
Photodynamic Priming Improves the Anti-Migratory Activity of Prostaglandin E Receptor 4 Antagonist in Cancer Cells In Vitro

 , , and
, , and 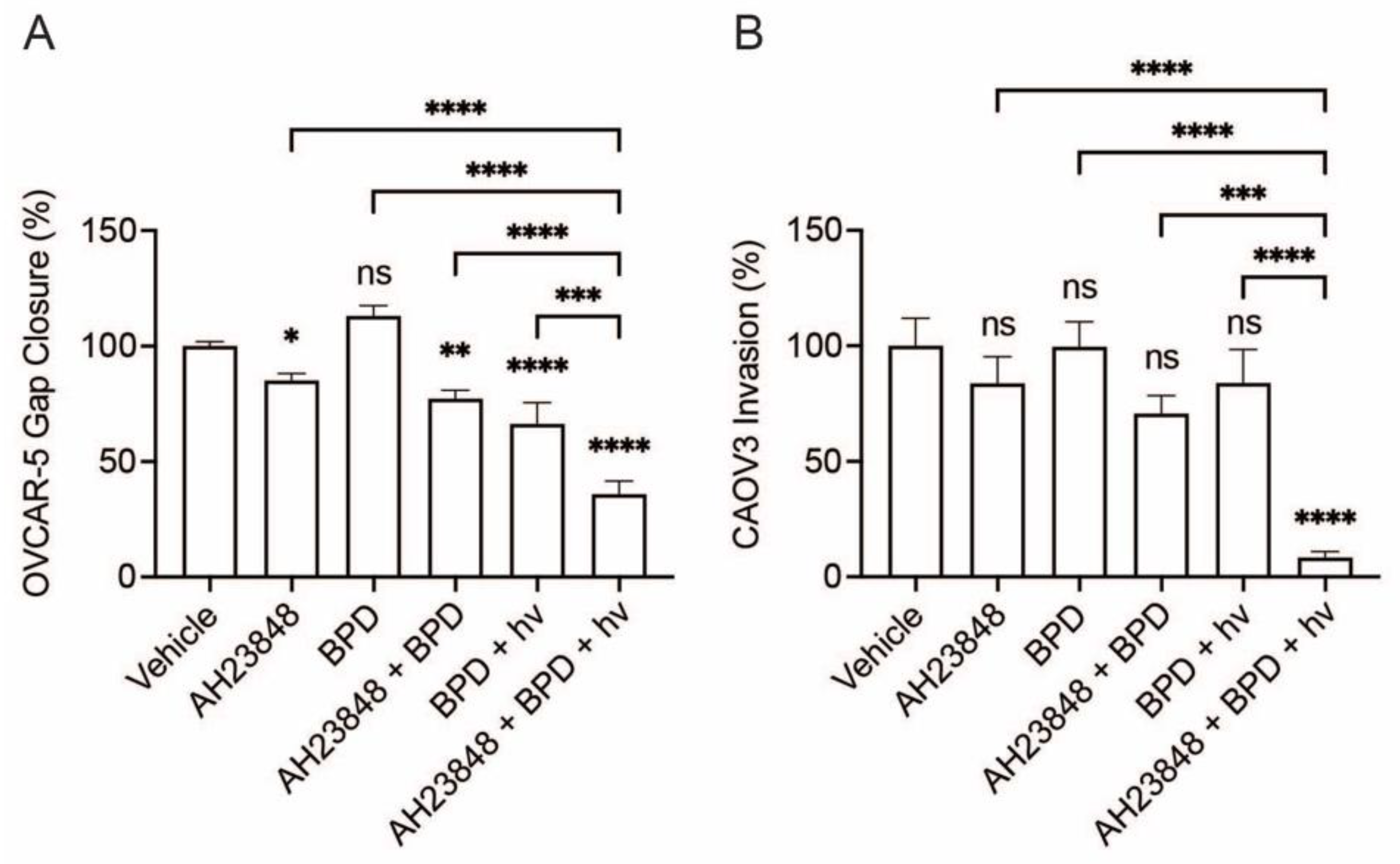{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Gap Closure and Metabolic Activity Studies
2.3. Lysate Collection and Western Blotting
2.4. Photoimmunoconjugate Synthesis and Drugs
2.5. Extraction Methods to Quantify Photosensitizer Uptake in Cells
2.6. Transwell Invasion Assay and PGE2 ELISA
2.7. Statistical Analysis
3. Results
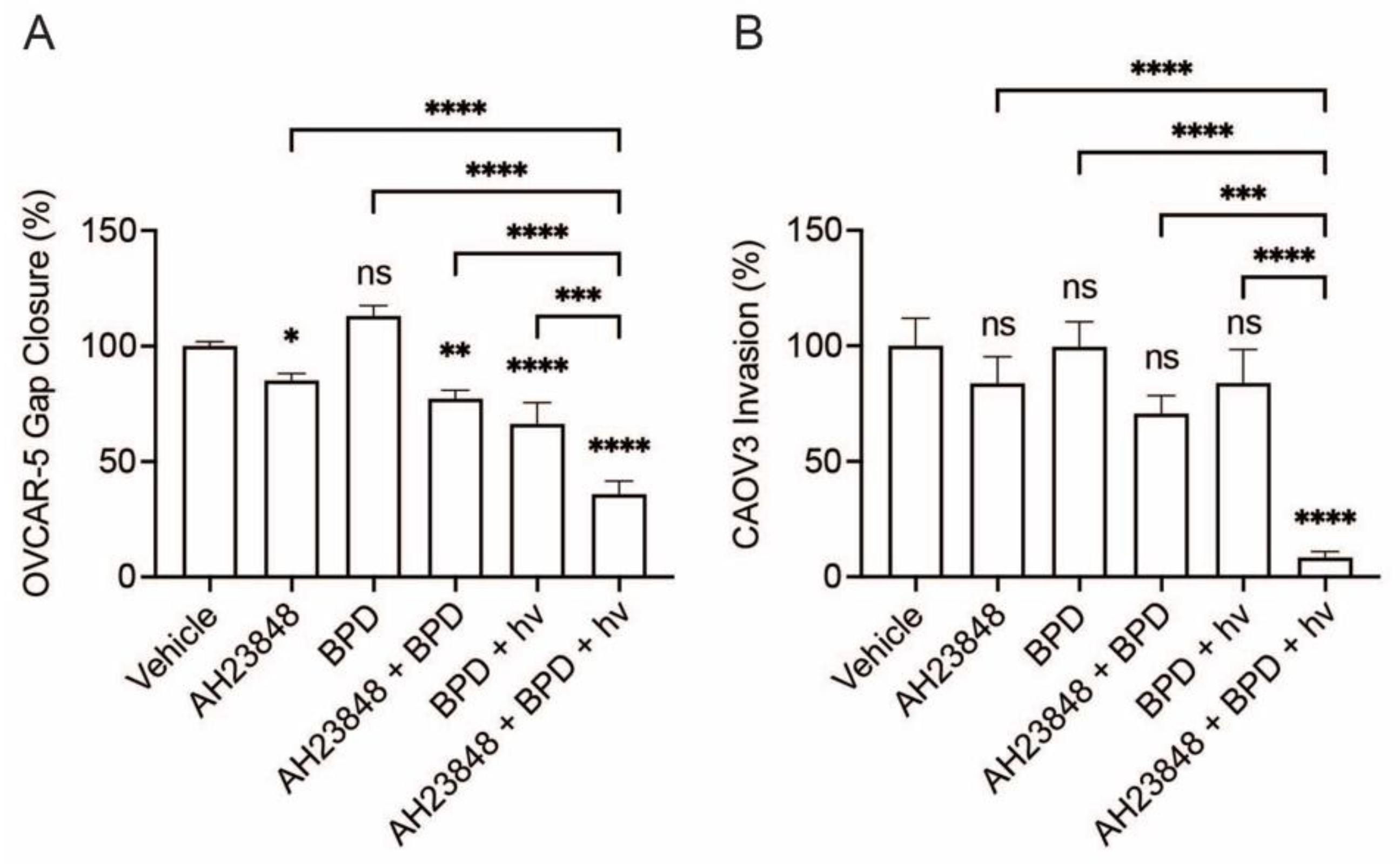
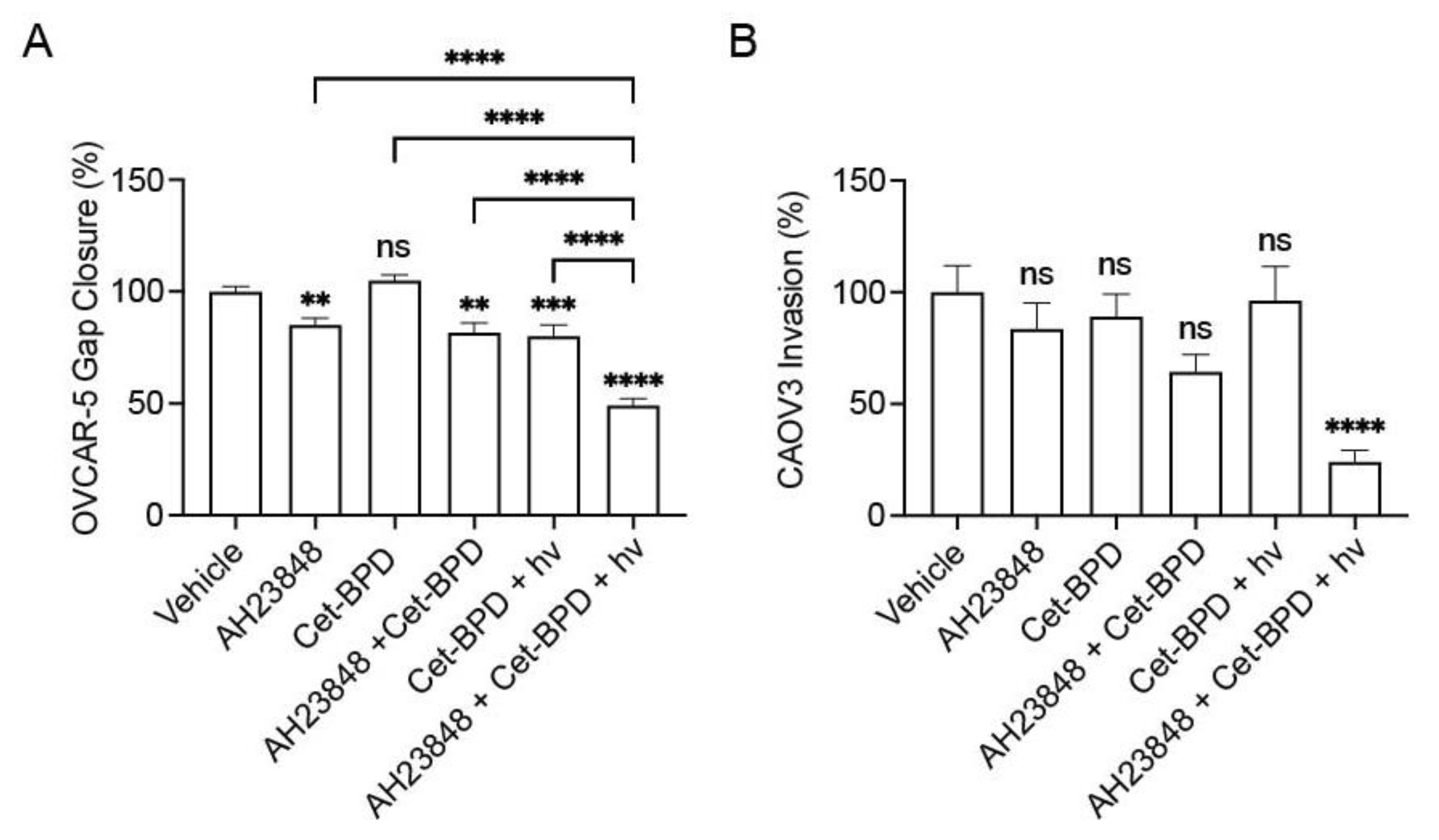
3.1. Combination of BPD-Based PDP and EP4 Inhibitor (AH23848) Decreases Ovarian Cancer Cell Migration and Invasion
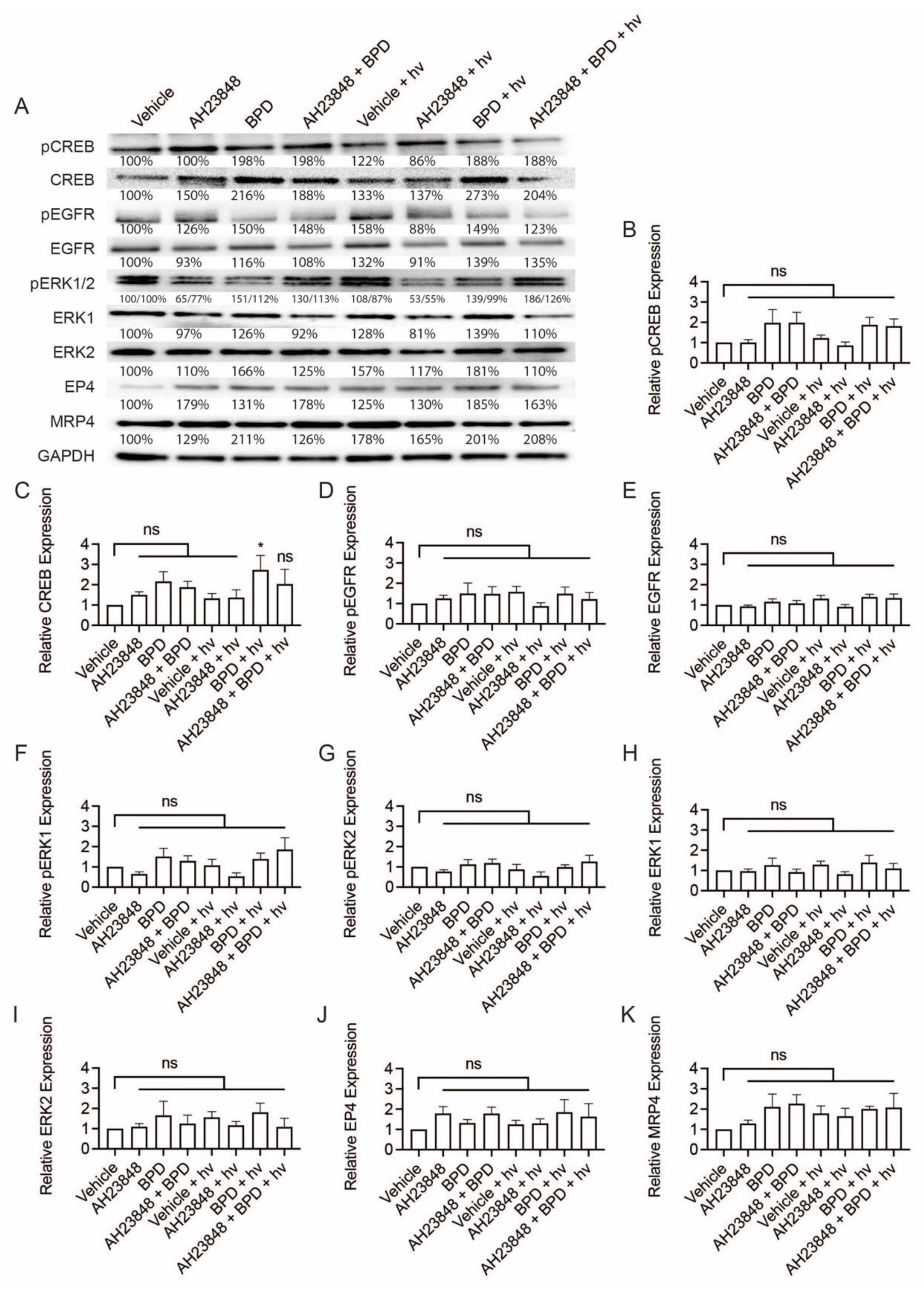
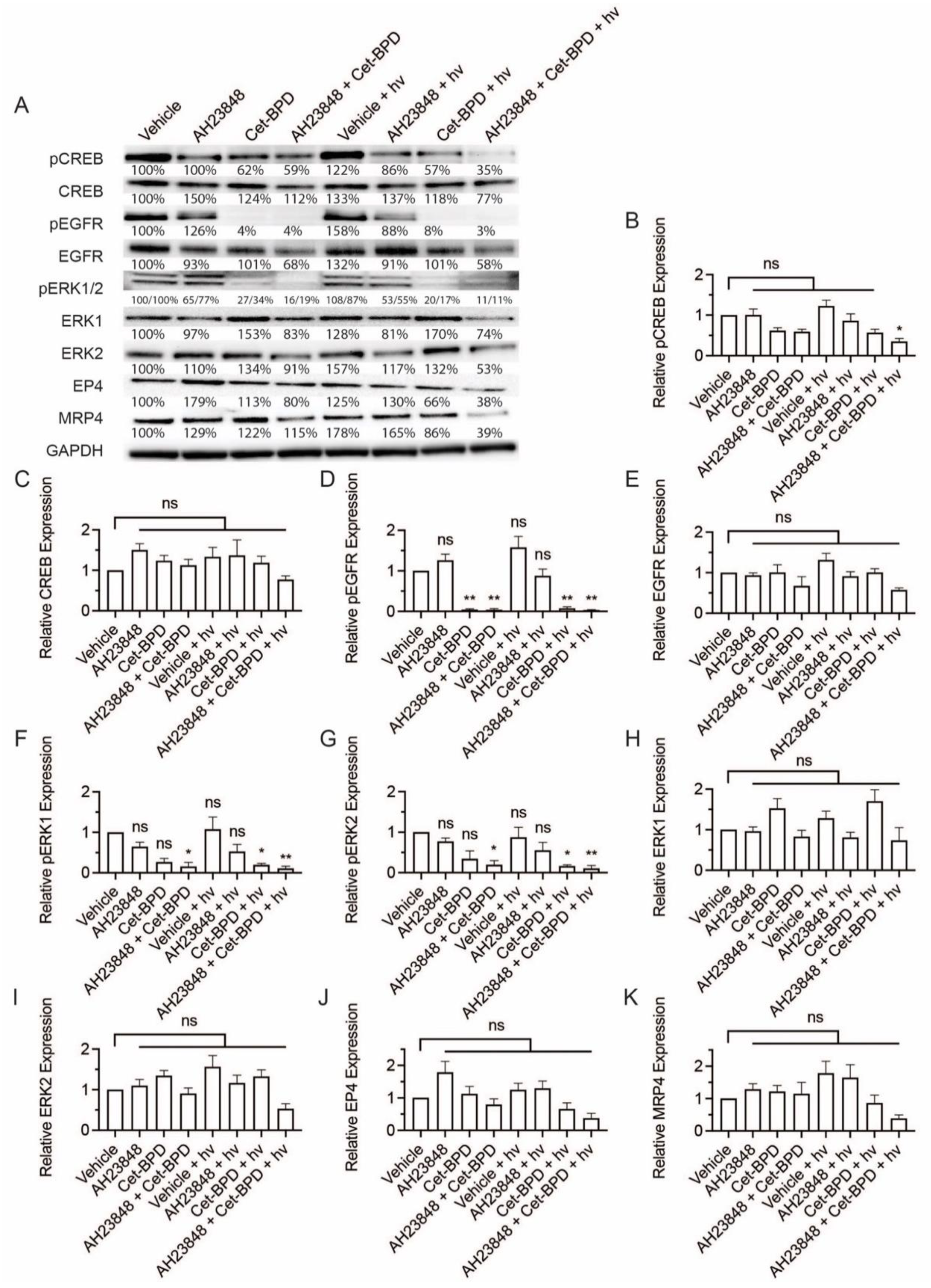
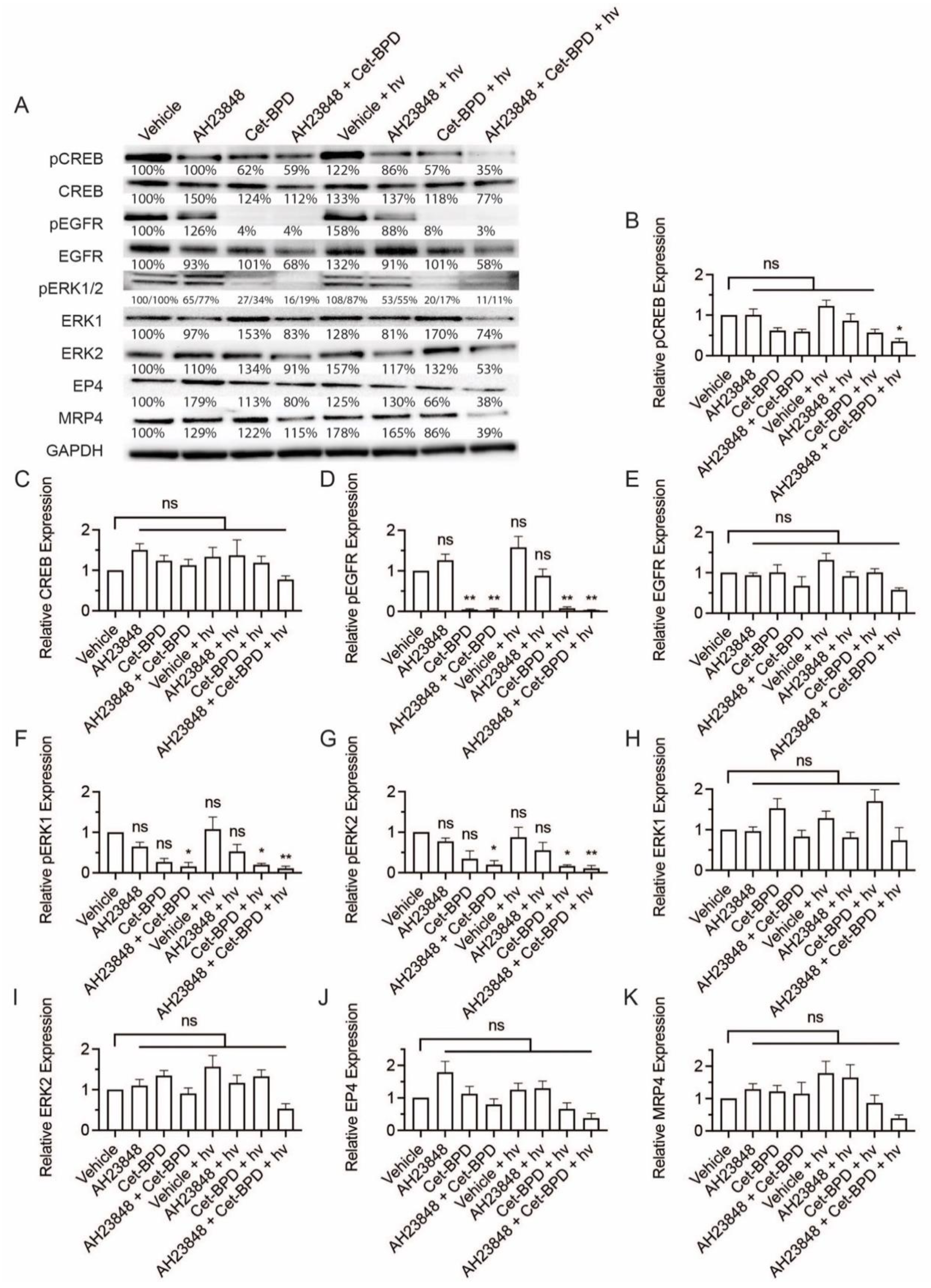
3.2. BPD-Based PDP Combined with EP4 Inhibition Does Not Attenuate Cell Signaling Pathways Linked to EP4 and EGFR
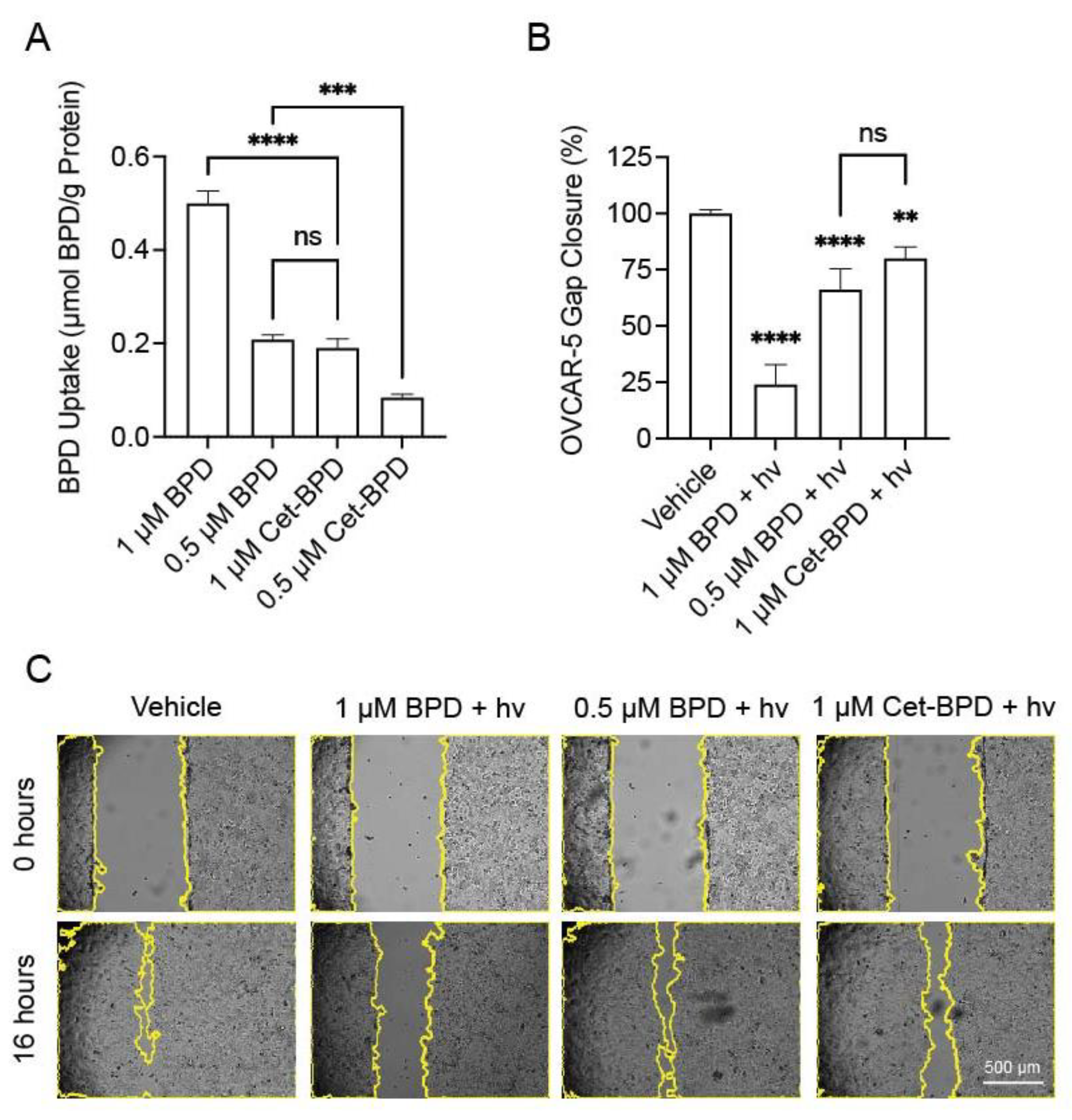
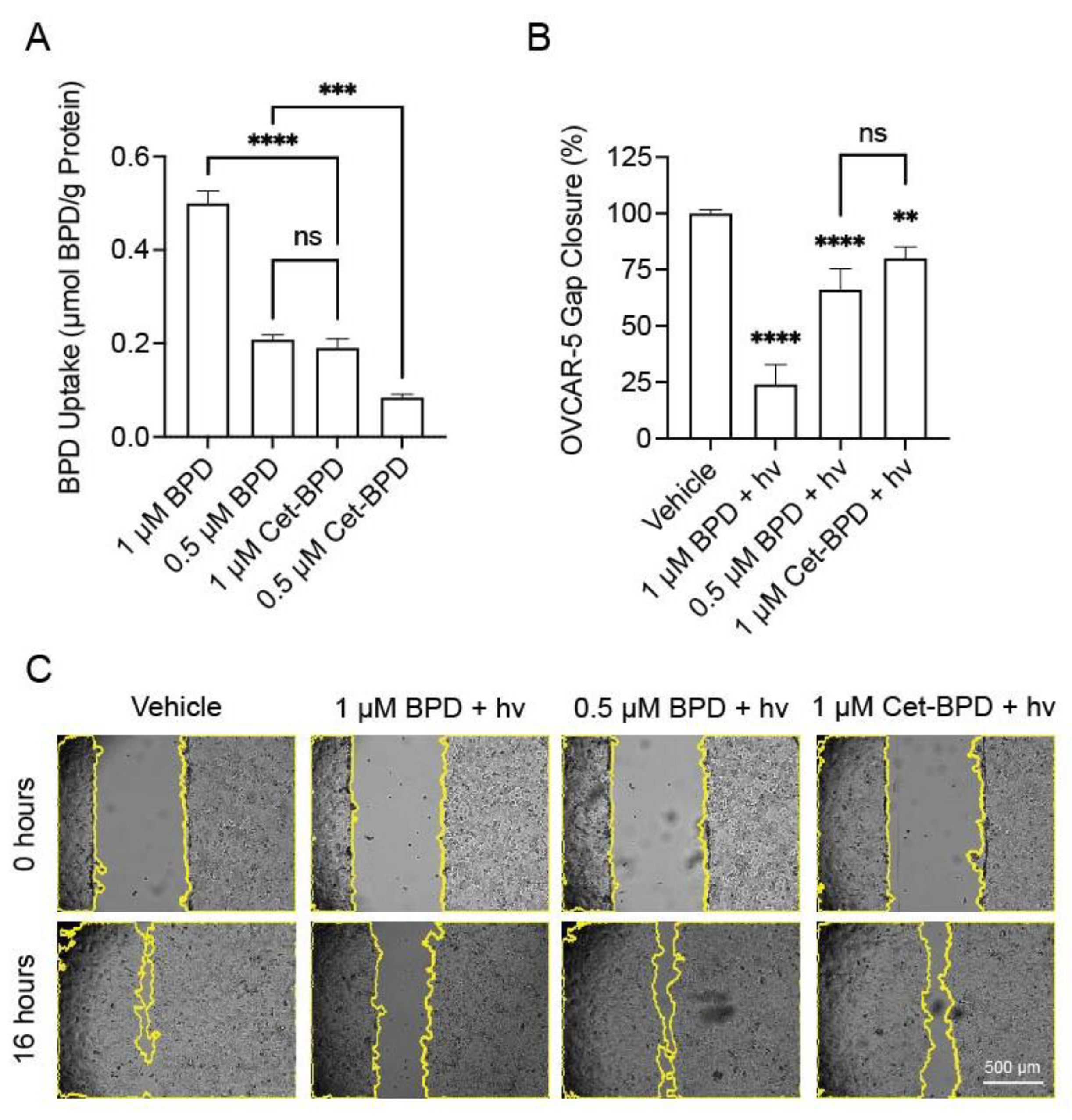
3.3. Cet-BPD-PDP and BPD-PDP Have Similar Effects on Gap Closure When Compared at Equivalent Intracellular Photosensitizer Concentrations
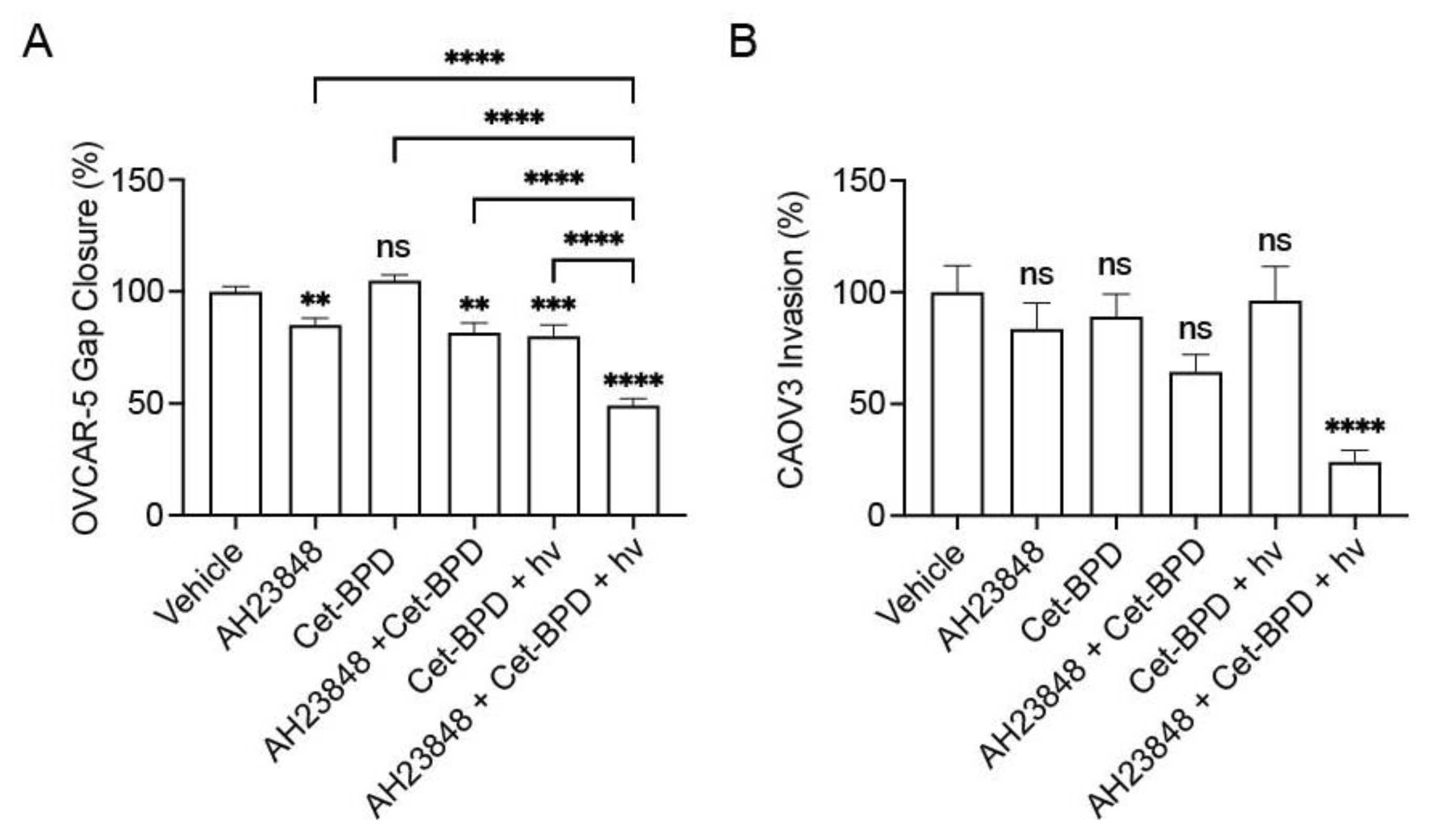
3.4. Cet-BPD-Based PDP Combined with EP4 Inhibition Attenuates Migration, Invasion, and Cell Signaling Linked to EP4 and EGFR
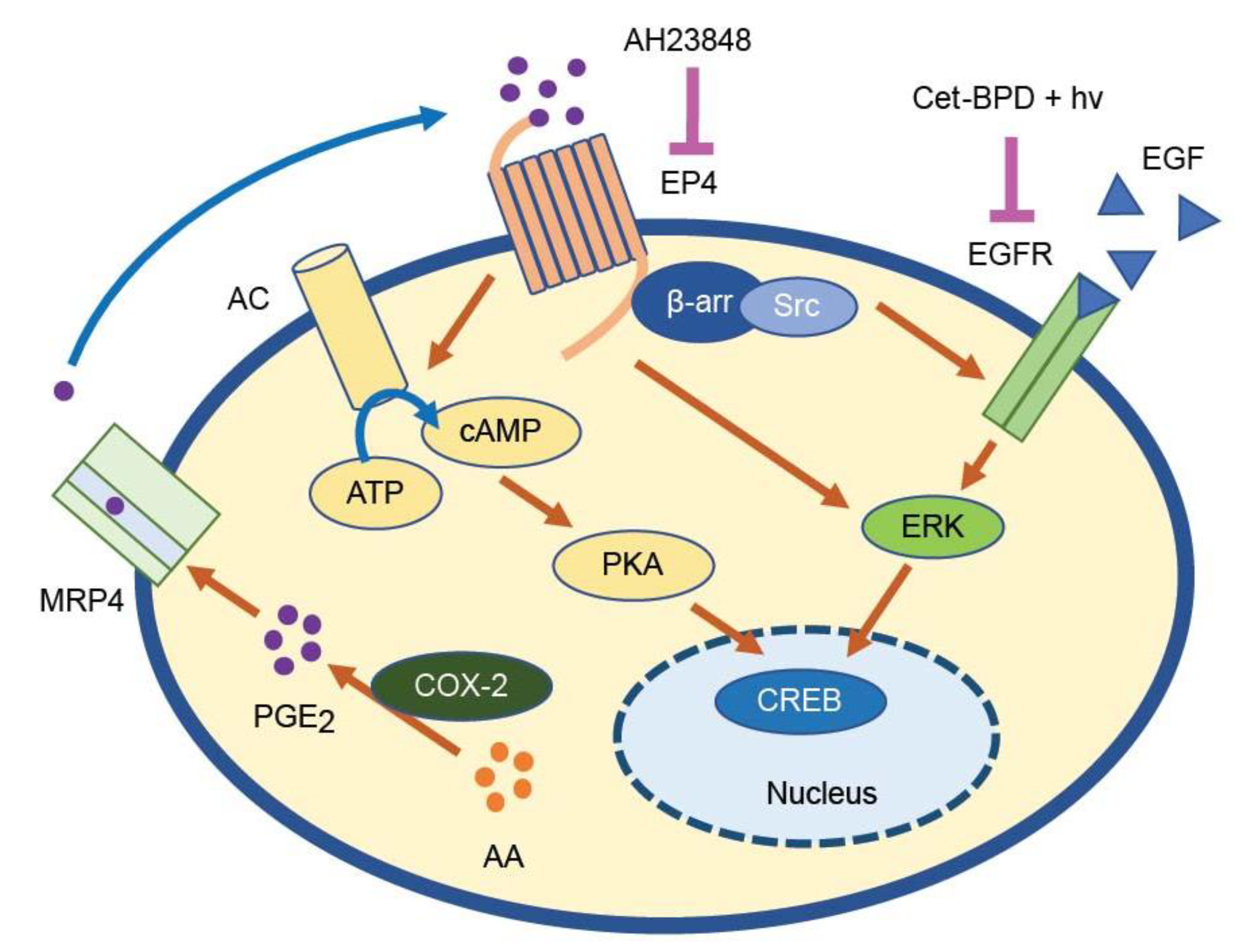
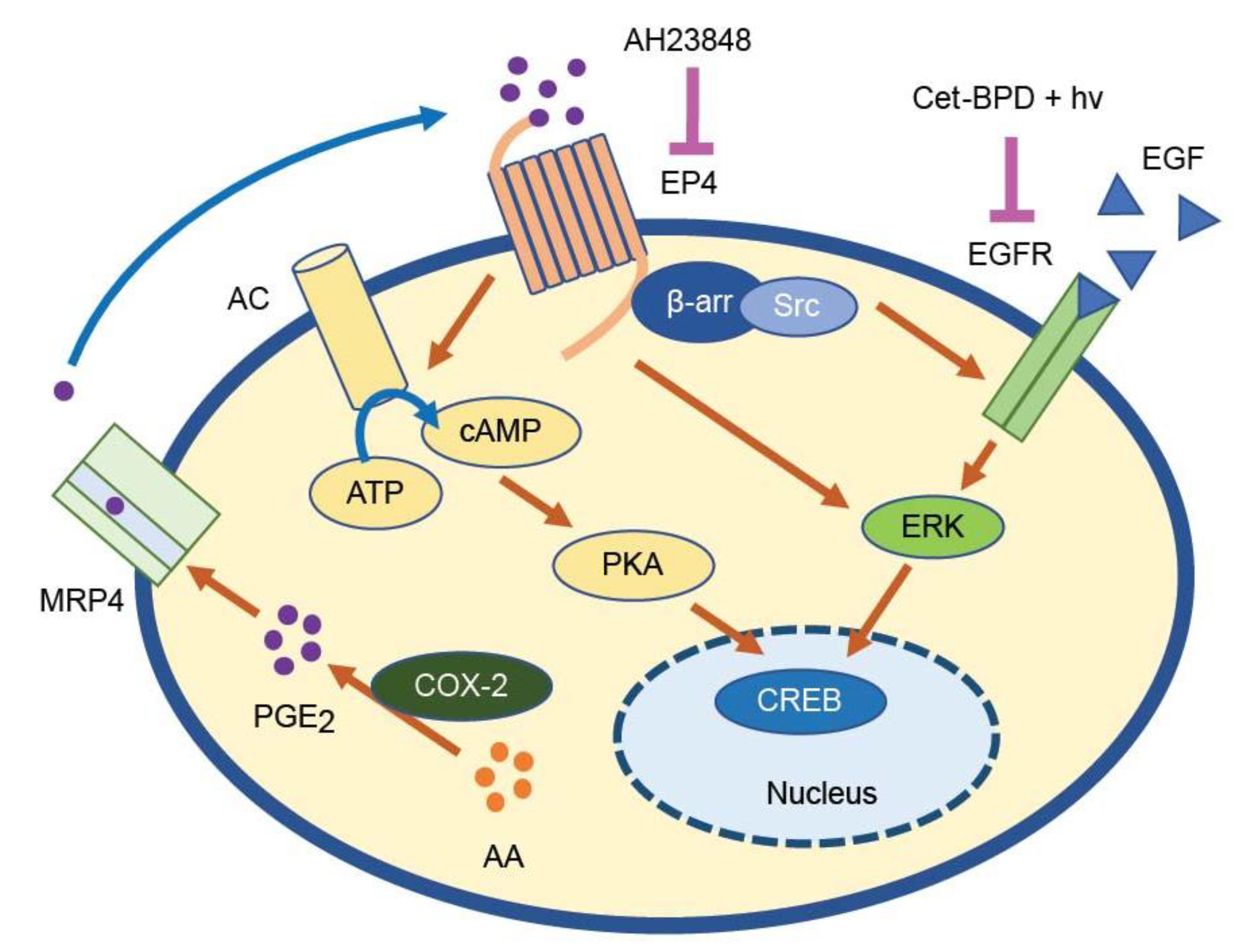
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benov, L. Photodynamic Therapy: Current Status and Future Directions. Med. Princ. Pract. 2015, 24 (Suppl. 1), 14–28. [Google Scholar] [CrossRef]
- De Silva, P.; Saad, M.A.; Thomsen, H.C.; Bano, S.; Ashraf, S.; Hasan, T. Photodynamic therapy, priming and optical imaging: Potential co-conspirators in treatment design and optimization—A Thomas Dougherty Award for Excellence in PDT paper. J. Porphyr. Phthalocyanines 2020, 24, 1320–1360. [Google Scholar] [CrossRef]
- Snyder, J.W.; Greco, W.R.; Bellnier, D.A.; Vaughan, L.; Henderson, B.W. Photodynamic Therapy: A Means to Enhanced Drug Delivery to Tumors. Cancer Res. 2003, 63, 8126–8131. [Google Scholar]
- Wang, Y.; Gonzalez, M.; Cheng, C.; Haouala, A.; Krueger, T.; Peters, S.; Decosterd, L.-A.; van den Bergh, H.; Perentes, J.Y.; Ris, H.-B.; et al. Photodynamic induced uptake of liposomal doxorubicin to rat lung tumors parallels tumor vascular density. Lasers Surg. Med. 2012, 44, 318–324. [Google Scholar] [CrossRef]
- Wang, X.; Gronchi, F.; Bensimon, M.; Mercier, T.; Decosterd, L.A.; Wagnières, G.; Debefve, E.; Ris, H.-B.; Letovanec, I.; Peters, S.; et al. Treatment of pleural malignancies by photo-induction combined to systemic chemotherapy: Proof of concept on rodent lung tumors and feasibility study on porcine chest cavities. Lasers Surg. Med. 2015, 47, 807–816. [Google Scholar] [CrossRef]
- Cheng, C.; Wang, Y.; Haouala, A.; Debefve, E.; Andrejevic Blant, S.; Krueger, T.; Gonzalez, M.; Ballini, J.P.; Peters, S.; Decosterd, L.; et al. Photodynamic therapy enhances liposomal doxorubicin distribution in tumors during isolated perfusion of rodent lungs. Eur. Surg. Res. 2011, 47, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Debefve, E.; Pegaz, B.; Ballini, J.P.; Konan, Y.N.; van den Bergh, H. Combination therapy using aspirin-enhanced photodynamic selective drug delivery. Vasc. Pharmacol. 2007, 46, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Debefve, E.; Pegaz, B.; Ballini, J.-P.; Van Den Bergh, H. Combination Therapy Using Verteporfin and Ranibizumab; Optimizing the Timing in the CAM Model. Photochem. Photobiol. 2009, 85, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Debefve, E.; Mithieux, F.; Perentes, J.Y.; Wang, Y.; Cheng, C.; Schaefer, S.C.; Ruffieux, C.; Ballini, J.-P.; Gonzalez, M.; van den Bergh, H.; et al. Leukocyte–endothelial cell interaction is necessary for photodynamic therapy induced vascular permeabilization. Lasers Surg. Med. 2011, 43, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Perentes, J.Y.; Wang, Y.; Wang, X.; Abdelnour, E.; Gonzalez, M.; Decosterd, L.; Wagnieres, G.; van den Bergh, H.; Peters, S.; Ris, H.-B.; et al. Low-Dose Vascular Photodynamic Therapy Decreases Tumor Interstitial Fluid Pressure, which Promotes Liposomal Doxorubicin Distribution in a Murine Sarcoma Metastasis Model. Transl. Oncol. 2014, 7, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-C.; Rizvi, I.; Liu, J.; Anbil, S.; Kalra, A.; Lee, H.; Baglo, Y.; Paz, N.; Hayden, D.; Pereira, S.; et al. Photodynamic Priming Mitigates Chemotherapeutic Selection Pressures and Improves Drug Delivery. Cancer Res. 2018, 78, 558–571. [Google Scholar] [CrossRef] [Green Version]
- Overchuk, M.; Harmatys, K.M.; Sindhwani, S.; Rajora, M.A.; Koebel, A.; Charron, D.M.; Syed, A.M.; Chen, J.; Pomper, M.G.; Wilson, B.C.; et al. Subtherapeutic Photodynamic Treatment Facilitates Tumor Nanomedicine Delivery and Overcomes Desmoplasia. Nano Lett. 2021, 21, 344–352. [Google Scholar] [CrossRef]
- Anbil, S.; Pigula, M.; Huang, H.-C.; Mallidi, S.; Broekgaarden, M.; Baglo, Y.; De Silva, P.; Simeone, D.M.; Mino-Kenudson, M.; Maytin, E.V.; et al. Vitamin D Receptor Activation and Photodynamic Priming Enables Durable Low-dose Chemotherapy. Mol. Cancer Ther. 2020, 19, 1308–1319. [Google Scholar] [CrossRef] [Green Version]
- Bulin, A.-L.; Broekgaarden, M.; Simeone, D.; Hasan, T. Low dose photodynamic therapy harmonizes with radiation therapy to induce beneficial effects on pancreatic heterocellular spheroids. Oncotarget 2019, 10, 2625–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, P.D.; Bano, S.; Pogue, B.W.; Wang, K.K.; Maytin, E.V.; Hasan, T. Photodynamic priming with triple-receptor targeted nanoconjugates that trigger T cell-mediated immune responses in a 3D in vitro heterocellular model of pancreatic cancer. Nanophotonics 2021, 10, 3199–3214. [Google Scholar] [CrossRef]
- Inglut, C.T.; Gray, K.M.; Vig, S.; Jung, J.W.; Stabile, J.; Zhang, Y.; Stroka, K.M.; Huang, H.C. Photodynamic Priming Modulates Endothelial Cell–Cell Junction Phenotype for Light-Activated Remote Control of Drug Delivery. IEEE J. Sel. Top. Quantum Electron. 2021, 27, 1–11. [Google Scholar] [CrossRef]
- Majumder, M.; Nandi, P.; Omar, A.; Ugwuagbo, K.C.; Lala, P.K. EP4 as a Therapeutic Target for Aggressive Human Breast Cancer. Int. J. Mol. Sci. 2018, 19, 1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonisen, F.; Perrin, L.; Bayarmagnai, B.; van den Dries, K.; Cambi, A.; Gligorijevic, B. EP4 receptor promotes invadopodia and invasion in human breast cancer. Eur. J. Cell. Biol. 2017, 96, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, U.; Iwatsubo, K.; Umemura, M.; Fujita, T.; Ishikawa, Y. The Prostanoid EP4 Receptor and Its Signaling Pathway. Pharmacol. Rev. 2013, 65, 1010–1052. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.G.; Roque, D.; Ching, M.M.; Fulton, A.; Rao, G.; Reader, J.C. The Role of Eicosanoids in Gynecological Malignancies. Front. Pharmacol. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- Ching, M.M.; Reader, J.; Fulton, A.M. Eicosanoids in Cancer: Prostaglandin E(2) Receptor 4 in Cancer Therapeutics and Immunotherapy. Front. Pharmacol. 2020, 11, 1–6. [Google Scholar] [CrossRef]
- Spinella, F.; Rosanò, L.; Di Castro, V.; Natali, P.G.; Bagnato, A. Endothelin-1-induced prostaglandin E2-EP2, EP4 signaling regulates vascular endothelial growth factor production and ovarian carcinoma cell invasion. J. Biol. Chem. 2004, 279, 46700–46705. [Google Scholar] [CrossRef] [Green Version]
- Konya, V.; Marsche, G.; Schuligoi, R.; Heinemann, A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol. Ther. 2013, 138, 485–502. [Google Scholar] [CrossRef] [Green Version]
- Gui, T.; Shen, K. The epidermal growth factor receptor as a therapeutic target in epithelial ovarian cancer. Cancer Epidemiol. 2012, 36, 490–496. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Parida, S.; Pal, I.; Parekh, A.; Thakur, B.; Bharti, R.; Das, S.; Mandal, M. GW627368X inhibits proliferation and induces apoptosis in cervical cancer by interfering with EP4/EGFR interactive signaling. Cell Death Dis. 2016, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ching, M.M.; Fan, C.; Roque, D.; Rao, G.; Staats, P.; Fulton, A.; Reader, J. Abstract A22: Functional analysis of PGE2 pathway members EP4 and MRP4 in ovarian cancer. Clin. Cancer Res. 2018, 24, A22. [Google Scholar] [CrossRef]
- Spring, B.Q.; Abu-Yousif, A.O.; Palanisami, A.; Rizvi, I.; Zheng, X.; Mai, Z.; Anbil, S.; Sears, R.B.; Mensah, L.B.; Goldschmidt, R.; et al. Selective treatment and monitoring of disseminated cancer micrometastases in vivo using dual-function, activatable immunoconjugates. Proc. Natl. Acad. Sci. USA 2014, 111, E933–E942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savellano, M.D.; Hasan, T. Targeting cells that overexpress the epidermal growth factor receptor with polyethylene glycolated BPD verteporfin photosensitizer immunoconjugates. Photochem. Photobiol. 2003, 77, 431–439. [Google Scholar] [CrossRef]
- Inglut, C.T.; Baglo, Y.; Liang, B.J.; Cheema, Y.; Stabile, J.; Woodworth, G.F.; Huang, H.C. Systematic Evaluation of Light-Activatable Biohybrids for Anti-Glioma Photodynamic Therapy. J. Clin. Med. 2019, 8, 1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, B.J.; Pigula, M.; Baglo, Y.; Najafali, D.; Hasan, T.; Huang, H.C. Breaking the selectivity-uptake trade-off of photoimmunoconjugates with nanoliposomal irinotecan for synergistic multi-tier cancer targeting. J. Nanobiotechnol. 2020, 18, 1–14. [Google Scholar] [CrossRef]
- Kobayashi, H.; Choyke, P.L. Near-Infrared Photoimmunotherapy of Cancer. Acc. Chem. Res. 2019, 52, 2332–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, H.; Furusawa, A.; Rosenberg, A.; Choyke, P.L. Near-infrared photoimmunotherapy of cancer: A new approach that kills cancer cells and enhances anti-cancer host immunity. Int. Immunol. 2021, 33, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.L.; Spirtos, N.M.; Enserro, D.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Kim, J.-W.; Park, S.-Y.; Kim, B.-G.; Nam, J.-H.; et al. Secondary Surgical Cytoreduction for Recurrent Ovarian Cancer. N. Engl. J. Med. 2019, 381, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.-S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. npj Precis. Oncol. 2018, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Baglo, Y.; Sorrin, A.J.; Liang, B.J.; Huang, H.C. Harnessing the Potential Synergistic Interplay Between Photosensitizer Dark Toxicity and Chemotherapy. Photochem. Photobiol. 2019, 96, 636–645. [Google Scholar] [CrossRef]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase Isozymes: The Biology of Prostaglandin Synthesis and Inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [Green Version]
- Kochel, T.J.; Fulton, A.M. Multiple Drug Resistance-Associated Protein 4 (MRP4), Prostaglandin Transporter (PGT), and 15-Hydroxyprostaglandin Dehydrogenase (15-PGDH) as Determinants of PGE2 Levels in Cancer. Prostaglandins Other Lipid Mediat. 2015, 116–117, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP–PKA–CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 1–13. [Google Scholar] [CrossRef]
- Krysan, K.; Reckamp, K.L.; Dalwadi, H.; Sharma, S.; Rozengurt, E.; Dohadwala, M.; Dubinett, S.M. Prostaglandin E2 Activates Mitogen-Activated Protein Kinase/Erk Pathway Signaling and Cell Proliferation in Non–Small Cell Lung Cancer Cells in an Epidermal Growth Factor Receptor–Independent Manner. Cancer Res. 2005, 65, 6275–6281. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Chopp, M.; Katakowski, M.; Cho, K.K.; Yang, X.; Hochbaum, N.; Tong, L.; Mikkelsen, T. Photodynamic Therapy with Photofrin Reduces Invasiveness of Malignant Human Glioma Cells. Lasers Med Sci. 2002, 17, 280–288. [Google Scholar] [CrossRef]
- Yang, T.-H.; Chen, C.-T.; Wang, C.-P.; Lou, P.-J. Photodynamic therapy suppresses the migration and invasion of head and neck cancer cells in vitro. Oral Oncol. 2007, 43, 358–365. [Google Scholar] [CrossRef]
- Coleman, R.A.; Grix, S.P.; Head, S.A.; Louttit, J.B.; Mallett, A.; Sheldrick, R.L. A novel inhibitory prostanoid receptor in piglet saphenous vein. Prostaglandins 1994, 47, 151–168. [Google Scholar] [CrossRef]
- Xin, X.; Majumder, M.; Girish, G.V.; Mohindra, V.; Maruyama, T.; Lala, P.K. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab. Investig. 2012, 92, 1115–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, M.; Xin, X.; Liu, L.; Girish, G.V.; Lala, P.K. Prostaglandin E2 receptor EP4 as the common target on cancer cells and macrophages to abolish angiogenesis, lymphangiogenesis, metastasis, and stem-like cell functions. Cancer Sci. 2014, 105, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhou, W.; Ge, J.; Zhang, Z. Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMP-PKA/PI3K-Akt signaling pathway. Mol. Med. Rep. 2018, 17, 4702–4712. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.; Harper, A.K.; Legesse, T.; Staats, P.N.; Goloubeva, O.; Rao, G.G.; Fulton, A.; Roque, D.M. EP4 and Class III β-Tubulin Expression in Uterine Smooth Muscle Tumors: Implications for Prognosis and Treatment. Cancers 2019, 11, 1590. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, F.G.; Gorden, D.L.; Matta, P.; Shi, Q.; Matrisian, L.M.; DuBois, R.N. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 1492–1497. [Google Scholar] [CrossRef] [Green Version]
- Abu-Yousif, A.O.; Moor, A.C.E.; Zheng, X.; Savellano, M.D.; Yu, W.; Selbo, P.K.; Hasan, T. Epidermal growth factor receptor-targeted photosensitizer selectively inhibits EGFR signaling and induces targeted phototoxicity in ovarian cancer cells. Cancer Lett. 2012, 321, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherukuri, D.P.; Chen, X.B.O.; Goulet, A.-C.; Young, R.N.; Han, Y.; Heimark, R.L.; Regan, J.W.; Meuillet, E.; Nelson, M.A. The EP4 receptor antagonist, L-161,982, blocks prostaglandin E2-induced signal transduction and cell proliferation in HCA-7 colon cancer cells. Exp. Cell Res. 2007, 313, 2969–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swarthout, J.T.; Tyson, D.R.; Jefcoat, S.C., Jr.; Partridge, N.C. Induction of Transcriptional Activity of the Cyclic Adenosine Monophosphate Response Element Binding Protein by Parathyroid Hormone and Epidermal Growth Factor in Osteoblastic Cells. J. Bone Miner. Res. 2002, 17, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Barlow, C.A.; Kitiphongspattana, K.; Siddiqui, N.; Roe, M.W.; Mossman, B.T.; Lounsbury, K.M. Protein kinase A-mediated CREB phosphorylation is an oxidant-induced survival pathway in alveolar type II cells. Apoptosis 2008, 13, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, A.; von Tiehl, K.; Wong, S.; Luna, M.; Gomer, C.J. Cyclooxygenase-2 Inhibitor Treatment Enhances Photodynamic Therapy-mediated Tumor Response. Cancer Res. 2002, 62, 3956–3961. [Google Scholar]
- Makowski, M.; Grzela, T.; Niderla, J.; Łazarczyk, M.; Mroz, P.; Kopee, M.; Legat, M.; Strusinska, K.; Koziak, K.; Nowis, D.; et al. Inhibition of cyclooxygenase-2 indirectly potentiates antitumor effects of photodynamic therapy in mice. Clin. Cancer Res. 2003, 9, 5417–5422. [Google Scholar] [PubMed]
- Hendrickx, N.; Volanti, C.; Moens, U.; Seternes, O.M.; de Witte, P.; Vandenheede, J.R.; Piette, J.; Agostinis, P. Up-regulation of Cyclooxygenase-2 and Apoptosis Resistance by p38 MAPK in Hypericin-mediated Photodynamic Therapy of Human Cancer Cells. J. Biol. Chem. 2003, 278, 52231–52239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, P.; Maeder, M.E.; Hunt, B.; Linn, B.; Mangels-Dick, T.; Hasan, T.; Wang, K.K.; Pogue, B.W. CT radiomic features of photodynamic priming in clinical pancreatic adenocarcinoma treatment. Phys. Med. Biol. 2021, 66, 1–10. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Le Bitoux, M.-A.; Wagnieres, G.; Vandenbergh, H.; Gonzalez, M.; Ris, H.-B.; Perentes, J.Y.; Krueger, T. Fluence plays a critical role on the subsequent distribution of chemotherapy and tumor growth delay in murine mesothelioma xenografts pre-treated by photodynamic therapy. Lasers Surg. Med. 2015, 47, 323–330. [Google Scholar] [CrossRef]
- Cormier, N.; Yeo, A.; Fiorentino, E.; Paxson, J. Optimization of the Wound Scratch Assay to Detect Changes in Murine Mesenchymal Stromal Cell Migration After Damage by Soluble Cigarette Smoke Extract. J. Vis. Exp. 2015, 1–9. [Google Scholar] [CrossRef] [Green Version]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Take, Y.; Koizumi, S.; Nagahisa, A. Prostaglandin E Receptor 4 Antagonist in Cancer Immunotherapy: Mechanisms of Action. Front. Immunol. 2020, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Holt, D.; Kundu, N.; Reader, J.; Goloubeva, O.; Take, Y.; Fulton, A.M. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. Oncoimmunology 2013, 2, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorrin, A.J.; Liu, C.; Cicalo, J.; Reader, J.; Najafali, D.; Zhang, Y.; Roque, D.M.; Huang, H.-C. Photodynamic Priming Improves the Anti-Migratory Activity of Prostaglandin E Receptor 4 Antagonist in Cancer Cells In Vitro. Cancers 2021, 13, 5259. https://doi.org/10.3390/cancers13215259
Sorrin AJ, Liu C, Cicalo J, Reader J, Najafali D, Zhang Y, Roque DM, Huang H-C. Photodynamic Priming Improves the Anti-Migratory Activity of Prostaglandin E Receptor 4 Antagonist in Cancer Cells In Vitro. Cancers. 2021; 13(21):5259. https://doi.org/10.3390/cancers13215259
Chicago/Turabian StyleSorrin, Aaron J., Cindy Liu, Julia Cicalo, Jocelyn Reader, Daniel Najafali, Yuji Zhang, Dana M. Roque, and Huang-Chiao Huang. 2021. "Photodynamic Priming Improves the Anti-Migratory Activity of Prostaglandin E Receptor 4 Antagonist in Cancer Cells In Vitro" Cancers 13, no. 21: 5259. https://doi.org/10.3390/cancers13215259
APA StyleSorrin, A. J., Liu, C., Cicalo, J., Reader, J., Najafali, D., Zhang, Y., Roque, D. M., & Huang, H.-C. (2021). Photodynamic Priming Improves the Anti-Migratory Activity of Prostaglandin E Receptor 4 Antagonist in Cancer Cells In Vitro. Cancers, 13(21), 5259. https://doi.org/10.3390/cancers13215259