
Germline Predisposition to Pediatric Cancer, from Next Generation Sequencing to Medical Care

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
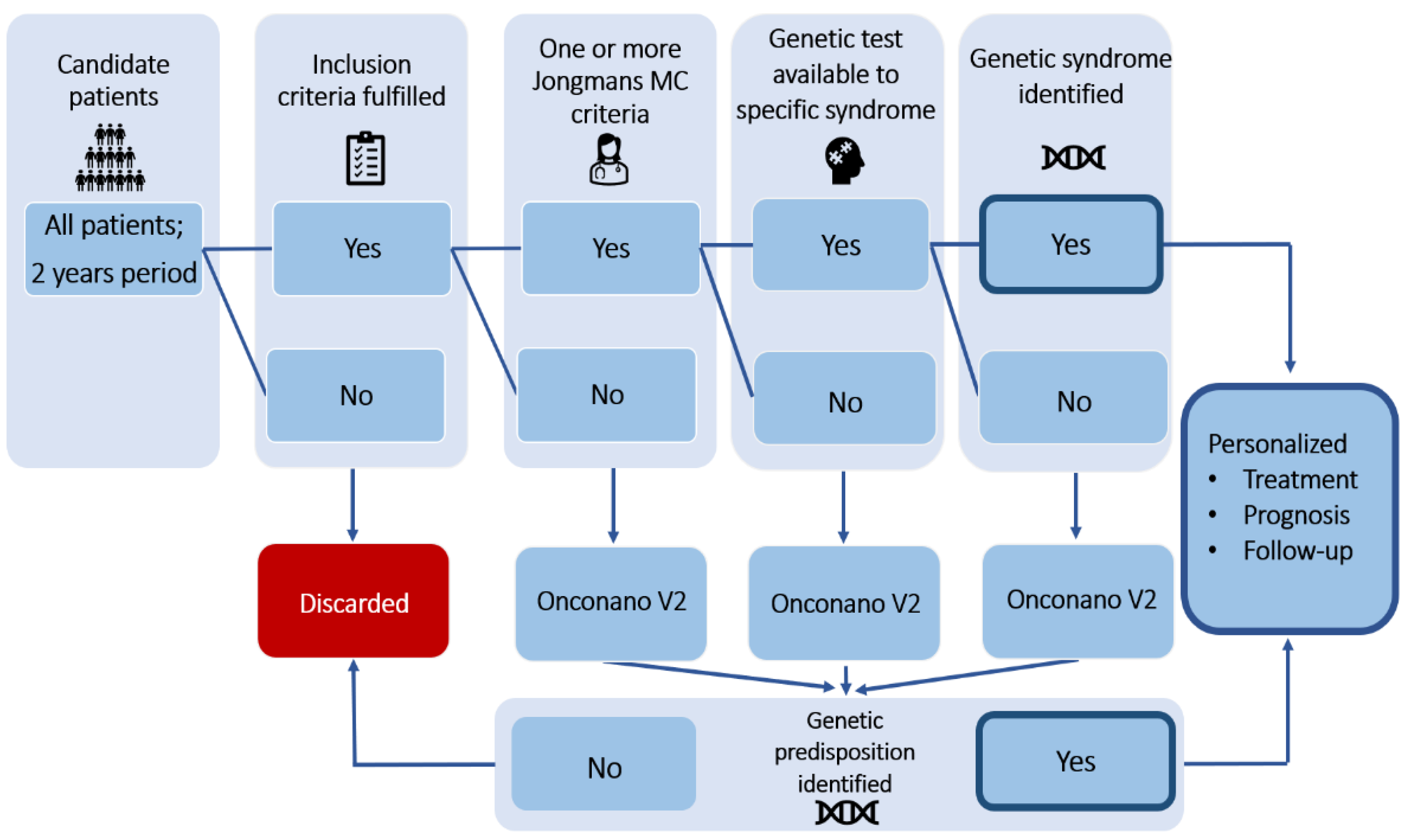
2. Materials and Methods
2.1. Patient Study Cohort
- −
- Age between 0 and 18 years old.
- −
- Final pathology diagnosis established.
- −
- Germline origin blood sample availability.
- −
- Patient clinical stability.
- −
- Patient voluntary agreement to participate having understood the information related to the study.
- −
- No exclusion criteria fulfilled.
- −
- Rejection of the study by the patient and/or family.
- −
- Unfavorable previous psychological evaluation.
- −
- Diagnosis of a benign tumor without any known genetic basis for its development.
2.2. NGS Panel, Sequencing and Analysis Features
3. Results
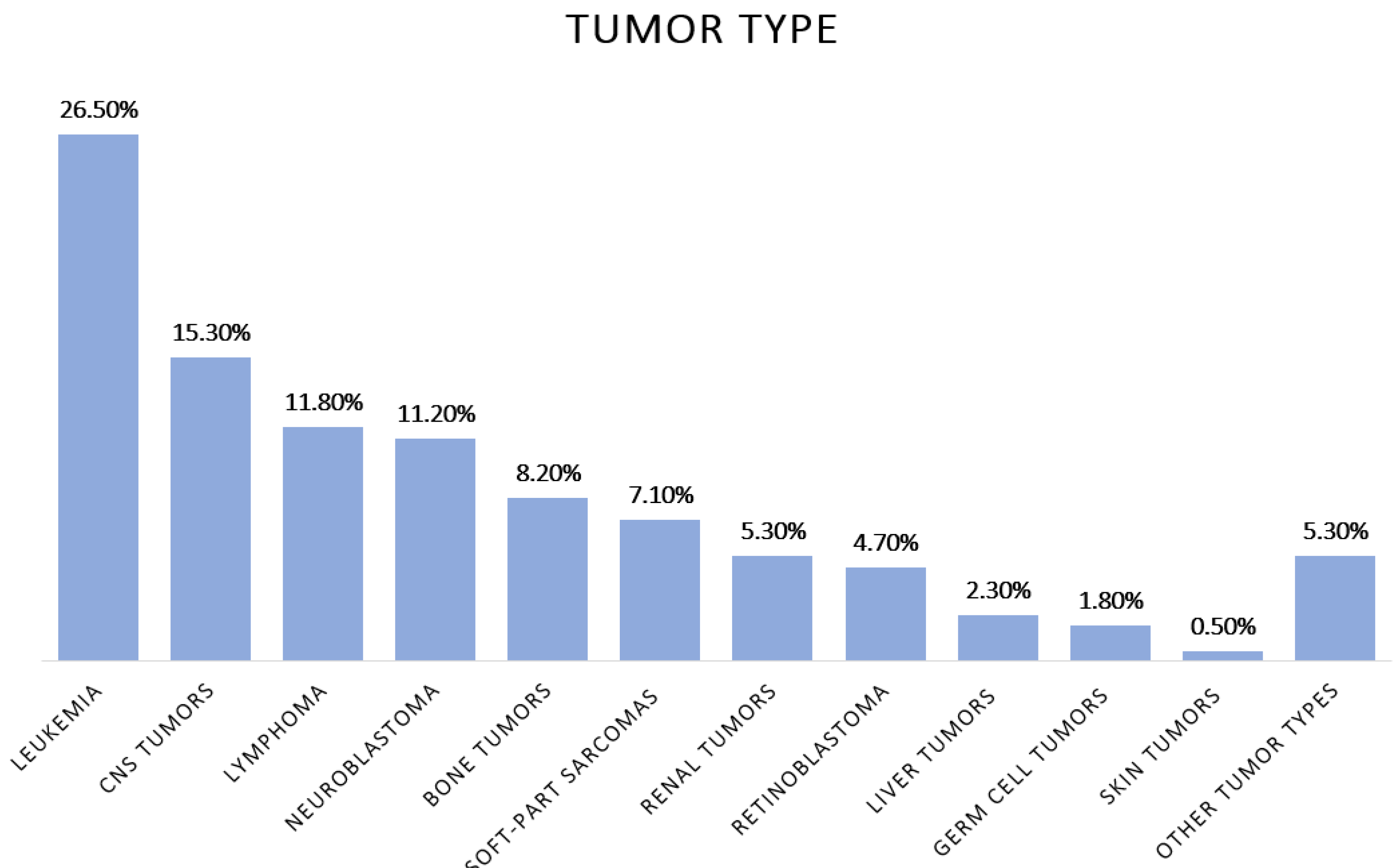
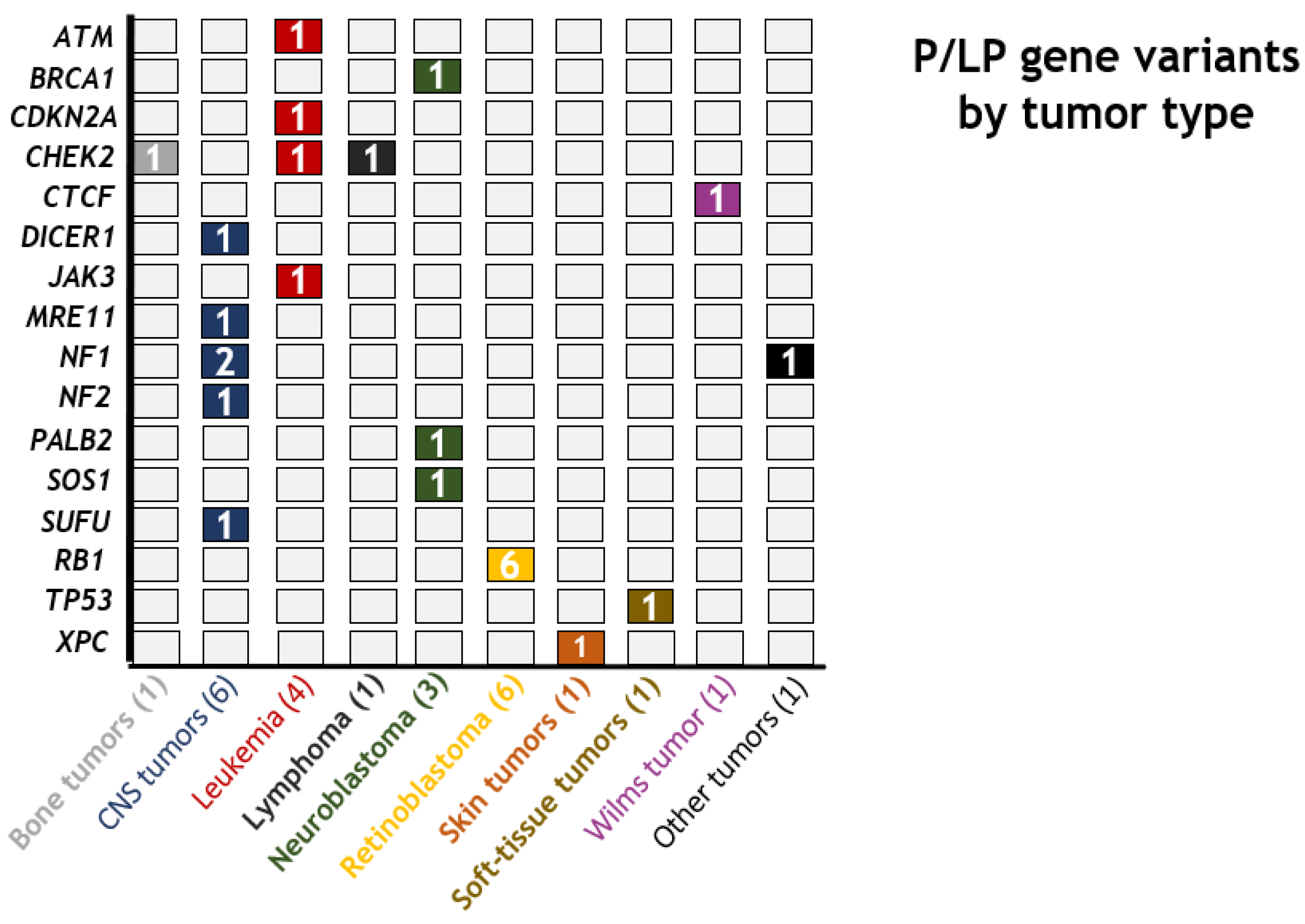
3.1. Patient Cohort and Genetic Variants Identified
3.2. Jongmans MC et al., 2016, Tool Evaluation
3.3. CTCF Variant c.1337-T>A and Wilms Predisposition
3.4. BRCA1 c.68_69del Variant and Neuroblastoma Susceptibility
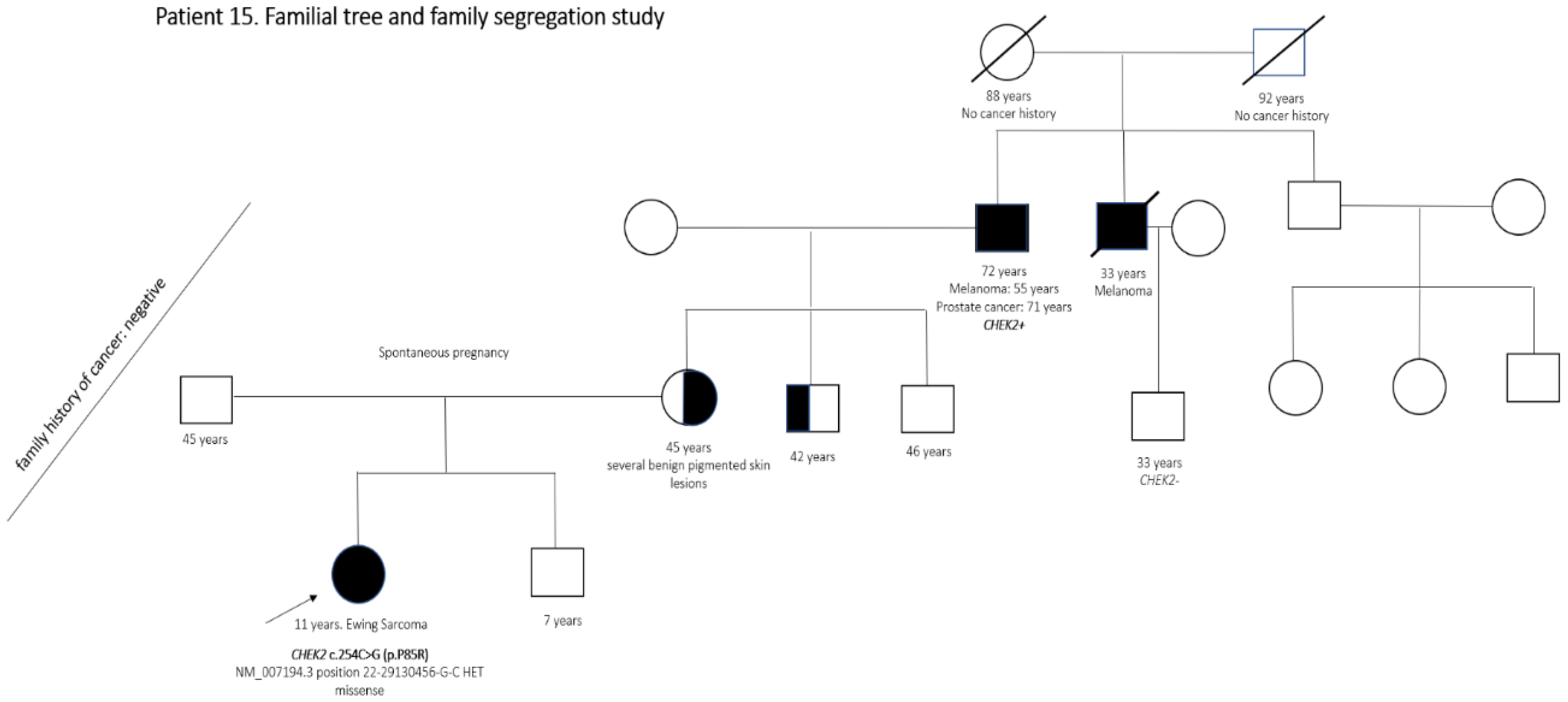
3.5. CHEK2 c.497A>G Variant and B-Cell ALL Risk
3.6. CDKN2A Deletion and Leukemia
3.7. JAK3 Mutations and Familial Leukemia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, K.L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic germline variants in 10, 389 adult cancers. Cell 2018, 173, 355–370. [Google Scholar] [PubMed] [Green Version]
- Scollon, S.; Anglin, A.K.; Thomas, M.; Turner, J.T.; Wolfe Schneider, K. A Comprehensive Review of Pediatric Tumors and Associated Cancer Predisposition Syndromes. J. Genet. Couns. 2017, 26, 387–434. [Google Scholar]
- Kentsis, A. Why do young people get cancer? Pediatr. Blood Cancer 2020, 67, e28335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [PubMed] [Green Version]
- Von Stedingk, K.; Stjernfelt, K.-J.; Kvist, A.; Wahlström, C.; Kristofersson, U.; Stenmark-Askmalm, M. Prevalence of germline pathogenic variants in 22 cancer susceptibility genes in Swedish pediatric cancer patients. Sci. Rep. 2021, 11, 5307. [Google Scholar] [CrossRef] [PubMed]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 15, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narod, S.A.; Stiller, C.; Lenoir, G.M. An estimate of the heritable fraction of childhood cancer. Br. J. Cancer 1991, 63, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Godley, L.A. Germline mutations in MDS/AML predisposition disorders. Curr. Opin. Hematol. 2021, 28, 86–93. [Google Scholar]
- Bloom, M.; Maciaszek, J.L.; Clark, M.E.; Pui, C.H.; Nichols, K.E. Recent advances in genetic predisposition to pediatric acute lymphoblastic leukemia. Expert Rev. Hematol. 2020, 13, 55–70. [Google Scholar] [PubMed]
- Ritenour, L.E.; Randall, M.P.; Bosse, K.R.; Diskin, S.J. Genetic susceptibility to neuroblastoma: Current knowledge and future directions. Cell Tissue Res. 2018, 372, 287–307. [Google Scholar]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugières, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar]
- Waszak, S.M.; Robinson, G.W.; Gudenas, B.L.; Smith, K.S.; Forget, A.; Kojic, M.; Garcia-Lopez, J.; Hadley, J.; Hamilton, K.V.; Indersie, E.; et al. Germline Elongator mutations in Sonic Hedgehog medulloblastoma. Nature 2020, 580, 396–401. [Google Scholar] [PubMed]
- Begemann, M.; Waszak, S.M.; Robinson, G.W.; Jäger, N.; Sharma, T.; Knopp, C.; Kraft, F.; Moser, O.; Mynarek, M.; Guerrini-Rousseau, L.; et al. Germline GPR161 Mutations Predispose to Pediatric Medulloblastoma. J. Clin. Oncol. 2020, 38, 43–50. [Google Scholar] [CrossRef]
- Mirabello, L.; Zhu, B.; Koster, R.; Karlins, E.; Dean, M.; Yeager, M.; Gianferante, M.; Spector, L.G.; Morton, L.M.; Karyadi, D.; et al. Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients with Osteosarcoma. JAMA Oncol. 2020, 6, 724–734. [Google Scholar] [PubMed]
- Benna, C.; Simioni, A.; Pasquali, S.; De Boni, D.; Rajendran, S.; Spiro, G.; Colombo, C.; Virgone, C.; DuBois, S.G.; Gronchi, A.; et al. Genetic susceptibility to bone and soft tissue sarcomas: A field synopsis and meta-analysis. Oncotarget 2018, 9, 18607–18626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargallo, P.; Yáñez, Y.; Juan, A.; Segura, V.; Balaguer, J.; Torres, B.; Oltra, S.; Castel, V.; Cañete, A. Review: Ewing Sarcoma Predisposition. Pathol. Oncol. Res. 2020, 26, 2057–2066. [Google Scholar] [PubMed]
- Capasso, M.; Montella, A.; Tirelli, M.; Maiorino, T.; Cantalupo, S.; Iolascon, A. Genetic Predisposition to Solid Pediatric Cancers. Front. Oncol. 2020, 10, 590033. [Google Scholar] [PubMed]
- Ripperger, T.; Bielack, S.S.; Borkhardt, A.; Brecht, I.B.; Burkhardt, B.; Calaminus, G.; Debatin, K.M.; Deubzer, H.; Dirksen, U.; Eckert, C.; et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am. J. Med. Genet. A 2017, 173, 1017–1037. [Google Scholar]
- Kha Nguyen, T.M.; Behnert, A.; Pietsch, T.; Vokuhl, C.; Kratz, C.P. Proportion of children with cancer that have an indication for genetic counseling and testing based on the cancer type irrespective of other features. Fam. Cancer 2021, 20, 273–277. [Google Scholar]
- Kratz, C.P.; Achatz, M.I.; Brugières, L.; Frebourg, T.; Garber, J.E.; Greer, M.C.; Hansford, J.R.; Janeway, K.A.; Kohlmann, W.K.; McGee, R.; et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin. Cancer Res. 2017, 23, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.C.; Druley, T.E.; Erez, A.; Kuiper, R.P.; Onel, K.; Schiffman, J.D.; Wolfe Schneider, K.; Scollon, S.R.; Scott, H.S.; Strong, L.C.; et al. Recommendations for Surveillance for Children with Leukemia-Predisposing Conditions. Clin. Cancer Res. 2017, 23, 14–22. [Google Scholar]
- Walsh, M.F.; Chang, V.Y.; Kohlmann, W.K.; Scott, H.S.; Cunniff, C.; Bourdeaut, F.; Molenaar, J.J.; Porter, C.C.; Sandlund, J.T.; Plon, S.E.; et al. Recommendations for Childhood Cancer Screening and Surveillance in DNA Repair Disorders. Clin. Cancer Res. 2017, 23, 23–31. [Google Scholar]
- Tabori, U.; Hansford, J.R.; Achatz, M.I.; Kratz, C.P.; Plon, S.E.; Frebourg, T.; Brugières, L. Clinical Management and Tumor Surveillance Recommendations of Inherited Mismatch Repair Deficiency in Childhood. Clin. Cancer Res. 2017, 23, 32–37. [Google Scholar]
- Evans, D.G.R.; Salvador, H.; Chang, V.Y.; Erez, A.; Voss, S.D.; Schneider, K.W.; Scott, H.S.; Plon, S.E.; Tabori, U. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis 1. Clin. Cancer Res. 2017, 23, 46–53. [Google Scholar]
- Evans, D.G.R.; Salvador, H.; Chang, V.Y.; Erez, A.; Voss, S.D.; Druker, H.; Scott, H.S.; Tabori, U. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis 2 and Related Disorders. Clin. Cancer Res. 2017, 23, 54–61. [Google Scholar]
- Foulkes, W.D.; Kamihara, J.; Evans, D.G.R.; Brugières, L.; Bourdeaut, F.; Molenaar, J.J.; Walsh, M.F.; Brodeur, G.M.; Diller, L. Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin. Cancer Res. 2017, 23, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Rednam, S.P.; Erez, A.; Druker, H.; Janeway, K.A.; Kamihara, J.; Kohlmann, W.K.; Nathanson, K.L.; States, L.J.; Tomlinson, G.E.; Villani, A.; et al. Von Hippel-Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin. Cancer Res. 2017, 23, 68–75. [Google Scholar]
- Schultz, K.A.P.; Rednam, S.P.; Kamihara, J.; Doros, L.; Achatz, M.I.; Wasserman, J.D.; Diller, L.R.; Brugières, L.; Druker, H.; Schneider, K.A.; et al. PTEN, DICER1, FH, and Their Associated Tumor Susceptibility Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin. Cancer Res. 2017, 23, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Villani, A.; Greer, M.C.; Kalish, J.M.; Nakagawara, A.; Nathanson, K.L.; Pajtler, K.W.; Pfister, S.M.; Walsh, M.F.; Wasserman, J.D.; Zelley, K.; et al. Recommendations for Cancer Surveillance in Individuals with RASopathies and Other Rare Genetic Conditions with Increased Cancer Risk. Clin. Cancer Res. 2017, 23, 83–90. [Google Scholar]
- Druker, H.; Zelley, K.; McGee, R.B.; Scollon, S.R.; Kohlmann, W.K.; Schneider, K.A. Genetic Counselor Recommendations for Cancer Predisposition Evaluation and Surveillance in the Pediatric Oncology Patient. Clin. Cancer Res. 2017, 23, 91–97. [Google Scholar]
- Kamihara, J.; Bourdeaut, F.; Foulkes, W.D.; Molenaar, J.J.; Mossé, Y.P.; Nakagawara, A.; Parareda, A.; Scollon, S.R.; Schneider, K.W.; Skalet, A.H.; et al. Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin. Cancer Res. 2017, 23, 98–106. [Google Scholar]
- Achatz, M.I.; Porter, C.C.; Brugières, L.; Druker, H.; Frebourg, T.; Foulkes, W.D.; Kratz, C.P.; Kuiper, R.P.; Hansford, J.R.; Hernandez, H.S.; et al. Cancer Screening Recommendations and Clinical Management of Inherited Gastrointestinal Cancer Syndromes in Childhood. Clin. Cancer Res. 2017, 23, 107–114. [Google Scholar]
- Kalish, J.M.; Doros, L.; Helman, L.J.; Hennekam, R.C.; Kuiper, R.P.; Maas, S.M.; Maher, E.R.; Nichols, K.E.; Plon, S.E.; Porter, C.C.; et al. Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin. Cancer Res. 2017, 23, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasserman, J.D.; Tomlinson, G.E.; Druker, H.; Kamihara, J.; Kohlmann, W.K.; Kratz, C.P.; Nathanson, K.L.; Pajtler, K.W.; Parareda, A.; Rednam, S.P.; et al. Multiple Endocrine Neoplasia and Hyperparathyroid-Jaw Tumor Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin. Cancer Res. 2017, 23, 123–132. [Google Scholar]
- Kesserwan, C.; Friedman Ross, L.; Bradbury, A.R.; Nichols, K.E. The Advantages and Challenges of Testing Children for Heritable Predisposition to Cancer. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 251–269. [Google Scholar]
- Jongmans, M.C.; Loeffen, J.L.; Waanders, E.; Hoogerbrugge, P.M.; Ligtenberg, M.J.; Kuiper, R.P.; Hoogerbrugge, N. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. Eur. J. Med. Genet. 2016, 59, 116–125. [Google Scholar] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar]
- Pardo Romaguera, E.; Muñoz López, A.; Valero Poveda, S.; Porta Cebolla, S.; Fernández-Delgado, R.; Barreda Reines, M.S.; Peris Bonet, R. Cáncer infantil en España. Estadísticas 1980–2017. In Registro Español de Tumores Infantiles (RETI-SEHOP); Universitat de València: Valencia, Spain, 2018. [Google Scholar]
- Konrad, E.D.H.; Nardini, N.; Caliebe, A.; Nagel, I.; Young, D.; Horvath, G. CTCF variants in 39 individuals with a variable neurodevelopmental disorder broaden the mutational and clinical spectrum. Genet Med. 2019, 21, 2723–2733. [Google Scholar] [PubMed] [Green Version]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar]
- Gocho, Y.; Yang, J.J. Genetic defects in hematopoietic transcription factors and predisposition to acute lymphoblastic leukemia. Blood 2019, 134, 793–797. [Google Scholar]
- Xu, H.; Zhang, H.; Yang, W.; Yadav, R.; Morrison, A.C.; Qian, M.; Devidas, M.; Liu, Y.; Perez-Andreu, V.; Zhao, X.; et al. Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nat. Commun. 2015, 6, 7553. [Google Scholar]
- Zhou, X.; Liao, F.; Zhang, J.; Qin, Y.; Xu, H.; Ding, Z.; Zhang, Y.; Zhang, F. Association of the independent polymorphisms in CDKN2A with susceptibility of acute lymphoblastic leukemia. Biosci. Rep. 2018, 38, BSR20180331. [Google Scholar] [PubMed]
- Gutierrez-Camino, A.; Martin-Guerrero, I.; Garcia de Andoin, N.; Sastre, A.; Carbone Bañeres, A.; Astigarraga, I.; Navajas, A.; Garcia-Orad, A. Confirmation of involvement of new variants at CDKN2A/B in pediatric acute lymphoblastic leukemia susceptibility in the Spanish population. PLoS ONE 2017, 12, e0177421. [Google Scholar]
- Desrichard, A.; Bidet, Y.; Uhrhammer, N.; Bignon, Y.-J. CHEK2 contribution to hereditary breast cancer in non-BRCA families. Breast Cancer Res. 2011, 13, R119. [Google Scholar] [PubMed] [Green Version]
- Fiala, E.M.; Jayakumaran, G.; Mauguen, A.; Kennedy, J.A.; Bouvier, N.; Kemel, Y.; Fleischut, M.H.; Maio, A.; Salo-Mullen, E.E.; Sheehan, M.; et al. Prospective pan-cancer germline testing using MSK-IMPACT informs clinical translation in 751 patients with pediatric solid tumors. Nat. Cancer 2021, 2, 357–365. [Google Scholar]
- Gargallo, P.; Yáñez, Y.; Segura, V.; Juan, A.; Torres, B.; Balaguer, J.; Oltra, S.; Castel, V.; Cañete, A. Li-Fraumeni syndrome heterogeneity. Clin. Transl. Oncol. 2020, 22, 978–988. [Google Scholar] [PubMed]
- Pondrom, M.; Bougeard, G.; Karanian, M.; Bonneau-Lagacherie, J.; Boulanger, C.; Boutroux, H.; Briandet, C.; Chevreau, C.; Corradini, N.; Coze, C.; et al. Rhabdomyosarcoma associated with germline TP53 alteration in children and adolescents: The French experience. Pediatr. Blood Cancer 2020, 67, e28486. [Google Scholar] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [PubMed]
- Resta, R.G. What have we been trying to do and have we been any good at it? A history of measuring the success of genetic counseling. Eur. J. Med. Genet. 2019, 62, 300–307. [Google Scholar]
- Harada, S.; Arend, R.; Dai, Q.; Levesque, J.A.; Winokur, T.S.; Guo, R.; Heslin, M.J.; Nabell, L.; Nabors, L.B.; Limdi, N.A.; et al. Implementation and utilization of the molecular tumor board to guide precision medicine. Oncotarget 2017, 8, 57845–57854. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number | Diagnosis | Gene Variant/Genomic Alteration | Categorization |
|---|---|---|---|
| 10 | Retinoblastoma (unilateral) | 13q12.13-q21.2 deletion | Pathogenic |
| 11 | Retinoblastoma (unilateral) | RB1 c.844G>T (p.E282*) | Pathogenic |
| 12 | Pilocytic astrocytoma | NF1 C.910C>T (p.R304*) | Pathogenic |
| 15 | Ewing sarcoma | CHEK2 c.254C>G (p.P85R) | Likely pathogenic |
| 31 | Neuroblastoma | SOS1 c.1300G>A (p.G434R) | Pathogenic |
| 36 | Pilocytic astrocytoma | MRE11 c.659+1G>A | Likely pathogenic |
| 39 | Neuroblastoma | PALB2 c.2747A>T (p.E916V) | Likely pathogenic |
| 44 | B-ALL | Trisomy 21 | Pathogenic |
| 51 | Retinoblastoma (bilateral) | RB1 c.2104 C>T (p.Q702*) | Pathogenic |
| 59 | Neuroblastoma | BRCA1 c.68_69del (p.E23Vfs*17) | Likely pathogenic |
| 64 | Retinoblastoma (unilateral) | 13q12q21 deletion | Pathogenic |
| 65 | Plexiform neurofibroma | NF1 c.4084C>T (p.R1362*) | Pathogenic |
| 66 | Retinoblastoma (bilateral) | RB1 c.224G>A (p.W75*) | Pathogenic |
| 89 | B-ALL | ATM c.1402_1403del (p.K468Efs*18) | Likely pathogenic |
| 103 | Cutaneous angiosarcoma | XPC c.1643_1644delTG (p.V548Afs*25) (homozygous) | Pathogenic |
| 105 | Wilms tumor | CTCF c.353T>A (p.I118K) | Likely pathogenic |
| 108 | Embryonal rhabdomyosarcoma | TP53 c.559G>A (p.G187S) | Pathogenic |
| 110 | B-ALL | CDKN2A deletion | Likely pathogenic |
| 113 | Medulloblastoma SHH | SUFU c.71dup (p.A25Gfs*23) | Pathogenic |
| 116 | B-ALL | JAK3 c.1465C>T (p.Q489*) and JAK3 c.1442-2A>G | Likely pathogenic |
| 118 | B-ALL | CHEK2 c.497A>G (p.N166S) | Likely pathogenic |
| 120 | Burkitt lymphoma | CHEK2 c.470T>C (p.I157T) | Likely pathogenic |
| 127 | Schwannoma CNS (NF1 phenotype) ** | NF1 c.2251+1 G>A | Pathogenic |
| 150 | Vestibular schwannoma (bilateral) | NF2 c.115-2A>G | Pathogenic |
| 156 | Pineoblastoma | DICER1 c.2026C>T (p.R676*) | Pathogenic |
| 169 | Retinoblastoma (bilateral) | RB1 c.2548_2552delCAGA-T (Q850Gfs*3) | Pathogenic |
| P/LP Variants Not Predisposing to Patient’s Tumor | ||
| Patient Number | Diagnosis | Gene Variant |
| 18 | Pilocytic astrocytoma | CEP57 c.241C>T (p.R81*) |
| 42 | Alveolar rhabdomyosarcoma | FANCL c.40del (p. L14Cfs*27) |
| 42 | Alveolar rhabdomyosarcoma | XPC c.1643_1644del (p. V548Afs*25) |
| 78 | Atypical teratoid rhabdoid tumor | NBN c.1648_1651del (p. K550Gfs*8) |
| 89 | B-ALL | RECQL4 c.2336_2357del (p.D779Cfs*57) |
| 111 | Lymphoblastic lymphoma | PIK3CG c.2340dup (p. E781Rfs*4) |
| 149 | Neuroblastoma | FANCM c.2161-1G>A |
| 151 | Ependymoma | XPC c.1643_1644del (p. V548Afs*25) |
| 159 | Ewing sarcoma | ERCC3 c.583C>T (p. R195T*) |
| VOPPS Variants | ||
| Patient Number | Diagnosis | Gene Variant |
| 48 | B-ALL | SH2B3 c.622G>C (p.E208Q) |
| 72 | B-ALL | RET c.2331C>A (p.N777K) |
| 78 | Atypical teratoid rhabdoid tumor | CHEK2 c.342G>T (p.W114C) |
| 81 | High grade glioma | FAT1 c.10990del (p.Q3664Sfs*10) |
| 119 | B-ALL | ALK c.3467G>A (p.C1156Y) |
| 139 | Wilms tumor | IGF1R c.3367A>G (p.M1123V) |
| 143 | Lymphoblastic lymphoma | FANCD2 c.2204G>A (p.R735Q) |
| 144 | Neuroblastoma | NF1 c.2998C>A (p.R1000S) |
| 153 | Carcinoid tumor | ING4 c.109+1G>C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargallo, P.; Oltra, S.; Yáñez, Y.; Juan-Ribelles, A.; Calabria, I.; Segura, V.; Lázaro, M.; Balaguer, J.; Tormo, T.; Dolz, S.; et al. Germline Predisposition to Pediatric Cancer, from Next Generation Sequencing to Medical Care. Cancers 2021, 13, 5339. https://doi.org/10.3390/cancers13215339
Gargallo P, Oltra S, Yáñez Y, Juan-Ribelles A, Calabria I, Segura V, Lázaro M, Balaguer J, Tormo T, Dolz S, et al. Germline Predisposition to Pediatric Cancer, from Next Generation Sequencing to Medical Care. Cancers. 2021; 13(21):5339. https://doi.org/10.3390/cancers13215339
Chicago/Turabian StyleGargallo, Pablo, Silvestre Oltra, Yania Yáñez, Antonio Juan-Ribelles, Inés Calabria, Vanessa Segura, Marián Lázaro, Julia Balaguer, Teresa Tormo, Sandra Dolz, and et al. 2021. "Germline Predisposition to Pediatric Cancer, from Next Generation Sequencing to Medical Care" Cancers 13, no. 21: 5339. https://doi.org/10.3390/cancers13215339
APA StyleGargallo, P., Oltra, S., Yáñez, Y., Juan-Ribelles, A., Calabria, I., Segura, V., Lázaro, M., Balaguer, J., Tormo, T., Dolz, S., Fernández, J. M., Fuentes, C., Torres, B., Andrés, M., Tasso, M., Castel, V., Font de Mora, J., & Cañete, A. (2021). Germline Predisposition to Pediatric Cancer, from Next Generation Sequencing to Medical Care. Cancers, 13(21), 5339. https://doi.org/10.3390/cancers13215339