
Identification of Biochemical and Molecular Markers of Early Aging in Childhood Cancer Survivors

 ,
,  , , , , , ,
, , , , , ,  , ,
, ,
Abstract
Simple Summary
Abstract
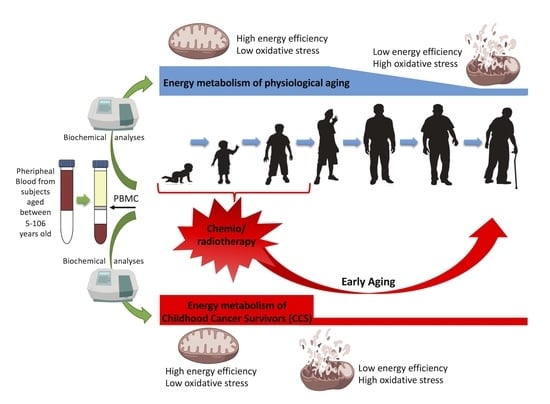
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Samples
2.3. Evaluation of Oxygen Consumption, ATP Synthesis, P/O Ratio, ATP/AMP Ratio, and Lactate Dehydrogenase Activity
2.4. ROS Production and Lipid Peroxidation Evaluation
2.5. Mitochondrial Trans Membrane Potential by Flow Cytometry and Confocal Microscopy
2.6. Proteomic Setup
2.7. qPCR Analyses
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
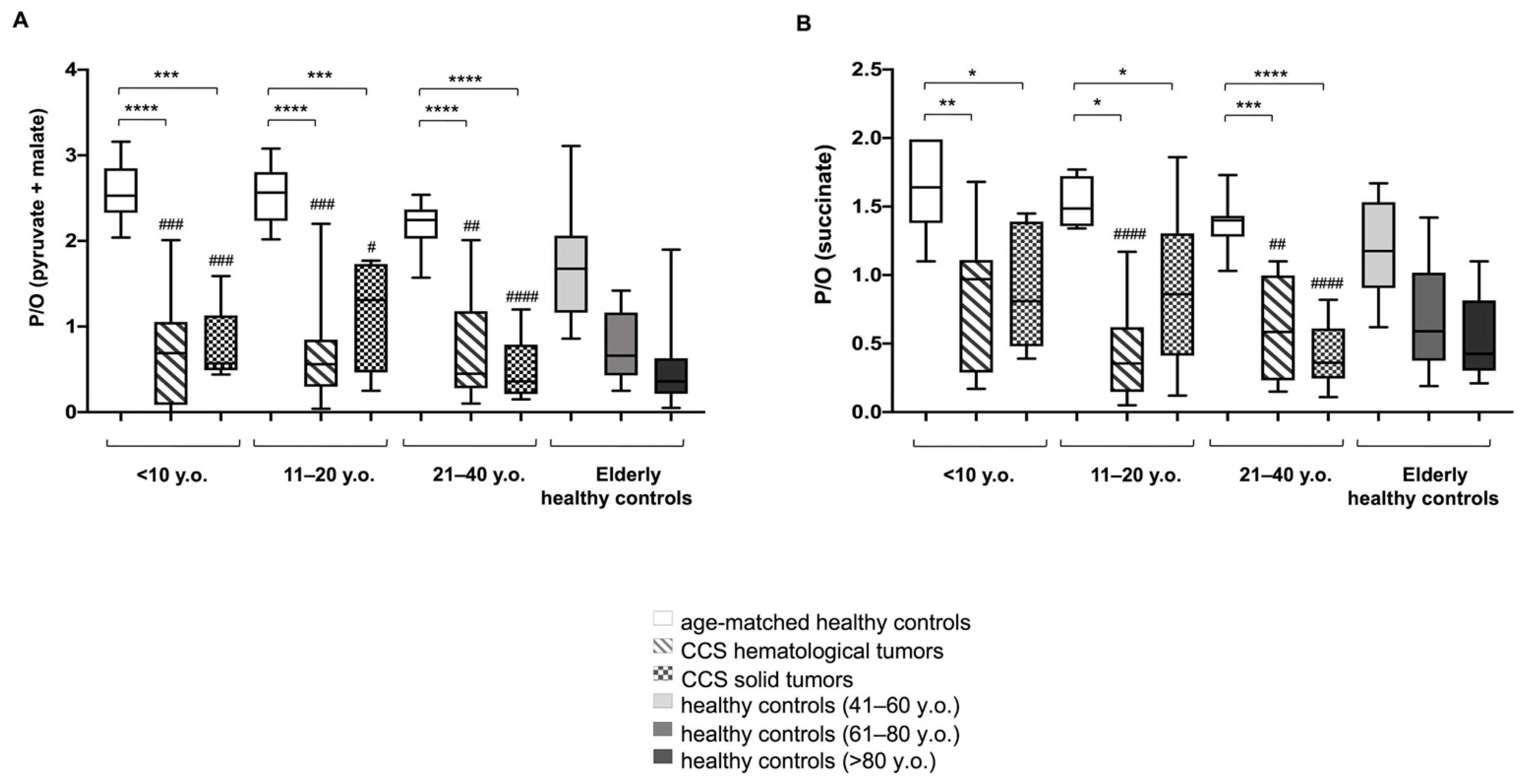
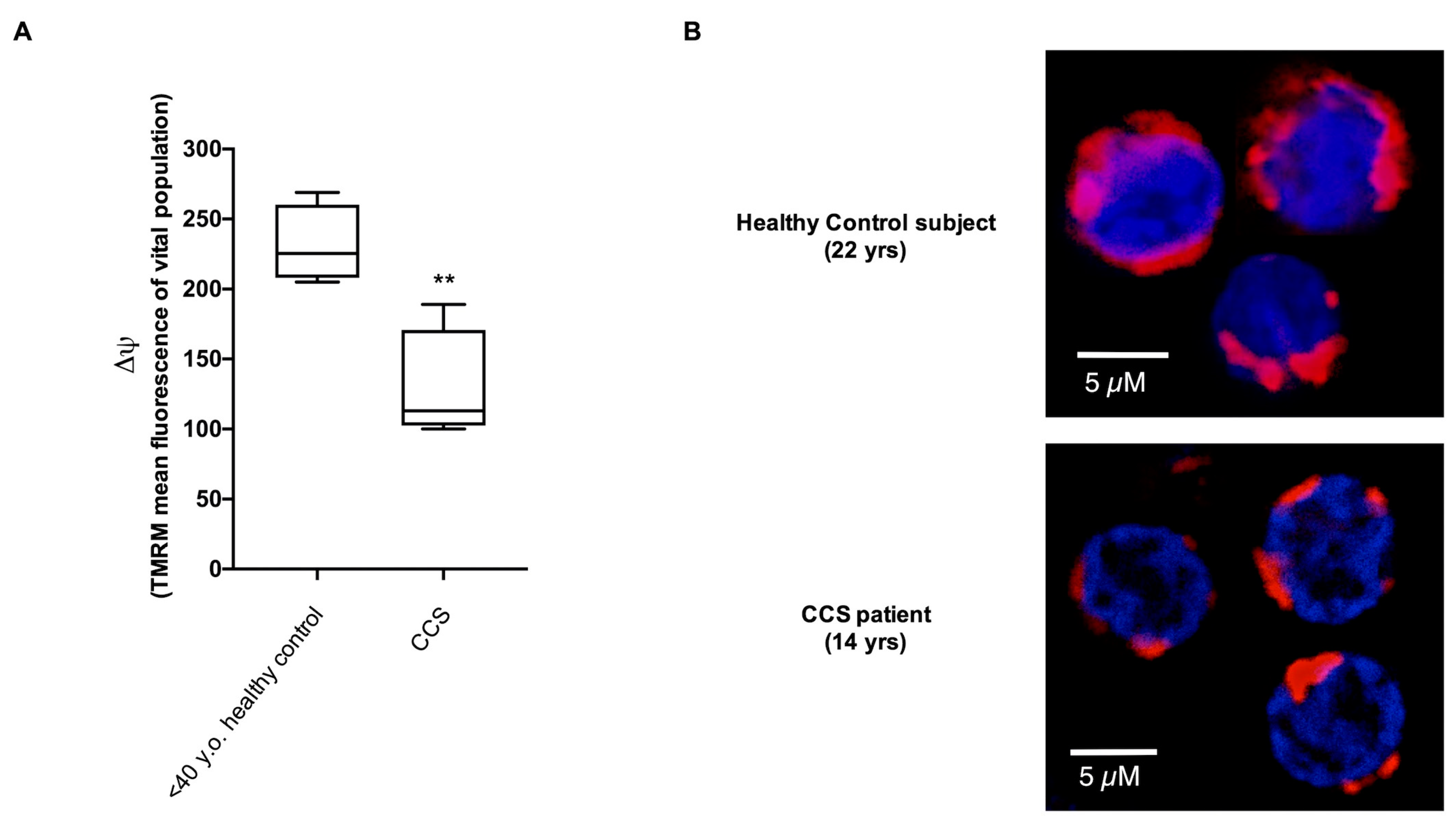
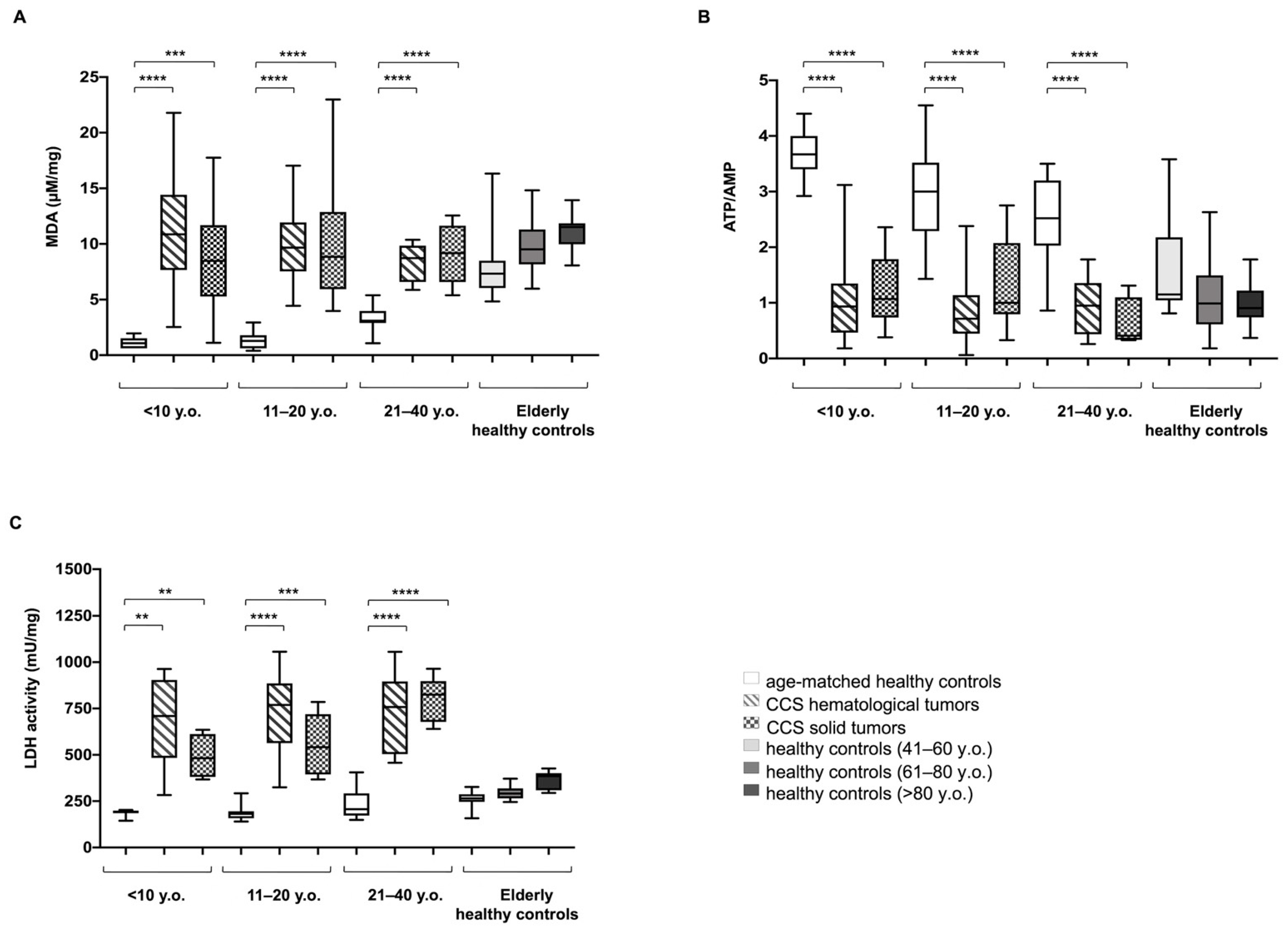
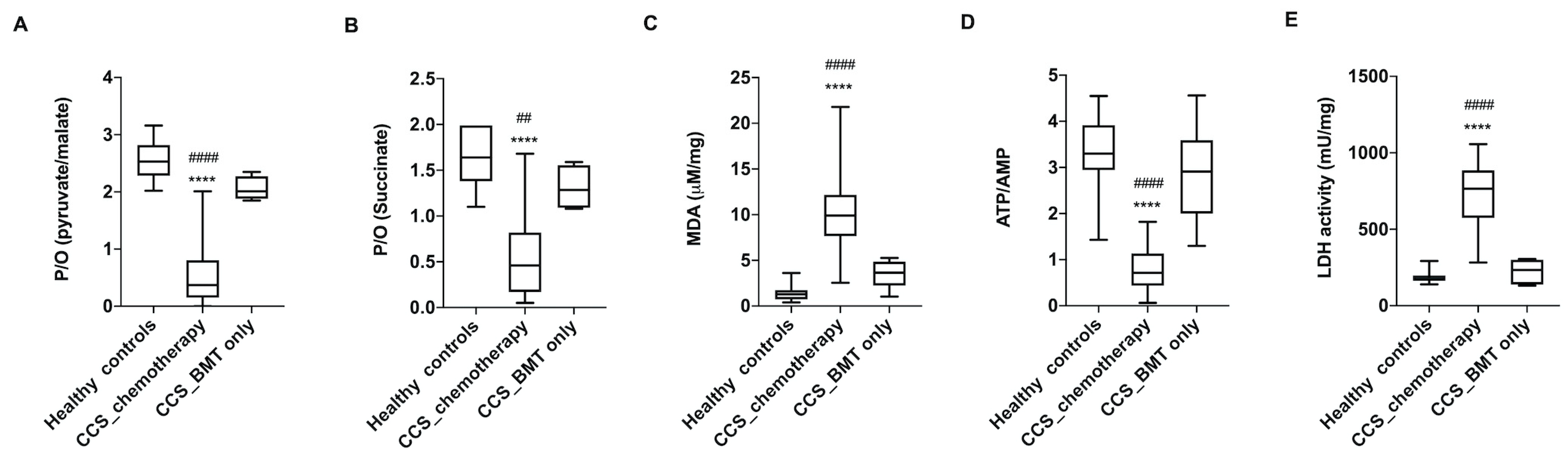
3.1. CCS-MNCs Showed Altered Glucose Metabolism and Increased Oxidative Stress Production in Comparison to the Age-Matched Control Cells
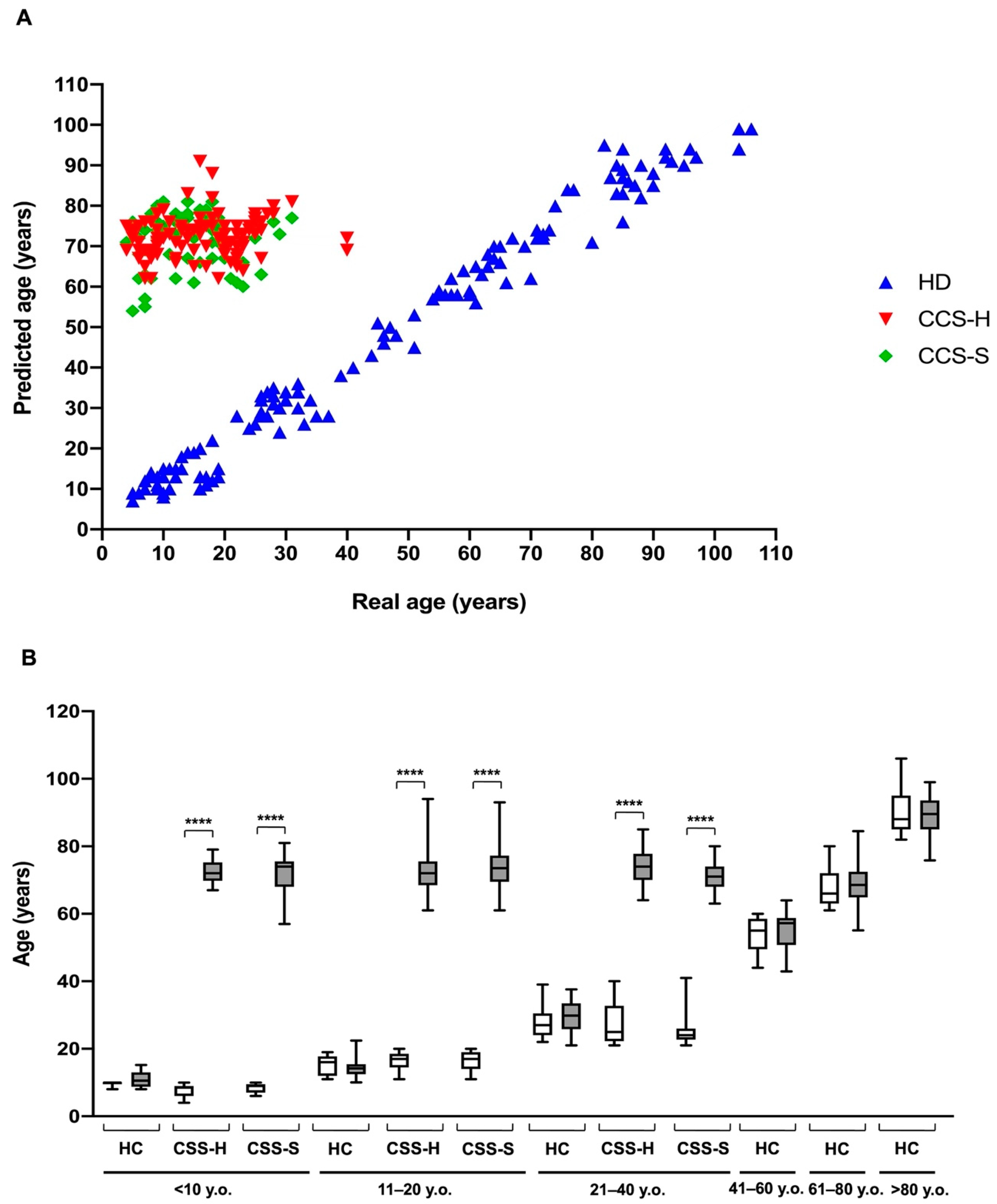
3.2. The Metabolic Alterations in CCS Are Suggestive of Premature Aging
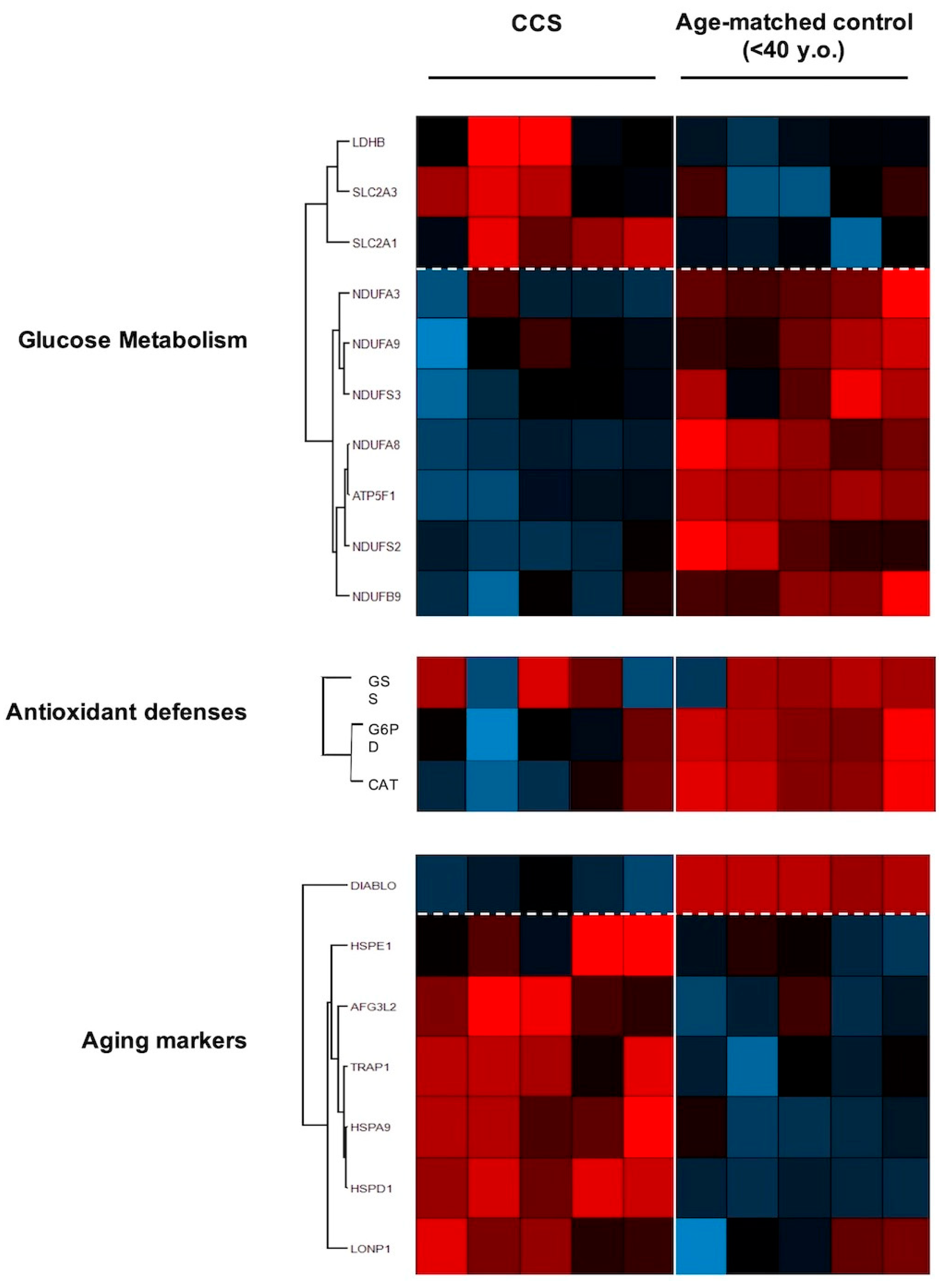
3.3. Proteomic Analysis Confirmed Altered Mitochondrial Metabolism
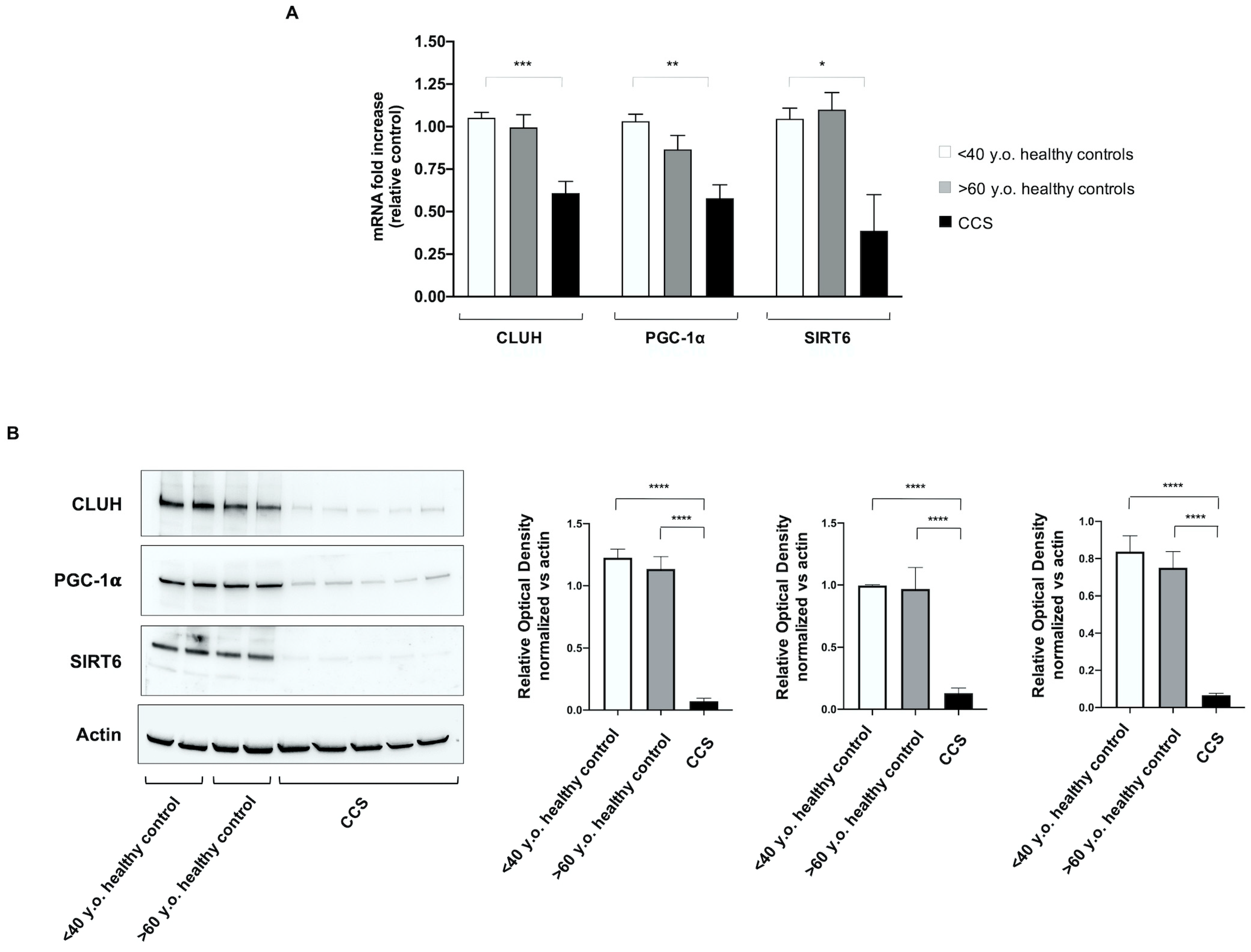
3.4. Genes Involved in Mitochondrial Biogenesis and Activity Regulation Are Altered in CCS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhatia, S.; Meadows, A.T. Long-term follow-up of childhood cancer survivors: Future directions for clinical care and research. Pediatr. Blood Cancer 2006, 46, 143–148. [Google Scholar] [CrossRef]
- Gatta, G.; Capocaccia, R.; Stiller, C.; Kaatsch, P.; Berrino, F.; Terenziani, M. Childhood cancer survival trends in Europe: A EUROCARE Working Group study. J. Clin. Oncol. 2005, 23, 3742–3751. [Google Scholar] [CrossRef]
- Shapiro, C.L. Cancer Survivorship. N. Engl. J. Med. 2018, 379, 2438–2450. [Google Scholar] [CrossRef]
- Nelson, M.B.; Meeske, K. Recognizing health risks in childhood cancer survivors. J. Am. Acad. Nurse Pract. 2005, 17, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Landier, W.; Skinner, R.; Wallace, W.H.; Hjorth, L.; Mulder, R.L.; Wong, F.L.; Yasui, Y.; Bhakta, N.; Constine, L.S.; Bhatia, S.; et al. Surveillance for Late Effects in Childhood Cancer Survivors. J. Clin. Oncol. 2018, 36, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Grabow, D.; Kaiser, M.; Hjorth, L.; Byrne, J.; Alessi, D.; Allodji, R.S.; Bagnasco, F.; Bárdi, E.; Bautz, A.; Bright, C.J.; et al. The PanCareSurFup cohort of 83,333 five-year survivors of childhood cancer: A cohort from 12 European countries. Eur. J. Epidemiol. 2018, 33, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.L.; Whitton, J.; Leisenring, W.; Mertens, A.C.; Hammond, S.; Stovall, M.; Donaldson, S.S.; Meadows, A.T.; Robison, L.L.; Neglia, J.P. Subsequent neoplasms in 5-year survivors of childhood cancer: The Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2010, 102, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.O.; Ness, K.K.; Cohen, H.J. Accelerated aging among cancer survivors: From pediatrics to geriatrics. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, e423–e430. [Google Scholar] [CrossRef] [PubMed]
- Ness, K.K.; Krull, K.R.; Jones, K.E.; Mulrooney, D.A.; Armstrong, G.T.; Green, D.M.; Chemaitilly, W.; Smith, W.A.; Wilson, C.L.; Sklar, C.A.; et al. Physiologic frailty as a sign of accelerated aging among adult survivors of childhood cancer: A report from the St Jude Lifetime cohort study. J. Clin. Oncol. 2013, 31, 4496–4503. [Google Scholar] [CrossRef]
- Armstrong, G.T.; Kawashima, T.; Leisenring, W.; Stratton, K.; Stovall, M.; Hudson, M.M.; Sklar, C.A.; Robison, L.L.; Oeffinger, K.C. Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J. Clin. Oncol. 2014, 32, 1218–1227. [Google Scholar] [CrossRef]
- Hartman, A.; van den Bos, C.; Stijnen, T.; Pieters, R. Decrease in peripheral muscle strength and ankle dorsiflexion as long-term side effects of treatment for childhood cancer. Pediatr. Blood Cancer 2008, 50, 833–837. [Google Scholar] [CrossRef]
- Ness, K.K.; Baker, K.S.; Dengel, D.R.; Youngren, N.; Sibley, S.; Mertens, A.C.; Gurney, J.G. Body composition, muscle strength deficits and mobility limitations in adult survivors of childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2007, 49, 975–981. [Google Scholar] [CrossRef]
- Krull, K.R.; Sabin, N.D.; Reddick, W.E.; Zhu, L.; Armstrong, G.T.; Green, D.M.; Arevalo, A.R.; Krasin, M.J.; Srivastava, D.K.; Robison, L.L.; et al. Neurocognitive function and CNS integrity in adult survivors of childhood hodgkin lymphoma. J. Clin. Oncol. 2012, 30, 3618–3624. [Google Scholar] [CrossRef]
- Schuitema, I.; Deprez, S.; van Hecke, W.; Daams, M.; Uyttebroeck, A.; Sunaert, S.; Barkhof, F.; van Dulmen-den Broeder, E.; van der Pal, H.J.; van den Bos, C.; et al. Accelerated aging, decreased white matter integrity, and associated neuropsychological dysfunction 25 years after pediatric lymphoid malignancies. J. Clin. Oncol. 2013, 31, 3378–3388. [Google Scholar] [CrossRef]
- Ness, K.K.; Armstrong, G.T.; Kundu, M.; Wilson, C.L.; Tchkonia, T.; Kirkland, J.L. Frailty in childhood cancer survivors. Cancer 2015, 121, 1540–1547. [Google Scholar] [CrossRef]
- Kadan-Lottick, N.S.; Zeltzer, L.K.; Liu, Q.; Yasui, Y.; Ellenberg, L.; Gioia, G.; Robison, L.L.; Krull, K.R. Neurocognitive functioning in adult survivors of childhood non-central nervous system cancers. J. Natl. Cancer Inst. 2010, 102, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Meeske, K.A.; Nelson, M.B. The role of the long-term follow-up clinic in discovering new emerging late effects in adult survivors of childhood cancer. J. Pediatr. Oncol. Nurs. 2008, 25, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Franco, V.I.; Miller, T.L.; Colan, S.D.; Sallan, S.E. Cardiovascular Disease in Adult Survivors of Childhood Cancer. Annu. Rev. Med. 2015, 66, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.T.; Liu, Q.; Yasui, Y.; Neglia, J.P.; Leisenring, W.; Robison, L.L.; Mertens, A.C. Late Mortality among 5-Year Survivors of Childhood Cancer: A Summary from the Childhood Cancer Survivor Study. J. Clin. Oncol. 2009, 27, 2328–2338. [Google Scholar] [CrossRef]
- Ness, K.K.; Kirkland, J.L.; Gramatges, M.M.; Wang, Z.; Kundu, M.; McCastlain, K.; Li-Harms, X.; Zhang, J.; Tchkonia, T.; Pluijm, S.M.F.; et al. Premature Physiologic Aging as a Paradigm for Understanding Increased Risk of Adverse Health Across the Lifespan of Survivors of Childhood Cancer. J. Clin. Oncol. 2018, 36, 2206–2215. [Google Scholar] [CrossRef]
- Gudmundsdottir, T.F.; Winther, J.; de Fine Licht, S.G.; Bonnesen, T.; H. Asdahl, P.; Tryggvadottir, L.; Anderson, H.; Wesenberg, F.; Malila, N.; Hasle, H.; et al. Cardiovascular disease in Adult Life after Childhood Cancer in Scandinavia: A population-based cohort study of 32,308 one-year survivors. Int. J. Cancer 2015, 137, 1176–1186. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Sastre, F.V.; Pallardo, J.J.; Pallardó, F.V.; Viña, J. Mitochondrial Oxidative Stress Plays a Key Role in Aging and Apoptosis. IUBMB Life 2000, 49, 427–435. [Google Scholar] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Fakouri, N.B.; Hou, Y.; Demarest, T.G.; Christiansen, L.S.; Okur, M.N.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. Toward understanding genomic instability, mitochondrial dysfunction and aging. FEBS J. 2019, 286, 1058–1073. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef]
- Ravera, S.; Podestà, M.; Sabatini, F.; Dagnino, M.; Cilloni, D.; Fiorini, S.; Barla, A.; Frassoni, F. Discrete Changes in Glucose Metabolism Define Aging. Sci. Rep. 2019, 9, 10347. [Google Scholar] [CrossRef]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Crawley, C.; Craig, J.; Scott, M.A.; Hodkinson, C.; et al. Leukemia-Associated Somatic Mutations Drive Distinct Patterns of Age-Related Clonal Hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef]
- Cappelli, E.; Cuccarolo, P.; Stroppiana, G.; Miano, M.; Bottega, R.; Cossu, V.; Degan, P.; Ravera, S. Defects in mitochondrial energetic function compels Fanconi Anaemia cells to glycolytic metabolism. Biochim. Biophys. Acta 2017, 1863, 1214–1221. [Google Scholar] [CrossRef]
- Hinkle, P.C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta 2005, 1706, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Podestà, M.; Sabatini, F.; Fresia, C.; Columbaro, M.; Bruno, S.; Fulcheri, E.; Ramenghi, L.A.; Frassoni, F. Mesenchymal stem cells from preterm to term newborns undergo a significant switch from anaerobic glycolysis to the oxidative phosphorylation. Cell. Mol. Life Sci. 2018, 75, 889–903. [Google Scholar] [CrossRef]
- Ravera, S.; Dufour, C.; Cesaro, S.; Bottega, R.; Faleschini, M.; Cuccarolo, P.; Corsolini, F.; Usai, C.; Columbaro, M.; Cipolli, M.; et al. Evaluation of energy metabolism and calcium homeostasis in cells affected by Shwachman-Diamond syndrome. Sci. Rep. 2016, 6, 25441. [Google Scholar] [CrossRef]
- Bruno, S.; Ghiotto, F.; Tenca, C.; Mazzarello, A.N.; Bono, M.; Luzzi, P.; Casciaro, S.; Recchia, A.; DeCensi, A.; Morabito, F.; et al. N-(4-hydroxyphenyl)retinamide promotes apoptosis of resting and proliferating B-cell chronic lymphocytic leukemia cells and potentiates fludarabine and ABT-737 cytotoxicity. Leukemia 2012, 26, 2260–2268. [Google Scholar] [CrossRef][Green Version]
- Cappelli, E.; Degan, P.; Bruno, S.; Pierri, F.; Miano, M.; Raggi, F.; Farruggia, P.; Mecucci, C.; Crescenzi, B.; Naim, V.; et al. The passage from bone marrow niche to bloodstream triggers the metabolic impairment in Fanconi Anemia mononuclear cells. Redox Biol. 2020, 36, 101618. [Google Scholar] [CrossRef]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Tiwari, B.S.; Belenghi, B.; Levine, A. Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol. 2002, 128, 1271–1281. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, N.; Liu, Q.; Ness, K.K.; Baassiri, M.; Eissa, H.; Yeo, F.; Chemaitilly, W.; Ehrhardt, M.J.; Bass, J.; Bishop, M.W.; et al. The cumulative burden of surviving childhood cancer: An initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet 2017, 390, 2569–2582. [Google Scholar] [CrossRef]
- Vassal, G.; Schrappe, M.; Pritchard-Jones, K.; Arnold, F.; Basset, L.; Biondi, A.; Bode, G.; Eggert, A.; Hjorth, L.; Kamerić, L.; et al. The SIOPE strategic plan: A European cancer plan for children and adolescents. J. Cancer Policy 2016, 8, 17–32. [Google Scholar] [CrossRef]
- Oeffinger, K.C.; Mertens, A.C.; Sklar, C.A.; Kawashima, T.; Hudson, M.M.; Meadows, A.T.; Friedman, D.L.; Marina, N.; Hobbie, W.; Kadan-Lottick, N.S.; et al. Chronic Health Conditions in Adult Survivors of Childhood Cancer. N. Engl. J. Med. 2006, 355, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Geenen, M.M.; Cardous-Ubbink, M.C.; Kremer, L.C.M.; van den Bos, C.; van der Pal, H.J.H.; Heinen, R.C.; Jaspers, M.W.M.; Koning, C.C.E.; Oldenburger, F.; Langeveld, N.E.; et al. Medical Assessment of Adverse Health Outcomes in Long-term Survivors of Childhood Cancer. JAMA 2007, 297, 2705. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.-F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.; Li, Z.; Hansen, J.; Hansen, J.J.; Nielsen, M.N.; Corydon, T.J.; Georgopoulos, C.; Ang, D.; Lundemose, J.B.; Niezen-Koning, K.; et al. Single-nucleotide variations in the genes encoding the mitochondrial Hsp60/Hsp10 chaperone system and their disease-causing potential. J. Hum. Genet. 2007, 52, 56–65. [Google Scholar] [CrossRef][Green Version]
- Bross, P.; Fernandez-Guerra, P. Disease-Associated Mutations in the HSPD1 Gene Encoding the Large Subunit of the Mitochondrial HSP60/HSP10 Chaperonin Complex. Front. Mol. Biosci. 2016, 3, 49. [Google Scholar] [CrossRef]
- Jadiya, P.; Tomar, D. Mitochondrial Protein Quality Control Mechanisms. Genes 2020, 11, 563. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef] [PubMed]
- Starenki, D.; Sosonkina, N.; Hong, S.-K.; Lloyd, R.V.; Park, J.-I. Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 2069. [Google Scholar] [CrossRef]
- Yoshida, S.; Tsutsumi, S.; Muhlebach, G.; Sourbier, C.; Lee, M.-J.; Lee, S.; Vartholomaiou, E.; Tatokoro, M.; Beebe, K.; Miyajima, N.; et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc. Natl. Acad. Sci. USA 2013, 110, E1604–E1612. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef]
- De Keizer, P.L.J. The Fountain of Youth by Targeting Senescent Cells? Trends Mol. Med. 2017, 23, 6–17. [Google Scholar] [CrossRef]
- Sanz, A.; Caro, P.; Gómez, J.; Barja, G. Testing the vicious cycle theory of mitochondrial ROS production: Effects of H2O2 and cumene hydroperoxide treatment on heart mitochondria. J. Bioenerg. Biomembr. 2006, 38, 121–127. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef]
- Hirst, J.; King, M.S.; Pryde, K.R. The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 2008, 36, 976–980. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef]
- Korge, P.; Calmettes, G.; Weiss, J.N. Increased reactive oxygen species production during reductive stress: The roles of mitochondrial glutathione and thioredoxin reductases. Biochim. Biophys. Acta 2015, 1847, 514–525. [Google Scholar] [CrossRef]
- Sociali, G.; Magnone, M.; Ravera, S.; Damonte, P.; Vigliarolo, T.; von Holtey, M.; Vellone, V.G.; Millo, E.; Caffa, I.; Cea, M.; et al. Pharmacological Sirt6 inhibition improves glucose tolerance in a type 2 diabetes mouse model. FASEB J. 2017, 31, 3138–3149. [Google Scholar] [CrossRef]
- Austin, S.; St-Pierre, J. PGC1a and mitochondrial metabolism-emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Schatton, D.; Martinelli, P.; Hansen, H.; Pla-Martin, D.; Barth, E.; Becker, C.; Altmueller, J.; Frommolt, P.; Sardiello, M.; et al. CLUH regulates mitochondrial biogenesis by binding mRNAs of nuclear-encoded mitochondrial proteins. J. Cell Biol. 2014, 207, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Schatton, D.; Pla-Martin, D.; Marx, M.-C.; Hansen, H.; Mourier, A.; Nemazanyy, I.; Pessia, A.; Zentis, P.; Corona, T.; Kondylis, V.; et al. CLUH regulates mitochondrial metabolism by controlling translation and decay of target mRNAs. J. Cell Biol. 2017, 216, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Papatsenko, D.; Niu, X.; Schaniel, C.; Moore, K. Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Rep. 2014, 2, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Lu, R. Sleeping Beauty Wakes Up the Clonal Succession Model for Homeostatic Hematopoiesis. Cell Stem Cell 2014, 15, 677–678. [Google Scholar] [CrossRef] [PubMed]
- Veltri, K.L.; Espiritu, M.; Singh, G. Distinct genomic copy number in mitochondria of different mammalian organs. J. Cell. Physiol. 1990, 143, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.A.; Fraser, C.; Muthalagu, N.; Bhola, P.D.; Chang, W.; McBrayer, S.K.; Cantlon, A.; Fisch, S.; Golomb-Mello, G.; Ryan, J.A.; et al. Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell 2017, 31, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Ariffin, H.; Azanan, M.S.; Abd Ghafar, S.S.; Oh, L.; Lau, K.H.; Thirunavakarasu, T.; Sedan, A.; Ibrahim, K.; Chan, A.; Chin, T.F.; et al. Young adult survivors of childhood acute lymphoblastic leukemia show evidence of chronic inflammation and cellular aging. Cancer 2017, 123, 4207–4214. [Google Scholar] [CrossRef]
- Berben, L.; Floris, G.; Wildiers, H.; Hatse, S. Cancer and Aging: Two Tightly Interconnected Biological Processes. Cancers 2021, 13, 1400. [Google Scholar] [CrossRef]
- Thomas, R.; Wong, W.S.W.; Saadon, R.; Vilboux, T.; Deeken, J.; Niederhuber, J.; Hourigan, S.K.; Yang, E. Gut microbial composition difference between pediatric ALL survivors and siblings. Pediatr. Hematol. Oncol. 2020, 37, 475–488. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Childhood Cancer Survivors | ||||||
|---|---|---|---|---|---|---|
| Age | Solid Tumors | Haematological Malignancies | ||||
| Years | Subjects N° | Female/Male | End of therapy (years) | Subjects N° | Female/Male | End of therapy (years) |
| <10 (Group 1) | 35 | 19/16 | 6 (1–9) | 32 | 25/7 | 2 (1–6) |
| 11–20 (Group 2) | 48 | 18/30 | 10 (3–17) | 38 | 16/22 | 4 (1–15) |
| 21–40 (Group 3) | 10 | 2/8 | 18 (4–25) | 33 | 21/12 | 9 (2–26) |
| Total number | 93 | 103 | ||||
| Controls | ||||||
| Age-Matched | Aged Controls | |||||
| Years | Subjects N° | Female/Male | Years | Subjects N° | Female/Male | |
| <10 | 29 | 14/15 | 41–60 | 27 | 22/5 | |
| 11–20 | 25 | 15/10 | 61–80 | 27 | 10/17 | |
| 21–40 | 25 | 15/10 | >80 | 25 | 17/8 | |
| Total number | 79 | 75 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravera, S.; Vigliarolo, T.; Bruno, S.; Morandi, F.; Marimpietri, D.; Sabatini, F.; Dagnino, M.; Petretto, A.; Bartolucci, M.; Muraca, M.; et al. Identification of Biochemical and Molecular Markers of Early Aging in Childhood Cancer Survivors. Cancers 2021, 13, 5214. https://doi.org/10.3390/cancers13205214
Ravera S, Vigliarolo T, Bruno S, Morandi F, Marimpietri D, Sabatini F, Dagnino M, Petretto A, Bartolucci M, Muraca M, et al. Identification of Biochemical and Molecular Markers of Early Aging in Childhood Cancer Survivors. Cancers. 2021; 13(20):5214. https://doi.org/10.3390/cancers13205214
Chicago/Turabian StyleRavera, Silvia, Tiziana Vigliarolo, Silvia Bruno, Fabio Morandi, Danilo Marimpietri, Federica Sabatini, Monica Dagnino, Andrea Petretto, Martina Bartolucci, Monica Muraca, and et al. 2021. "Identification of Biochemical and Molecular Markers of Early Aging in Childhood Cancer Survivors" Cancers 13, no. 20: 5214. https://doi.org/10.3390/cancers13205214
APA StyleRavera, S., Vigliarolo, T., Bruno, S., Morandi, F., Marimpietri, D., Sabatini, F., Dagnino, M., Petretto, A., Bartolucci, M., Muraca, M., Biasin, E., Haupt, R., Zecca, M., Fagioli, F., Cilloni, D., Podestà, M., & Frassoni, F. (2021). Identification of Biochemical and Molecular Markers of Early Aging in Childhood Cancer Survivors. Cancers, 13(20), 5214. https://doi.org/10.3390/cancers13205214