaga, O.; et al. Mutation in Hepatocyte Nuclear Factor-1 Beta Gene (TCF2) Associated with MODY. Nat. Genet. 1997, 17, 384–385. [Google Scholar] [CrossRef]
- Furuta, H.; Iwasaki, N.; Oda, N.; Hinokio, Y.; Horikawa, Y.; Yamagata, K.; Yano, N.; Sugahiro, J.; Ogata, M.; Ohgawara, H.; et al. Organization and Partial Sequence of the Hepatocyte Nuclear Factor-4 Alpha/MODY1 Gene and Identification of a Missense Mutation, R127W, in a Japanese Family with MODY. Diabetes 1997, 46, 1652–1657. [Google Scholar] [CrossRef]
- Jiang, G.; Sladek, F.M. The DNA Binding Domain of Hepatocyte Nuclear Factor 4 Mediates Cooperative, Specific Binding to DNA and Heterodimerization with the Retinoid X Receptor α. J. Biol. Chem. 1997, 272, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.L.; Zhuo, Z.; Ren, E.C. HNF4α Combinatorial Isoform Heterodimers Activate Distinct Gene Targets That Differ from Their Corresponding Homodimers. Cell Rep. 2019, 26, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Vető, B.; Bojcsuk, D.; Bacquet, C.; Kiss, J.; Sipeki, S.; Martin, L.; Buday, L.; Bálint, B.L.; Arányi, T. The Transcriptional Activity of Hepatocyte Nuclear Factor 4 Alpha is Inhibited via Phosphorylation by ERK1/2. PLoS ONE 2017, 12, e0172020. [Google Scholar] [CrossRef]
- Chen, W.S.; Manova, K.; Weinstein, D.C.; Duncan, S.A.; Plump, A.S.; Prezioso, V.R.; Bachvarova, R.F.; Darnell, J.E. Disruption of the HNF-4 Gene, Expressed in Visceral Endoderm, Leads to Cell Death in Embryonic Ectoderm and Impaired Gastrulation of Mouse Embryos. Genes Dev. 1994, 8, 2466–2477. [Google Scholar] [CrossRef]
- Li, J.; Ning, G.; Duncan, S.A. Mammalian Hepatocyte Differentiation Requires the Transcription Factor HNF-4alpha. Genes Dev. 2000, 14, 464–474. [Google Scholar]
- Chandra, V.; Huang, P.; Potluri, N.; Wu, D.; Kim, Y.; Rastinejad, F. Multidomain Integration in the Structure of the HNF-4α Nuclear Receptor Complex. Nature 2013, 495, 394–398. [Google Scholar] [CrossRef]
- Huang, J.; Levitsky, L.L.; Rhoads, D.B. Novel P2 Promoter-Derived HNF4α Isoforms with Different N-Terminus Generated by Alternate Exon Insertion. Exp. Cell Res. 2009, 315, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Fujimoto, A.; Kono, H.; Furuta, M.; Fujita, M.; Nakagawa, H. Loss-of-Function Mutations in Zn-Finger DNA-Binding Domain of HNF4A Cause Aberrant Transcriptional Regulation in Liver Cancer. Oncotarget 2018, 9, 26144–26156. [Google Scholar] [CrossRef] [PubMed]
- Hwang-Verslues, W.W.; Sladek, F.M. HNF4α—Role in Drug Metabolism and Potential Drug Target? Curr. Opin. Pharmacol. 2010, 10, 698–705. [Google Scholar] [CrossRef]
- Eeckhoute, J.; Formstecher, P.; Laine, B. Hepatocyte Nuclear Factor 4α Enhances the Hepatocyte Nuclear Factor 1α-Mediated Activation of Transcription. Nucleic Acids Res. 2004, 32, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Torres-Padilla, M.E.; Sladek, F.M.; Weiss, M.C. Developmentally Regulated N-Terminal Variants of the Nuclear Receptor Hepatocyte Nuclear Factor 4alpha Mediate Multiple Interactions through Coactivator and Corepressor-Histone Deacetylase Complexes. J. Biol. Chem. 2002, 277, 44677–44687. [Google Scholar] [CrossRef] [PubMed]
- Walesky, C.; Gunewardena, S.; Terwilliger, E.F.; Edwards, G.; Borude, P.; Apte, U. Hepatocyte-Specific Deletion of Hepatocyte Nuclear Factor-4α in Adult Mice Results in Increased Hepatocyte Proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 304, G26–G37. [Google Scholar] [CrossRef]
- Bonzo, J.A.; Ferry, C.H.; Matsubara, T.; Kim, J.-H.; Gonzalez, F.J. Suppression of Hepatocyte Proliferation by Hepatocyte Nuclear Factor 4α in Adult Mice. J. Biol. Chem. 2012, 287, 7345–7356. [Google Scholar] [CrossRef] [PubMed]
- Sel, S.; Ebert, T.; Ryffel, G.U.; Drewes, T. Human Renal Cell Carcinogenesis is Accompanied by a Coordinate Loss of the Tissue Specific Transcription Factors HNF4 Alpha and HNF1 Alpha. Cancer Lett. 1996, 101, 205–210. [Google Scholar] [CrossRef]
- Hoskins, J.W.; Jia, J.; Flandez, M.; Parikh, H.; Xiao, W.; Collins, I.; Emmanuel, M.A.; Ibrahim, A.; Powell, J.; Zhang, L.; et al. Transcriptome Analysis of Pancreatic Cancer Reveals a Tumor Suppressor Function for HNF1A. Carcinogenesis 2014, 35, 2670–2678. [Google Scholar] [CrossRef]
- Saandi, T.; Baraille, F.; Derbal-Wolfrom, L.; Cattin, A.-L.; Benahmed, F.; Martin, E.; Cardot, P.; Duclos, B.; Ribeiro, A.; Freund, J.-N.; et al. Regulation of the Tumor Suppressor Homeogene Cdx2 by HNF4α in Intestinal Cancer. Oncogene 2013, 32, 3782–3788. [Google Scholar] [CrossRef]
- Ahn, S.-H.; Shah, Y.M.; Inoue, J.; Morimura, K.; Kim, I.; Yim, S.; Lambert, G.; Kurotani, R.; Nagashima, K.; Gonzalez, F.J.; et al. Hepatocyte Nuclear Factor 4alpha in the Intestinal Epithelial Cells Protects against Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2008, 14, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Babeu, J.-P.; Boudreau, F. Hepatocyte Nuclear Factor 4-Alpha Involvement in Liver and Intestinal Inflammatory Networks. World J. Gastroenterol. 2014, 20, 22–30. [Google Scholar] [CrossRef]
- Hatziapostolou, M.; Polytarchou, C.; Aggelidou, E.; Drakaki, A.; Poultsides, G.A.; Jaeger, S.A.; Ogata, H.; Karin, M.; Struhl, K.; Hadzopoulou-Cladaras, M.; et al. An HNF4α-MiRNA Inflammatory Feedback Circuit Regulates Hepatocellular Oncogenesis. Cell 2011, 147, 1233–1247. [Google Scholar] [CrossRef]
- Yang, M.; Li, S.-N.; Anjum, K.M.; Gui, L.-X.; Zhu, S.-S.; Liu, J.; Chen, J.-K.; Liu, Q.-F.; Ye, G.-D.; Wang, W.-J.; et al. A Double-Negative Feedback Loop between Wnt-β-Catenin Signaling and HNF4α Regulates Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. J. Cell Sci. 2013, 126, 5692–5703. [Google Scholar] [CrossRef]
- Cicchini, C.; Amicone, L.; Alonzi, T.; Marchetti, A.; Mancone, C.; Tripodi, M. Molecular Mechanisms Controlling the Phenotype and the EMT/MET Dynamics of Hepatocyte. Liver Int. 2015, 35, 302–310. [Google Scholar] [CrossRef]
- Lazarevich, N.L.; Shavochkina, D.A.; Fleishman, D.I.; Kustova, I.F.; Morozova, O.V.; Chuchuev, E.S.; Patyutko, Y.I. Deregulation of Hepatocyte Nuclear Factor 4 (HNF4)as a Marker of Epithelial Tumors Progression. Exp. Oncol. 2010, 32, 167–171. [Google Scholar] [PubMed]
- Boj, S.F.; Petrov, D.; Ferrer, J. Epistasis of Transcriptomes Reveals Synergism between Transcriptional Activators Hnf1alpha and Hnf4alpha. PLoS Genet. 2010, 6, e1000970. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, E.; Mellottee, L.; Bioulac-Sage, P.; Balabaud, C.; Scoazec, J.-Y.; Tran van Nhieu, J.; Bacq, Y.; Michalak, S.; Buob, D.; Laurent-Puig, P.; et al. Spectrum of HNF1A Somatic Mutations in Hepatocellular Adenoma Differs from That in Patients with MODY3 and Suggests Genotoxic Damage. Diabetes 2010, 59, 1836–1844. [Google Scholar] [CrossRef]
- Cereghini, S. Liver-Enriched Transcription Factors and Hepatocyte Differentiation. FASEB J. 1996, 10, 267–282. [Google Scholar] [CrossRef]
- Mendel, D.B.; Hansen, L.P.; Graves, M.K.; Conley, P.B.; Crabtree, G.R. HNF-1 Alpha and HNF-1 Beta (VHNF-1) Share Dimerization and Homeo Domains, but Not Activation Domains, and Form Heterodimers in Vitro. Genes Dev. 1991, 5, 1042–1056. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Willson, J.S.B.; Godwin, T.D.; Wiggins, G.a.R.; Guilford, P.J.; McCall, J.L. Primary Hepatocellular Neoplasms in a MODY3 Family with a Novel HNF1A Germline Mutation. J. Hepatol. 2013, 59, 904–907. [Google Scholar] [CrossRef]
- Chi, Y.-I.; Frantz, J.D.; Oh, B.-C.; Hansen, L.; Dhe-Paganon, S.; Shoelson, S.E. Diabetes Mutations Delineate an Atypical POU Domain in HNF-1alpha. Mol. Cell 2002, 10, 1129–1137. [Google Scholar] [CrossRef]
- Luo, Z.; Li, Y.; Wang, H.; Fleming, J.; Li, M.; Kang, Y.; Zhang, R.; Li, D. Hepatocyte Nuclear Factor 1A (HNF1A) as a Possible Tumor Suppressor in Pancreatic Cancer. PLoS ONE 2015, 10, e0121082. [Google Scholar] [CrossRef]
- Patitucci, C.; Couchy, G.; Bagattin, A.; Cañeque, T.; de Reyniès, A.; Scoazec, J.-Y.; Rodriguez, R.; Pontoglio, M.; Zucman-Rossi, J.; Pende, M.; et al. Hepatocyte Nuclear Factor 1α Suppresses Steatosis-Associated Liver Cancer by Inhibiting PPARγ Transcription. J. Clin. Investig. 2017, 127, 1873–1888. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, L.; Rebouissou, S.; Paris, A.; Rathahao-Paris, E.; Perdu, E.; Bioulac-Sage, P.; Imbeaud, S.; Zucman-Rossi, J. Loss of Hepatocyte Nuclear Factor 1alpha Function in Human Hepatocellular Adenomas Leads to Aberrant Activation of Signaling Pathways Involved in Tumorigenesis. Hepatology 2010, 51, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Deng, X.; Huang, Z.-W.; Wei, J.; Ding, C.-H.; Feng, R.-X.; Zeng, X.; Chen, Y.-X.; Ding, J.; Qiu, L.; et al. An HNF1α-Regulated Feedback Circuit Modulates Hepatic Fibrogenesis via the Crosstalk between Hepatocytes and Hepatic Stellate Cells. Cell Res. 2015, 25, 930–945. [Google Scholar] [CrossRef]
- Hatzis, P.; Talianidis, I. Regulatory Mechanisms Controlling Human Hepatocyte Nuclear Factor 4α Gene Expression. Mol. Cell. Biol. 2001, 21, 7320–7330. [Google Scholar] [CrossRef] [PubMed]
- Ktistaki, E.; Talianidis, I. Modulation of Hepatic Gene Expression by Hepatocyte Nuclear Factor 1. Science 1997, 277, 109–112. [Google Scholar] [CrossRef]
- Kuo, C.J.; Mendel, D.B.; Hansen, L.P.; Crabtree, G.R. Independent Regulation of HNF-1 Alpha and HNF-1 Beta by Retinoic Acid in F9 Teratocarcinoma Cells. EMBO J. 1991, 10, 2231–2236. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.K.; Párrizas, M.; Jensen, M.L.; Pruhova, S.; Ek, J.; Boj, S.F.; Johansen, A.; Maestro, M.A.; Rivera, F.; Eiberg, H.; et al. Genetic Evidence That HNF-1alpha-Dependent Transcriptional Control of HNF-4alpha is Essential for Human Pancreatic Beta Cell Function. J. Clin. Investig. 2002, 110, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Kyithar, M.P.; Bonner, C.; Bacon, S.; Kilbride, S.M.; Schmid, J.; Graf, R.; Prehn, J.H.M.; Byrne, M.M. Effects of Hepatocyte Nuclear Factor-1A and -4A on Pancreatic Stone Protein/Regenerating Protein and C-Reactive Protein Gene Expression: Implications for Maturity-Onset Diabetes of the Young. J. Transl. Med. 2013, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.-X.; Ling, Y.; Wang, H.-Y. Role of Nonresolving Inflammation in Hepatocellular Carcinoma Development and Progression. NPJ Precis. Oncol. 2018, 2. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-Signaling Pathway in Cancer. OncoTargets Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.-X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef]
- O’Rourke, J.M.; Sagar, V.M.; Shah, T.; Shetty, S. Carcinogenesis on the Background of Liver Fibrosis: Implications for the Management of Hepatocellular Cancer. World J. Gastroenterol. 2018, 24, 4436–4447. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Maiese, K. The Src Homology 2 Domain Tyrosine Phosphatases SHP-1 and SHP-2: Diversified Control of Cell Growth, Inflammation, and Injury. Histol. Histopathol. 2007, 22, 1251–1267. [Google Scholar] [CrossRef]
- Bao, C.; Li, Y.; Huan, L.; Zhang, Y.; Zhao, F.; Wang, Q.; Liang, L.; Ding, J.; Liu, L.; Chen, T.; et al. NF-ΚB Signaling Relieves Negative Regulation by MiR-194 in Hepatocellular Carcinoma by Suppressing the Transcription Factor HNF-1α. Sci. Signal. 2015, 8, ra75. [Google Scholar] [CrossRef]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-Redundant Functions of EMT Transcription Factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Liu, Y.; Xue, M.; Liu, H.; Du, S.; Zhang, L.; Wang, P. Synergistic Action of Master Transcription Factors Controls Epithelial-to-Mesenchymal Transition. Nucleic Acids Res. 2016, 44, 2514–2527. [Google Scholar] [CrossRef]
- Santangelo, L.; Marchetti, A.; Cicchini, C.; Conigliaro, A.; Conti, B.; Mancone, C.; Bonzo, J.A.; Gonzalez, F.J.; Alonzi, T.; Amicone, L.; et al. The Stable Repression of Mesenchymal Program is Required for Hepatocyte Identity: A Novel Role for Hepatocyte Nuclear Factor 4α. Hepatology 2011, 53, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, A.; Kannari, M.; Tsuchida, Y.; Sasaki, S.; Saito, C.; Matsuta, T.; Maeda, T.; Akiyama, M.; Nakamura, T.; Sakaguchi, M.; et al. An HNF4α-MicroRNA-194/192 Signaling Axis Maintains Hepatic Cell Function. J. Biol. Chem. 2017, 292, 10574–10585. [Google Scholar] [CrossRef]
- Pelletier, L.; Rebouissou, S.; Vignjevic, D.; Bioulac-Sage, P.; Zucman-Rossi, J. HNF1α Inhibition Triggers Epithelial-Mesenchymal Transition in Human Liver Cancer Cell Lines. BMC Cancer 2011, 11, 427. [Google Scholar] [CrossRef]
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; de Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The Snail Repressor Recruits EZH2 to Specific Genomic Sites through the Enrollment of the LncRNA HOTAIR in Epithelial-to-Mesenchymal Transition. Oncogene 2017, 36, 942–955. [Google Scholar] [CrossRef]
- Bisceglia, F.; Battistelli, C.; Noce, V.; Montaldo, C.; Zammataro, A.; Strippoli, R.; Tripodi, M.; Amicone, L.; Marchetti, A. TGFβ Impairs HNF1α Functional Activity in Epithelial-to-Mesenchymal Transition Interfering with the Recruitment of CBP/P300 Acetyltransferases. Front. Pharmacol. 2019, 10, 942. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the Global Map of Adipogenesis and Beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef]
- Panasyuk, G.; Espeillac, C.; Chauvin, C.; Pradelli, L.A.; Horie, Y.; Suzuki, A.; Annicotte, J.-S.; Fajas, L.; Foretz, M.; Verdeguer, F.; et al. PPARγ Contributes to PKM2 and HK2 Expression in Fatty Liver. Nat. Commun. 2012, 3, 672. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and Liver Cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome Activation and Function in Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Schnabl, B. Intestinal Microbiota and Nonalcoholic Steatohepatitis. Curr. Opin. Gastroenterol. 2017, 33, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The Immunology of Hepatocellular Carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.; Akiyama, T.E.; Kudo, G.; Gonzalez, F.J. Hepatic Expression of Cytochrome P450s in Hepatocyte Nuclear Factor 1-Alpha (HNF1alpha)-Deficient Mice. Biochem. Pharmacol. 2003, 66, 2011–2020. [Google Scholar] [CrossRef]
- Shih, D.Q.; Bussen, M.; Sehayek, E.; Ananthanarayanan, M.; Shneider, B.L.; Suchy, F.J.; Shefer, S.; Bollileni, J.S.; Gonzalez, F.J.; Breslow, J.L.; et al. Hepatocyte Nuclear Factor-1alpha is an Essential Regulator of Bile Acid and Plasma Cholesterol Metabolism. Nat. Genet. 2001, 27, 375–382. [Google Scholar] [CrossRef]
- Ni, Q.; Ding, K.; Wang, K.-Q.; He, J.; Yin, C.; Shi, J.; Zhang, X.; Xie, W.-F.; Shi, Y.-Q. Deletion of HNF1α in Hepatocytes Results in Fatty Liver-Related Hepatocellular Carcinoma in Mice. FEBS Lett. 2017, 591, 1947–1957. [Google Scholar] [CrossRef]
- Hayhurst, G.P.; Lee, Y.H.; Lambert, G.; Ward, J.M.; Gonzalez, F.J. Hepatocyte Nuclear Factor 4alpha (Nuclear Receptor 2A1) is Essential for Maintenance of Hepatic Gene Expression and Lipid Homeostasis. Mol. Cell. Biol. 2001, 21, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Ma, H.; Ge, X.; Edwards, P.A.; Zhang, Y. Hepatic Hepatocyte Nuclear Factor 4α is Essential for Maintaining Triglyceride and Cholesterol Homeostasis. Arter. Thromb. Vasc. Biol. 2011, 31, 328–336. [Google Scholar] [CrossRef]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A Metabolic Stress-Inducible MiR-34a-HNF4α Pathway Regulates Lipid and Lipoprotein Metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef]
- Fernández-Hernando, C.; Suárez, Y.; Rayner, K.J.; Moore, K.J. MicroRNAs in Lipid Metabolism. Curr. Opin. Lipidol. 2011, 22, 86–92. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-Genome Mutational Landscape and Characterization of Noncoding and Structural Mutations in Liver Cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Bacq, Y.; Jacquemin, E.; Balabaud, C.; Jeannot, E.; Scotto, B.; Branchereau, S.; Laurent, C.; Bourlier, P.; Pariente, D.; de Muret, A.; et al. Familial Liver Adenomatosis Associated with Hepatocyte Nuclear Factor 1alpha Inactivation. Gastroenterology 2003, 125, 1470–1475. [Google Scholar] [CrossRef]
- Bluteau, O.; Jeannot, E.; Bioulac-Sage, P.; Marqués, J.M.; Blanc, J.-F.; Bui, H.; Beaudoin, J.-C.; Franco, D.; Balabaud, C.; Laurent-Puig, P.; et al. Bi-Allelic Inactivation of TCF1 in Hepatic Adenomas. Nat. Genet. 2002, 32, 312–315. [Google Scholar] [CrossRef]
- Hechtman, J.F.; Abou-Alfa, G.K.; Stadler, Z.K.; Mandelker, D.L.; Roehrl, M.H.A.; Zehir, A.; Vakiani, E.; Middha, S.; Klimstra, D.S.; Shia, J. Somatic HNF1A Mutations in the Malignant Transformation of Hepatocellular Adenomas: A Retrospective Analysis of Data from MSK-IMPACT and TCGA. Hum. Pathol. 2019, 83, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.-H.; Deng, L.-F.; Chen, F.; Ding, K.; Chen, W.-S.; Xie, W.-F.; Zhang, X.P. Q511L Mutation of HNF1α in Hepatocellular Carcinoma Suppresses the Transcriptional Activity and the Anti-Tumor Effect of HNF1α. Biochem. Biophys. Res. Commun. 2018, 495, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Rebouissou, S.; Vasiliu, V.; Thomas, C.; Bellanné-Chantelot, C.; Bui, H.; Chrétien, Y.; Timsit, J.; Rosty, C.; Laurent-Puig, P.; Chauveau, D.; et al. Germline Hepatocyte Nuclear Factor 1alpha and 1beta Mutations in Renal Cell Carcinomas. Hum. Mol. Genet. 2005, 14, 603–614. [Google Scholar] [CrossRef]
- Laurent-Puig, P.; Plomteux, O.; Bluteau, O.; Zinzindohoué, F.; Jeannot, E.; Dahan, K.; Kartheuser, A.; Chapusot, C.; Cugnenc, P.-H.; Zucman-Rossi, J. Frequent Mutations of Hepatocyte Nuclear Factor 1 in Colorectal Cancer with Microsatellite Instability. Gastroenterology 2003, 124, 1311–1314. [Google Scholar] [CrossRef]
- Raft, M.B.; Jørgensen, E.N.; Vainer, B. Gene Mutations in Hepatocellular Adenomas. Histopathology 2015, 66, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Bioulac-Sage, P.; Zucman-Rossi, J. Hepatocellular Benign Tumors-from Molecular Classification to Personalized Clinical Care. Gastroenterology 2013, 144, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Tward, A.D.; Jones, K.D.; Yant, S.; Cheung, S.T.; Fan, S.T.; Chen, X.; Kay, M.A.; Wang, R.; Bishop, J.M. Distinct Pathways of Genomic Progression to Benign and Malignant Tumors of the Liver. Proc. Natl. Acad. Sci. USA 2007, 104, 14771–14776. [Google Scholar] [CrossRef]
- Ng, N.H.J.; Jasmen, J.B.; Lim, C.S.; Lau, H.H.; Krishnan, V.G.; Kadiwala, J.; Kulkarni, R.N.; Ræder, H.; Vallier, L.; Hoon, S.; et al. HNF4A Haploinsufficiency in MODY1 Abrogates Liver and Pancreas Differentiation from Patient-Derived Induced Pluripotent Stem Cells. iScience 2019, 16, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Thymiakou, E.; Othman, A.; Hornemann, T.; Kardassis, D. Defects in High Density Lipoprotein Metabolism and Hepatic Steatosis in Mice with Liver-Specific Ablation of Hepatocyte Nuclear Factor 4A. Metabolism 2020, 110, 154307. [Google Scholar] [CrossRef]
- Huck, I.; Gunewardena, S.; Espanol-Suner, R.; Willenbring, H.; Apte, U. Hepatocyte Nuclear Factor 4 Alpha Activation is Essential for Termination of Liver Regeneration in Mice. Hepatology 2019, 70, 666–681. [Google Scholar] [CrossRef]
- Dubois, V.; Gheeraert, C.; Vankrunkelsven, W.; Dubois-Chevalier, J.; Dehondt, H.; Bobowski-Gerard, M.; Vinod, M.; Zummo, F.P.; Güiza, F.; Ploton, M.; et al. Endoplasmic Reticulum Stress Actively Suppresses Hepatic Molecular Identity in Damaged Liver. Mol. Syst. Biol. 2020, 16, e9156. [Google Scholar] [CrossRef]
- Joo, M.S.; Koo, J.H.; Kim, T.H.; Kim, Y.S.; Kim, S.G. LRH1-Driven Transcription Factor Circuitry for Hepatocyte Identity: Super-Enhancer Cistromic Analysis. EBioMedicine 2019, 40, 488–503. [Google Scholar] [CrossRef]
- Aydin, Y.; Kurt, R.; Song, K.; Lin, D.; Osman, H.; Youngquist, B.; Scott, J.W.; Shores, N.J.; Thevenot, P.; Cohen, A.; et al. Hepatic Stress Response in HCV Infection Promotes STAT3-Mediated Inhibition of HNF4A-MiR-122 Feedback Loop in Liver Fibrosis and Cancer Progression. Cancers 2019, 11, 1407. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, I.; Dafou, D.; Vassilaki, N.; Mavromara, P.; Hadzopoulou-Cladaras, M. Hepatitis C Virus Suppresses Hepatocyte Nuclear Factor 4 Alpha, a Key Regulator of Hepatocellular Carcinoma. Int. J. Biochem. Cell Biol. 2016, 78, 315–326. [Google Scholar] [CrossRef]
- Coulouarn, C.; Factor, V.M.; Andersen, J.B.; Durkin, M.E.; Thorgeirsson, S.S. Loss of MiR-122 Expression in Liver Cancer Correlates with Suppression of the Hepatic Phenotype and Gain of Metastatic Properties. Oncogene 2009, 28, 3526–3536. [Google Scholar] [CrossRef]
- Piccolo, P.; Annunziata, P.; Soria, L.R.; Attanasio, S.; Barbato, A.; Castello, R.; Carissimo, A.; Quagliata, L.; Terracciano, L.M.; Brunetti-Pierri, N. Down-Regulation of Hepatocyte Nuclear Factor-4α and Defective Zonation in Livers Expressing Mutant Z A1-Antitrypsin. Hepatology 2017, 66, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.-F.; Ding, J.; Yin, C.; Zhong, W.; Wu, K.; Zeng, X.; Yang, W.; Chen, Y.-X.; Zhang, J.-P.; Zhang, X.; et al. Hepatocyte Nuclear Factor 4 Alpha Suppresses the Development of Hepatocellular Carcinoma. Cancer Res. 2010, 70, 7640–7651. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-W.; Reebye, V.; Czysz, K.; Ciriello, S.; Dorman, S.; Reccia, I.; Lai, H.-S.; Peng, L.; Kostomitsopoulos, N.; Nicholls, J.; et al. Liver Activation of Hepatocellular Nuclear Factor-4α by Small Activating RNA Rescues Dyslipidemia and Improves Metabolic Profile. Mol. Ther. Nucleic Acids 2020, 19, 361–370. [Google Scholar] [CrossRef]
- Tung, J.Y.-L.; Boodhansingh, K.; Stanley, C.A.; de León, D.D. Clinical Heterogeneity of Hyperinsulinism Due to HNF1A and HNF4A Mutations. Pediatr. Diabetes 2018, 19, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, K.; Gupta, S. Cancer Epigenetics: An Introduction. Methods Mol. Biol. 2015, 1238, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Adams, A.; Roberts, B.; O’Neil, M.; Vittal, A.; Schmitt, T.; Kumer, S.; Cox, J.; Li, Z.; Weinman, S.A.; et al. Protein Arginine Methyl Transferase 1- and Jumonji C Domain-Containing Protein 6-Dependent Arginine Methylation Regulate Hepatocyte Nuclear Factor 4 Alpha Expression and Hepatocyte Proliferation in Mice. Hepatology 2018, 67, 1109–1126. [Google Scholar] [CrossRef]
- Zheng, B.-N.; Ding, C.-H.; Chen, S.-J.; Zhu, K.; Shao, J.; Feng, J.; Xu, W.-P.; Cai, L.-Y.; Zhu, C.-P.; Duan, W.; et al. Targeting PRMT5 Activity Inhibits the Malignancy of Hepatocellular Carcinoma by Promoting the Transcription of HNF4α. Theranostics 2019, 9, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Ruoß, M.; Damm, G.; Vosough, M.; Ehret, L.; Grom-Baumgarten, C.; Petkov, M.; Naddalin, S.; Ladurner, R.; Seehofer, D.; Nussler, A.; et al. Epigenetic Modifications of the Liver Tumor Cell Line HepG2 Increase Their Drug Metabolic Capacity. Int. J. Mol. Sci. 2019, 20, 347. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Wong, J.C.H.; Wang, E.Y.; Lotto, J.; Kim, D.; Cheng, J.-C.; Mingay, M.; Cullum, R.; Moudgil, V.; Ahmed, N.; et al. Hepatocyte Nuclear Factor 4-Alpha is Essential for the Active Epigenetic State at Enhancers in Mouse Liver. Hepatology 2019, 70, 1360–1376. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Xi, Y.; Zhu, W.-N.; Zeng, C.; Zhang, Z.-Q.; Guo, Z.-C.; Hao, D.-L.; Liu, G.; Feng, L.; Chen, H.-Z.; et al. Positive Regulation of Hepatic MiR-122 Expression by HNF4α. J. Hepatol. 2011, 55, 602–611. [Google Scholar] [CrossRef]
- Yang, G.; Zhang, M.; Zhao, Y.; Pan, Y.; Kan, M.; Li, J.; He, K.; Zhang, X. HNF-4α Inhibits Hepatocellular Carcinoma Cell Proliferation through Mir-122-Adam17 Pathway. PLoS ONE 2020, 15, e0230450. [Google Scholar] [CrossRef]
- Wang, S.-C.; Lin, X.-L.; Li, J.; Zhang, T.-T.; Wang, H.-Y.; Shi, J.-W.; Yang, S.; Zhao, W.-T.; Xie, R.-Y.; Wei, F.; et al. MicroRNA-122 Triggers Mesenchymal-Epithelial Transition and Suppresses Hepatocellular Carcinoma Cell Motility and Invasion by Targeting RhoA. PLoS ONE 2014, 9, e101330. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, H.-O.; Liu, Y.-D.; Liu, W.-S.; Pan, D.; Zhang, W.-J.; Yang, L.; Fu, Q.; Xu, J.-J.; Gu, J.-X. Decreased Expression of Hepatocyte Nuclear Factor 4α (Hnf4α)/MicroRNA-122 (MiR-122) Axis in Hepatitis B Virus-Associated Hepatocellular Carcinoma Enhances Potential Oncogenic GALNT10 Protein Activity. J. Biol. Chem. 2015, 290, 1170–1185. [Google Scholar] [CrossRef]
- Yang, Y.M.; Lee, C.G.; Koo, J.H.; Kim, T.H.; Lee, J.M.; An, J.; Kim, K.M.; Kim, S.G. Gα12 Overexpressed in Hepatocellular Carcinoma Reduces MicroRNA-122 Expression via HNF4α Inactivation, Which Causes c-Met Induction. Oncotarget 2015, 6, 19055–19069. [Google Scholar] [CrossRef]
- Yin, C.; Wang, P.-Q.; Xu, W.-P.; Yang, Y.; Zhang, Q.; Ning, B.-F.; Zhang, P.-P.; Zhou, W.-P.; Xie, W.-F.; Chen, W.-S.; et al. Hepatocyte Nuclear Factor-4α Reverses Malignancy of Hepatocellular Carcinoma through Regulating MiR-134 in the DLK1-DIO3 Region. Hepatology 2013, 58, 1964–1976. [Google Scholar] [CrossRef]
- Ning, B.-F.; Ding, J.; Liu, J.; Yin, C.; Xu, W.-P.; Cong, W.-M.; Zhang, Q.; Chen, F.; Han, T.; Deng, X.; et al. Hepatocyte Nuclear Factor 4α-Nuclear Factor-ΚB Feedback Circuit Modulates Liver Cancer Progression. Hepatology 2014, 60, 1607–1619. [Google Scholar] [CrossRef]
- Gougelet, A.; Sartor, C.; Bachelot, L.; Godard, C.; Marchiol, C.; Renault, G.; Tores, F.; Nitschke, P.; Cavard, C.; Terris, B.; et al. Antitumour Activity of an Inhibitor of MiR-34a in Liver Cancer with β-Catenin-Mutations. Gut 2016, 65, 1024–1034. [Google Scholar] [CrossRef]
- Li, Z.; Chen, H. MiR-34a Inhibits Proliferation, Migration and Invasion of Paediatric Neuroblastoma Cells via Targeting HNF4α. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3072–3078. [Google Scholar] [CrossRef]
- Deng, D.; Yang, S.; Wang, X. Long Non-Coding RNA SNHG16 Regulates Cell Behaviors through MiR-542-3p/HNF4α Axis via RAS/RAF/MEK/ERK Signaling Pathway in Pediatric Neuroblastoma Cells. Biosci. Rep. 2020, 40, BSR20200723. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Dai, R.; Chen, Y.; Guo, H.; Han, Y.; Zhang, Y. LncRNA LINP1 Regulates Acute Myeloid Leukemia Progression via HNF4α/AMPK/WNT5A Signaling Pathway. Hematol. Oncol. 2019, 37, 474–482. [Google Scholar] [CrossRef]
- Cicchini, C.; de Nonno, V.; Battistelli, C.; Cozzolino, A.M.; de Santis Puzzonia, M.; Ciafrè, S.A.; Brocker, C.; Gonzalez, F.J.; Amicone, L.; Tripodi, M. Epigenetic Control of EMT/MET Dynamics: HNF4α Impacts DNMT3s through MiRs-29. Biochim. Biophys. Acta 2015, 1849, 919–929. [Google Scholar] [CrossRef]
- Battistelli, C.; Sabarese, G.; Santangelo, L.; Montaldo, C.; Gonzalez, F.J.; Tripodi, M.; Cicchini, C. The LncRNA HOTAIR Transcription is Controlled by HNF4α-Induced Chromatin Topology Modulation. Cell Death Differ. 2019, 26, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Battistelli, C.; Garbo, S.; Riccioni, V.; Montaldo, C.; Santangelo, L.; Vandelli, A.; Strippoli, R.; Tartaglia, G.G.; Tripodi, M.; Cicchini, C. Design and Functional Validation of a Mutant Variant of the LncRNA HOTAIR to Counteract Snail Function in Epithelial-to-Mesenchymal Transition. Cancer Res. 2021, 81, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.-H.; Yin, C.; Chen, S.-J.; Wen, L.-Z.; Ding, K.; Lei, S.-J.; Liu, J.-P.; Wang, J.; Chen, K.-X.; Jiang, H.-L.; et al. The HNF1α-Regulated LncRNA HNF1A-AS1 Reverses the Malignancy of Hepatocellular Carcinoma by Enhancing the Phosphatase Activity of SHP-1. Mol. Cancer 2018, 17, 63. [Google Scholar] [CrossRef]
- Ladeiro, Y.; Couchy, G.; Balabaud, C.; Bioulac-Sage, P.; Pelletier, L.; Rebouissou, S.; Zucman-Rossi, J. MicroRNA Profiling in Hepatocellular Tumors is Associated with Clinical Features and Oncogene/Tumor Suppressor Gene Mutations. Hepatology 2008, 47, 1955–1963. [Google Scholar] [CrossRef]
- Zeng, X.; Lin, Y.; Yin, C.; Zhang, X.; Ning, B.-F.; Zhang, Q.; Zhang, J.-P.; Qiu, L.; Qin, X.-R.; Chen, Y.-X.; et al. Recombinant Adenovirus Carrying the Hepatocyte Nuclear Factor-1alpha Gene Inhibits Hepatocellular Carcinoma Xenograft Growth in Mice. Hepatology 2011, 54, 2036–2047. [Google Scholar] [CrossRef]
- Krützfeldt, J.; Rösch, N.; Hausser, J.; Manoharan, M.; Zavolan, M.; Stoffel, M. MicroRNA-194 is a Target of Transcription Factor 1 (Tcf1, HNF1α) in Adult Liver and Controls Expression of Frizzled-6. Hepatology 2012, 55, 98–107. [Google Scholar] [CrossRef]
- Petruzziello, A. Epidemiology of Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) Related Hepatocellular Carcinoma. Open Virol. J. 2018, 12, 26–32. [Google Scholar] [CrossRef]
- Honda, M.; Shirasaki, T.; Terashima, T.; Kawaguchi, K.; Nakamura, M.; Oishi, N.; Wang, X.; Shimakami, T.; Okada, H.; Arai, K.; et al. Hepatitis B Virus (HBV) Core-Related Antigen During Nucleos(t)Ide Analog Therapy is Related to Intra-Hepatic HBV Replication and Development of Hepatocellular Carcinoma. J. Infect. Dis. 2016, 213, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, J.; Kasai, H.; Yoshimura, K.; Otoguro, T.; Watashi, K.; Wakita, T.; Yamashita, A.; Tanaka, T.; Takeda, S.; Moriishi, K. Hepatitis B Virus Prevents Excessive Viral Production via Reduction of Cell Death-Inducing DFF45-like Effectors. J. Gen. Virol. 2017, 98, 1762–1773. [Google Scholar] [CrossRef]
- Park, S.; Ha, Y.N.; Dezhbord, M.; Lee, A.R.; Park, E.-S.; Park, Y.K.; Won, J.; Kim, N.Y.; Choo, S.Y.; Shin, J.J.; et al. Suppression of Hepatocyte Nuclear Factor 4 α by Long-Term Infection of Hepatitis B Virus Contributes to Tumor Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 948. [Google Scholar] [CrossRef]
- Yu, X.; Mertz, J.E. Distinct Modes of Regulation of Transcription of Hepatitis B Virus by the Nuclear Receptors HNF4alpha and COUP-TF1. J. Virol. 2003, 77, 2489–2499. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, J.; Ou, J. Regulation of Hepatitis B Virus Core Promoter by Transcription Factors HNF1 and HNF4 and the Viral X Protein. J. Virol. 2004, 78, 6908–6914. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Chen, E.; Liu, C.; Huang, F.; Zhou, T.; He, F.; Liu, L.; Liu, F.; Tang, H. The Correlation of Hepatocyte Nuclear Factor 4 Alpha and 3 Beta with Hepatitis B Virus Replication in the Liver of Chronic Hepatitis B Patients. J. Viral Hepat. 2009, 16, 537–546. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Chen, E.-Q.; Liu, L.; Zhou, T.-Y.; Liu, C.; Cheng, X.; Liu, F.-J.; Tang, H. Inhibition of Hepatitis B Virus Replication by Hepatocyte Nuclear Factor 4-Alpha Specific Short Hairpin RNA. Liver Int. 2012, 32, 742–751. [Google Scholar] [CrossRef]
- Wang, S.-H.; Yeh, S.-H.; Lin, W.-H.; Yeh, K.-H.; Yuan, Q.; Xia, N.-S.; Chen, D.-S.; Chen, P.-J. Estrogen Receptor α Represses Transcription of HBV Genes via Interaction with Hepatocyte Nuclear Factor 4α. Gastroenterology 2012, 142, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.-Q.; Sun, H.; Feng, P.; Gong, D.-Y.; Liu, C.; Bai, L.; Yang, W.-B.; Lei, X.-Z.; Chen, L.-Y.; Huang, F.-J.; et al. Study of the Expression Levels of Hepatocyte Nuclear Factor 4 Alpha and 3 Beta in Patients with Different Outcome of HBV Infection. Virol. J. 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Nong, Y.; Shi, Y.; Liu, M.; Yan, L.; Shang, J.; Huang, F.; Lin, Y.; Tang, H. Luteolin Inhibits Hepatitis B Virus Replication through Extracellular Signal-Regulated Kinase-Mediated Down-Regulation of Hepatocyte Nuclear Factor 4α Expression. Mol. Pharm. 2016, 13, 568–577. [Google Scholar] [CrossRef]
- Park, Y.K.; Park, E.-S.; Kim, D.H.; Ahn, S.H.; Park, S.H.; Lee, A.R.; Park, S.; Kang, H.S.; Lee, J.-H.; Kim, J.M.; et al. Cleaved C-FLIP Mediates the Antiviral Effect of TNF-α against Hepatitis B Virus by Dysregulating Hepatocyte Nuclear Factors. J. Hepatol. 2016, 64, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.-Q.; Cai, W.-T.; Wu, X.; Chen, Y.; Han, F.-M. Protocatechuic Acid Inhibits Hepatitis B Virus Replication by Activating ERK1/2 Pathway and down-Regulating HNF4α and HNF1α in Vitro. Life Sci. 2017, 180, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Y.; Li, Y.-Q.; Guo, Z.-W.; Zhou, X.-H.; Lu, M.-D.; Xue, T.-C.; Gao, B. ERK1/2-HNF4α Axis is Involved in Epigallocatechin-3-Gallate Inhibition of HBV Replication. Acta Pharmacol. Sin. 2020, 41, 278–285. [Google Scholar] [CrossRef]
- Li, J.; Liu, F.-W.; Wu, D.-B.; Chen, E.-Q.; Chen, X.-J.; Chen, S.-C.; Liu, C.; Zhao, L.-S.; Tang, H.; Zhou, T.-Y. TRAIL Inhibits HBV Replication and Expression by Down-Regulating Liver-Enriched Transcription Factors. Arab. J. Gastroenterol. 2020, 21, 169–173. [Google Scholar] [CrossRef]
- Lin, J.; Gu, C.; Shen, Z.; Liu, Y.; Wang, W.; Tao, S.; Cui, X.; Liu, J.; Xie, Y. Hepatocyte Nuclear Factor 1α Downregulates HBV Gene Expression and Replication by Activating the NF-ΚB Signaling Pathway. PLoS ONE 2017, 12, e0174017. [Google Scholar] [CrossRef]
- Zhou, D.X.; Yen, T.S. The Ubiquitous Transcription Factor Oct-1 and the Liver-Specific Factor HNF-1 are Both Required to Activate Transcription of a Hepatitis B Virus Promoter. Mol. Cell. Biol. 1991, 11, 1353–1359. [Google Scholar] [CrossRef]
- Pan, Y.; Ke, Z.; Ye, H.; Sun, L.; Ding, X.; Shen, Y.; Zhang, R.; Yuan, J. Saikosaponin C Exerts Anti-HBV Effects by Attenuating HNF1α and HNF4α Expression to Suppress HBV PgRNA Synthesis. Inflamm. Res. 2019, 68, 1025–1034. [Google Scholar] [CrossRef]
- Xia, C.; Tang, W.; Geng, P.; Zhu, H.; Zhou, W.; Huang, H.; Zhou, P.; Shi, X. Baicalin Down-Regulating Hepatitis B Virus Transcription Depends on the Liver-Specific HNF4α-HNF1α Axis. Toxicol. Appl. Pharmacol. 2020, 403, 115131. [Google Scholar] [CrossRef]
- Wang, Z.; Ceniccola, K.; Florea, L.; Wang, B.-D.; Lee, N.H.; Kumar, A. Viral Non-Coding RNA Inhibits HNF4α Expression in HCV Associated Hepatocellular Carcinoma. Infect. Agent Cancer 2015, 10, 19. [Google Scholar] [CrossRef]
- Levy, G.; Habib, N.; Guzzardi, M.A.; Kitsberg, D.; Bomze, D.; Ezra, E.; Uygun, B.E.; Uygun, K.; Trippler, M.; Schlaak, J.F.; et al. Nuclear Receptors Control Pro-Viral and Antiviral Metabolic Responses to Hepatitis C Virus Infection. Nat. Chem. Biol. 2016, 12, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, H.; Qu, L.; Yao, W.; Cai, H.; Chen, L.; Peng, T. Hepatocyte Nuclear Factor 4α and Downstream Secreted Phospholipase A2 GXIIB Regulate Production of Infectious Hepatitis C Virus. J. Virol. 2014, 88, 612–627. [Google Scholar] [CrossRef] [PubMed]
- Foka, P.; Karamichali, E.; Dalagiorgou, G.; Serti, E.; Doumba, P.P.; Pissas, G.; Kakkanas, A.; Kazazi, D.; Kochlios, E.; Gaitanou, M.; et al. Hepatitis C Virus Modulates Lipid Regulatory Factor Angiopoietin-like 3 Gene Expression by Repressing HNF-1α Activity. J. Hepatol. 2014, 60, 30–38. [Google Scholar] [CrossRef]
- Matsui, C.; Shoji, I.; Kaneda, S.; Sianipar, I.R.; Deng, L.; Hotta, H. Hepatitis C Virus Infection Suppresses GLUT2 Gene Expression via Downregulation of Hepatocyte Nuclear Factor 1α. J. Virol. 2012, 86, 12903–12911. [Google Scholar] [CrossRef]
- Matsui, C.; Rosalyn Sianipar, I.; Minami, N.; Deng, L.; Hotta, H.; Shoji, I. A Single-Amino-Acid Mutation in Hepatitis C Virus NS5A Disrupts Physical and Functional Interaction with the Transcription Factor HNF-1α. J. Gen. Virol. 2015, 96, 2200–2205. [Google Scholar] [CrossRef] [PubMed]
- Matsui, C.; Deng, L.; Minami, N.; Abe, T.; Koike, K.; Shoji, I. Hepatitis C Virus NS5A Protein Promotes the Lysosomal Degradation of Hepatocyte Nuclear Factor 1α via Chaperone-Mediated Autophagy. J. Virol. 2018, 92, e00639-18. [Google Scholar] [CrossRef]
- Tanaka, T.; Jiang, S.; Hotta, H.; Takano, K.; Iwanari, H.; Sumi, K.; Daigo, K.; Ohashi, R.; Sugai, M.; Ikegame, C.; et al. Dysregulated Expression of P1 and P2 Promoter-Driven Hepatocyte Nuclear Factor-4alpha in the Pathogenesis of Human Cancer. J. Pathol. 2006, 208, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Kishimoto, T.; Nagai, Y.; Tanizawa, T.; Nakatani, Y.; Miyazaki, M.; Ishikura, H. The Expression of Hepatocyte Nuclear Factor-4alpha, a Developmental Regulator of Visceral Endoderm, Correlates with the Intestinal Phenotype of Gastric Adenocarcinomas. Pathology 2006, 38, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Kawasaki, T.; Ohashi, R.; Hasegawa, G.; Jiang, S.; Umezu, H.; Aoyagi, Y.; Iwanari, H.; Tanaka, T.; Hamakubo, T.; et al. Downregulated P1 Promoter-Driven Hepatocyte Nuclear Factor-4alpha Expression in Human Colorectal Carcinoma is a New Prognostic Factor against Liver Metastasis. Pathol. Int. 2007, 57, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, K.; Jankova, L.; Schnabl, J.M.; Pan, S.; Brelivet, Y.; Fung, C.L.-S.; Chan, C.; Dent, O.F.; Clarke, S.J.; Robertson, G.R.; et al. Src Tyrosine Kinase Phosphorylation of Nuclear Receptor HNF4α Correlates with Isoform-Specific Loss of HNF4α in Human Colon Cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2302–2307. [Google Scholar] [CrossRef] [PubMed]
- Qinyu, L.; Long, C.; Zhen-dong, D.; Min-min, S.; Wei-ze, W.; Wei-ping, Y.; Cheng-hong, P. FOXO6 Promotes Gastric Cancer Cell Tumorigenicity via Upregulation of C-Myc. FEBS Lett. 2013, 587, 2105–2111. [Google Scholar] [CrossRef]
- Chang, H.R.; Nam, S.; Kook, M.-C.; Kim, K.-T.; Liu, X.; Yao, H.; Jung, H.R.; Lemos, R.; Seo, H.H.; Park, H.S.; et al. HNF4α is a Therapeutic Target That Links AMPK to WNT Signalling in Early-Stage Gastric Cancer. Gut 2016, 65, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wei, X.; Wu, Z. HNF-4α Promotes Multidrug Resistance of Gastric Cancer Cells through the Modulation of Cell Apoptosis. Oncol. Lett. 2017, 14, 6477–6484. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xu, W.; Ji, S.; Qin, Y.; Liu, W.; Hu, Q.; Zhang, Z.; Liu, M.; Yu, X.; Xu, X. Role of Hepatocyte Nuclear Factor 4 Alpha in Cell Proliferation and Gemcitabine Resistance in Pancreatic Adenocarcinoma. Cancer Cell Int. 2019, 19, 49. [Google Scholar] [CrossRef]
- Cattin, A.-L.; Le Beyec, J.; Barreau, F.; Saint-Just, S.; Houllier, A.; Gonzalez, F.J.; Robine, S.; Pinçon-Raymond, M.; Cardot, P.; Lacasa, M.; et al. Hepatocyte Nuclear Factor 4alpha, a Key Factor for Homeostasis, Cell Architecture, and Barrier Function of the Adult Intestinal Epithelium. Mol. Cell. Biol. 2009, 29, 6294–6308. [Google Scholar] [CrossRef]
- Yao, H.S.; Wang, J.; Zhang, X.P.; Wang, L.Z.; Wang, Y.; Li, X.X.; Jin, K.Z.; Hu, Z.Q.; Wang, W.J. Hepatocyte Nuclear Factor 4α Suppresses the Aggravation of Colon Carcinoma. Mol. Carcinog. 2016, 55, 458–472. [Google Scholar] [CrossRef]
- Jin, L.; Pan, Y.-L.; Zhang, J.; Cao, P.-G. LncRNA HOTAIR Recruits SNAIL to Inhibit the Transcription of HNF4α and Promote the Viability, Migration, Invasion and EMT of Colorectal Cancer. Transl. Oncol. 2021, 14, 101036. [Google Scholar] [CrossRef]
- Schwartz, B.; Algamas-Dimantov, A.; Hertz, R.; Nataf, J.; Kerman, A.; Peri, I.; Bar-Tana, J. Inhibition of Colorectal Cancer by Targeting Hepatocyte Nuclear Factor-4alpha. Int. J. Cancer 2009, 124, 1081–1089. [Google Scholar] [CrossRef]
- Darsigny, M.; Babeu, J.-P.; Seidman, E.G.; Gendron, F.-P.; Levy, E.; Carrier, J.; Perreault, N.; Boudreau, F. Hepatocyte Nuclear Factor-4alpha Promotes Gut Neoplasia in Mice and Protects against the Production of Reactive Oxygen Species. Cancer Res. 2010, 70, 9423–9433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic Characterization of Human Colon and Rectal Cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef]
- Hu, Q.; Li, L.; Zou, X.; Xu, L.; Yi, P. Berberine Attenuated Proliferation, Invasion and Migration by Targeting the AMPK/HNF4α/WNT5A Pathway in Gastric Carcinoma. Front. Pharmacol. 2018, 9, 1150. [Google Scholar] [CrossRef]
- He, X.-X.; Luo, S.-S.; Qin, H.-Q.; Mo, X.-W. MicroRNA-766-3p-Mediated Downregulation of HNF4G Inhibits Proliferation in Colorectal Cancer Cells through the PI3K/AKT Pathway. Cancer Gene Ther. 2021. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.W.; Cowley, G.S.; Weir, B.A.; Boehm, J.S.; Rusin, S.; Scott, J.A.; East, A.; Ali, L.D.; Lizotte, P.H.; Wong, T.C.; et al. Systematic Investigation of Genetic Vulnerabilities across Cancer Cell Lines Reveals Lineage-Specific Dependencies in Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 12372–12377. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [CrossRef]
- Lu, Y.; Yu, Y.; Liu, F.; Han, Y.; Xue, H.; Sun, X.; Jiang, Y.; Tian, Z. LINC00511-Dependent Inhibition of IL-24 Contributes to the Oncogenic Role of HNF4α in Colorectal Cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G338–G350. [Google Scholar] [CrossRef]
- Brex, D.; Barbagallo, C.; Mirabella, F.; Caponnetto, A.; Battaglia, R.; Barbagallo, D.; Caltabiano, R.; Broggi, G.; Memeo, L.; Di Pietro, C.; et al. LINC00483 Has a Potential Tumor-Suppressor Role in Colorectal Cancer Through Multiple Molecular Axes. Front. Oncol. 2020, 10, 614455. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wu, K.; Zhang, L.; Zheng, S.; Wang, X.; Zuo, H.; Wu, X.; Tao, G.; Jiang, B.; Zhang, L. Long Non-Coding RNA LINC00858 Exerts a Tumor-Promoting Role in Colon Cancer via HNF4α and WNK2 Regulation. Cell. Oncol. 2020, 43, 297–310. [Google Scholar] [CrossRef]
- Babeu, J.-P.; Wilson, S.D.; Lambert, É.; Lévesque, D.; Boisvert, F.-M.; Boudreau, F. Quantitative Proteomics Identifies DNA Repair as a Novel Biological Function for Hepatocyte Nuclear Factor 4α in Colorectal Cancer Cells. Cancers 2019, 11, 626. [Google Scholar] [CrossRef]
- Vuong, L.M.; Chellappa, K.; Dhahbi, J.M.; Deans, J.R.; Fang, B.; Bolotin, E.; Titova, N.V.; Hoverter, N.P.; Spindler, S.R.; Waterman, M.L.; et al. Differential Effects of Hepatocyte Nuclear Factor 4α Isoforms on Tumor Growth and T-Cell Factor 4/AP-1 Interactions in Human Colorectal Cancer Cells. Mol. Cell. Biol. 2015, 35, 3471–3490. [Google Scholar] [CrossRef]
- Babeu, J.-P.; Jones, C.; Geha, S.; Carrier, J.C.; Boudreau, F. P1 Promoter-Driven HNF4α Isoforms are Specifically Repressed by β-Catenin Signaling in Colorectal Cancer Cells. J. Cell Sci. 2018, 131, jcs.214734. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.-D.; Zhou, L.-Y.; Tang, H. Hepatocyte Nuclear Factor 4α and Cancer-Related Cell Signaling Pathways: A Promising Insight into Cancer Treatment. Exp. Mol. Med. 2021, 53, 8–18. [Google Scholar] [CrossRef]
- Yuan, X.; Ta, T.C.; Lin, M.; Evans, J.R.; Dong, Y.; Bolotin, E.; Sherman, M.A.; Forman, B.M.; Sladek, F.M. Identification of an Endogenous Ligand Bound to a Native Orphan Nuclear Receptor. PLoS ONE 2009, 4, e5609. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiao, X.; Feng, K.; Wang, T.; Li, W.; Yuan, T.; Sun, X.; Sun, Q.; Xiang, H.; Wang, H. Berberine Moderates Glucose and Lipid Metabolism through Multipathway Mechanism. Evid. Based Complement. Altern. Med. 2011, 2011, 1–10. [Google Scholar] [CrossRef]
- Azmi, A.S.; Bao, G.W.; Gao, J.; Mohammad, R.M.; Sarkar, F.H. Network Insights into the Genes Regulated by Hepatocyte Nuclear Factor 4 in Response to Drug Induced Perturbations: A Review. Curr. Drug Discov. Technol. 2013, 10, 147–154. [Google Scholar] [CrossRef]
- Oggu, G.S.; Sasikumar, S.; Reddy, N.; Ella, K.K.R.; Rao, C.M.; Bokara, K.K. Gene Delivery Approaches for Mesenchymal Stem Cell Therapy: Strategies to Increase Efficiency and Specificity. Stem Cell Rev. 2017, 13, 725–740. [Google Scholar] [CrossRef]
- Wu, N.; Zhang, Y.-L.; Wang, H.-T.; Li, D.-W.; Dai, H.-J.; Zhang, Q.-Q.; Zhang, J.; Ma, Y.; Xia, Q.; Bian, J.-M.; et al. Overexpression of Hepatocyte Nuclear Factor 4α in Human Mesenchymal Stem Cells Suppresses Hepatocellular Carcinoma Development through Wnt/β-Catenin Signaling Pathway Downregulation. Cancer Biol. Ther. 2016, 17, 558–565. [Google Scholar] [CrossRef]
- Amer, M.H. Gene Therapy for Cancer: Present Status and Future Perspective. Mol. Cell. Ther. 2014, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Horisawa, K.; Udono, M.; Ohkawa, Y.; Suzuki, A. Prolonged Inhibition of Hepatocellular Carcinoma Cell Proliferation by Combinatorial Expression of Defined Transcription Factors. Cancer Sci. 2018, 109, 3543–3553. [Google Scholar] [CrossRef]
- Ding, K.; Wu, S.; Ying, W.; Pan, Q.; Li, X.; Zhao, D.; Li, X.; Zhao, Q.; Zhu, Y.; Ren, H.; et al. Leveraging a Multi-Omics Strategy for Prioritizing Personalized Candidate Mutation-Driver Genes: A Proof-of-Concept Study. Sci. Rep. 2015, 5, 17564. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teeli, A.S.; Łuczyńska, K.; Haque, E.; Gayas, M.A.; Winiarczyk, D.; Taniguchi, H. Disruption of Tumor Suppressors HNF4α/HNF1α Causes Tumorigenesis in Liver. Cancers 2021, 13, 5357. https://doi.org/10.3390/cancers13215357
Teeli AS, Łuczyńska K, Haque E, Gayas MA, Winiarczyk D, Taniguchi H. Disruption of Tumor Suppressors HNF4α/HNF1α Causes Tumorigenesis in Liver. Cancers. 2021; 13(21):5357. https://doi.org/10.3390/cancers13215357
Chicago/Turabian StyleTeeli, Aamir Salam, Kamila Łuczyńska, Effi Haque, Mohmmad Abrar Gayas, Dawid Winiarczyk, and Hiroaki Taniguchi. 2021. "Disruption of Tumor Suppressors HNF4α/HNF1α Causes Tumorigenesis in Liver" Cancers 13, no. 21: 5357. https://doi.org/10.3390/cancers13215357
APA StyleTeeli, A. S., Łuczyńska, K., Haque, E., Gayas, M. A., Winiarczyk, D., & Taniguchi, H. (2021). Disruption of Tumor Suppressors HNF4α/HNF1α Causes Tumorigenesis in Liver. Cancers, 13(21), 5357. https://doi.org/10.3390/cancers13215357