
Potential of miRNA-Based Nanotherapeutics for Uveal Melanoma

Abstract
Simple Summary
Abstract

1. Introduction
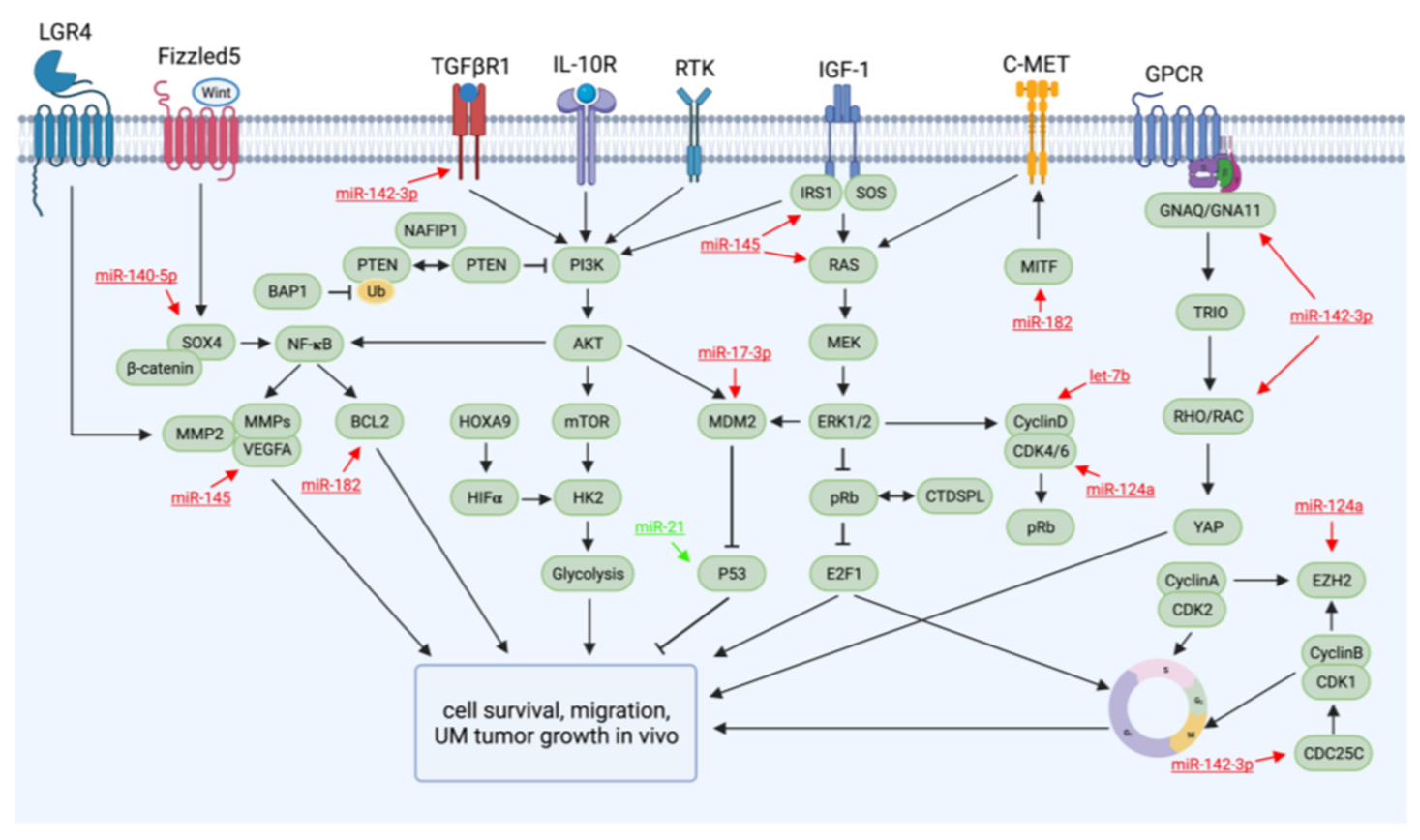
2. Dysregulated Pathways and Molecules Involved in UM Tumorigenesis and Metastasis
3. miRNAs with Therapeutic Potential for UM, Identified in Preclinical Studies
4. Approaches of Therapeutic Targeting of miRNAs and Limitations of miRNAs in Translational Therapeutics
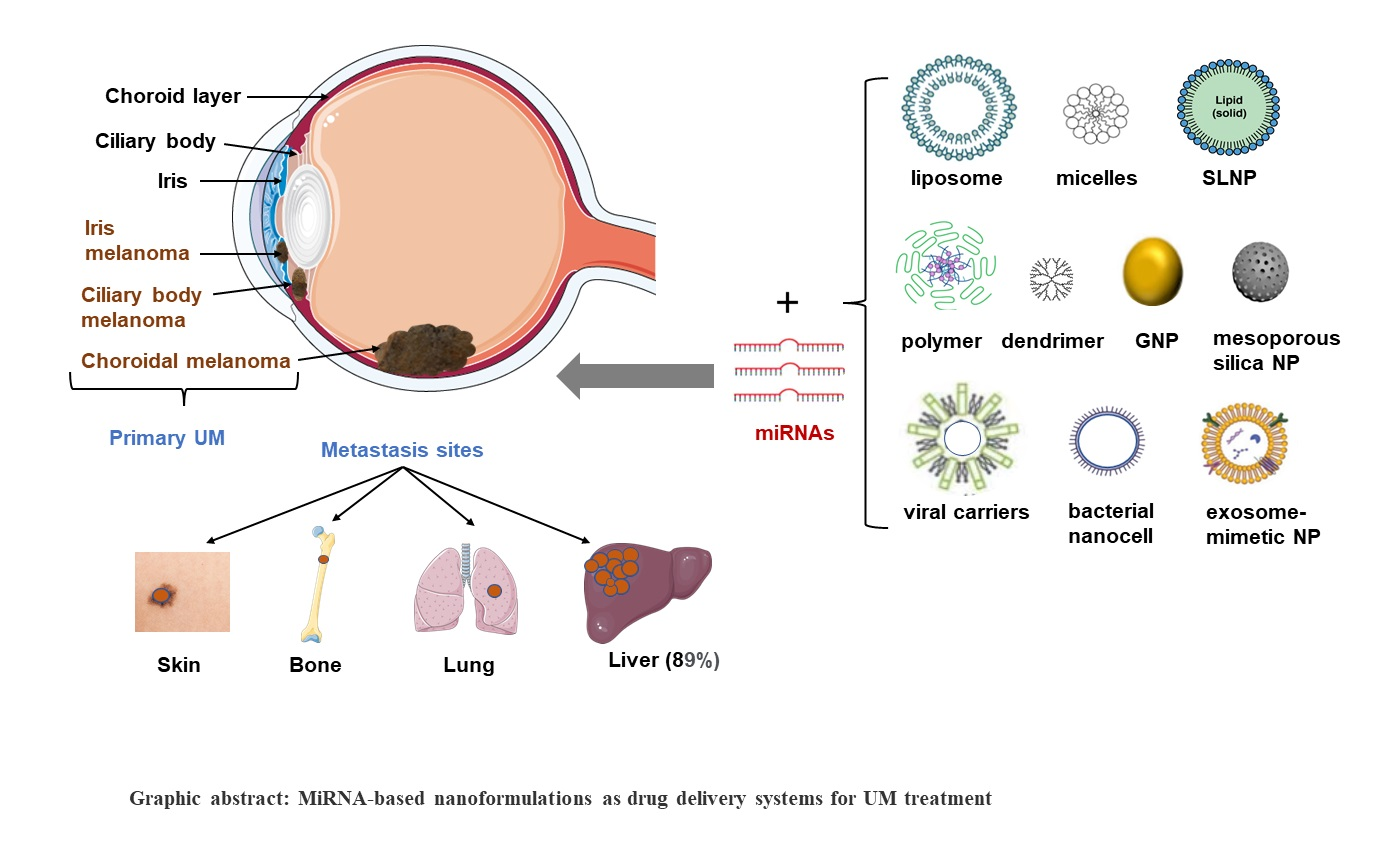
5. Nanotechnology-Based miRNA Delivery Systems
5.1. Nanodelivery Systems for miRNA Therapeutics
5.2. The Developed Nanocarriers/NPs Relevant to the Potential Therapeutic miRNAs for UM
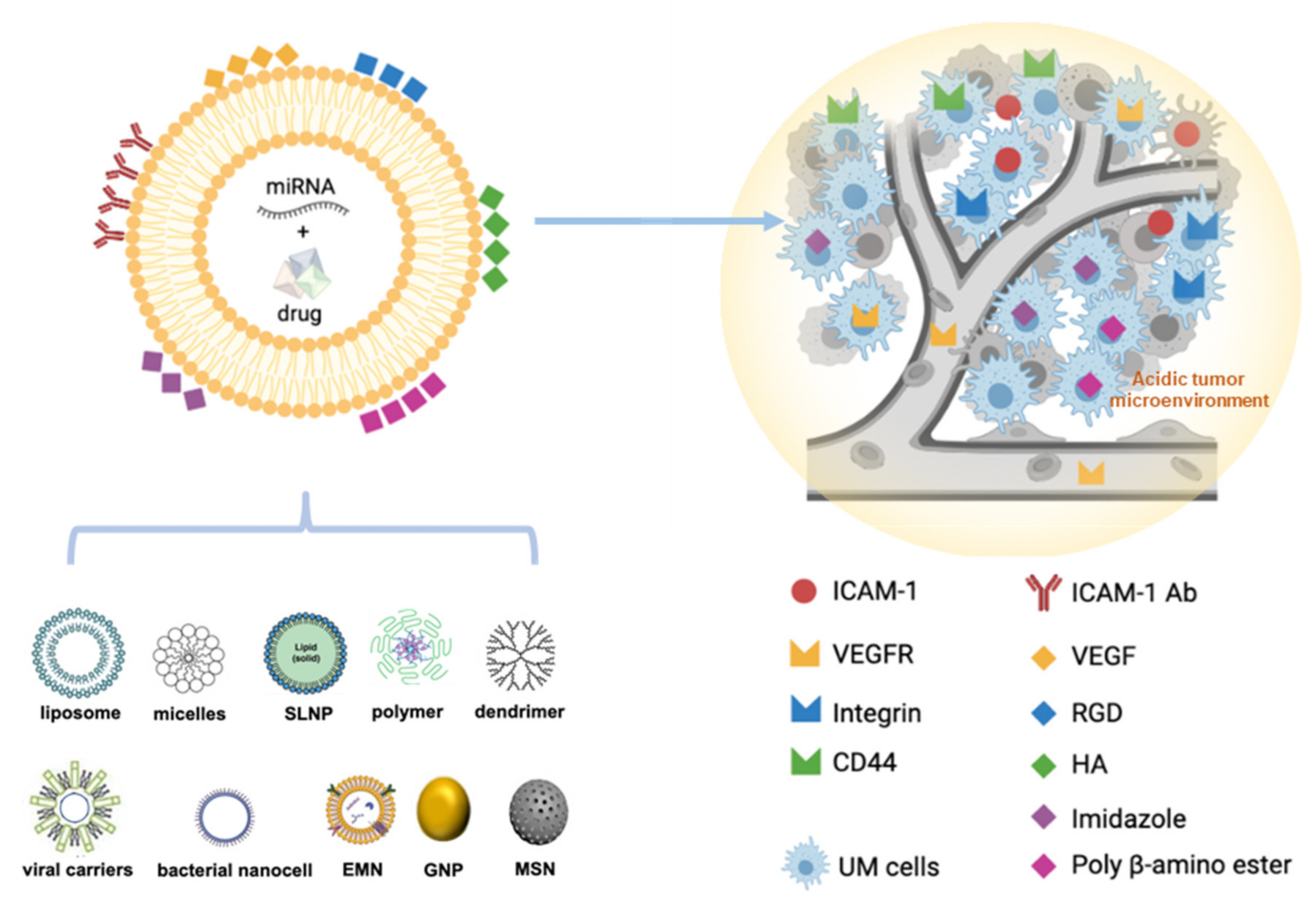
5.3. Modification of NP Surfaces for Improving Biocompatibility and Active Targeting
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| antimiRs | miRNA antisense oligonucleotides |
| BAP1 | BRCA associated protein 1 |
| BCL2 | B-cell lymphoma 2 |
| CDC25C | Cell division cycle 25 homolog c |
| CDK | Cyclin-dependent kinase |
| DOTAP | 1,2-dioleoyl-3-trimethylammonium-propane |
| EIF1AX | Eukaryotic translation initiation factor 1A X-linked |
| ERK | Extracellular-signal-regulated kinase |
| EZH2 | Enhancer of zeste homolog 2 |
| GNAQ | Guanine nucleotide-binding protein alpha-Q |
| GNAQ/11 | Guanine nucleotide-binding protein alpha-Q and subunit alpha-11 |
| GNPs | Gold nanoparticles |
| HA | Hyaluronic acid |
| HGF | Hepatocyte growth factor |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IRS-1 | Insulin receptor substrate-1 |
| LASP1 | LIM and SH3 protein 1 |
| LNA | Locked nucleic acid oligonucleotide |
| MAPK | Mitogen-activated protein kinase |
| MDM2 | Murine double-minute clone 2 oncoprotein |
| miRNA | MicroRNA |
| MITF | Melanogenesis-associated transcription factor |
| MMPs | Matrix metalloproteinases |
| mUM | Metastatic uveal melanoma |
| NPs | Nanoparticles |
| PAMAM | Polyamidoamine |
| PDX | Patient-derived xenograft |
| PEG | Polyethylene glycol |
| PEI | Polyethyleneimine |
| PI3K | Phosphatidylinositol 3-kinase |
| PLGA | Polylactide-co-glycolide |
| PNIPAM | Poly N-isopropylacrylamide |
| PTEN | Phosphatase and tensin homolog |
| RAC1 | ras-related c3 botulinum toxin substrate 1 |
| RB | Retinoblastoma protein |
| RGD | Arginylglycylaspartic acid |
| SF3B1 | Splicing factor 3b subunit 1 |
| SLNPs | Solid lipid nanoparticles |
| TGFβR1 | Transforming growth factor beta receptor 1 |
| UM | Uveal melanoma |
| VEGF | Vascular endothelial growth factor |
| VEGFR | VEGF receptor |
| WASL | Wiskott–Aldrich-syndrome-like |
References
- Basile, M.S.; Mazzon, E.; Fagone, P.; Longo, A.; Russo, A.; Fallico, M.; Bonfiglio, V.; Nicoletti, F.; Avitabile, T.; Reibaldi, M. Immunobiology of Uveal Melanoma: State of the Art and Therapeutic Targets. Front. Oncol. 2019, 9, 1145. [Google Scholar] [CrossRef] [PubMed]
- Vsan den Bosch, T.; Kilic, E.; Paridaens, D.; de Klein, A. Genetics of uveal melanoma and cutaneous melanoma: Two of a kind? Dermatol. Res. Pract. 2010, 2010, 360136. [Google Scholar] [CrossRef] [PubMed]
- Berus, T.; Halon, A.; Markiewicz, A.; Orlowska-Heitzman, J.; Romanowska-Dixon, B.; Donizy, P. Clinical, Histopathological and Cytogenetic Prognosticators in Uveal Melanoma—A Comprehensive Review. Anticancer. Res. 2017, 37, 6541–6549. [Google Scholar] [PubMed]
- Messineo, D.; Barile, G.; Morrone, S.; La Torre, G.; Turchetti, P.; Accetta, L.; Trovato Battagliola, E.; Agostinelli, E.; Pacella, F. Meta-analysis on the utility of radiotherapy for the treatment of Ocular Melanoma. La Clin. Ter. 2020, 170, e89–e98. [Google Scholar]
- Krantz, B.A.; Dave, N.; Komatsubara, K.M.; Marr, B.P.; Carvajal, R.D. Uveal melanoma: Epidemiology, etiology, and treatment of primary disease. Clin. Ophthalmol. 2017, 11, 279–289. [Google Scholar] [CrossRef]
- Rantala, E.S.; Hernberg, M.; Kivelä, T.T. Overall survival after treatment for metastatic uveal melanoma: A systematic review and meta-analysis. Melanoma Res. 2019, 29, 561–568. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; García-Honduvilla, N.; Coca, S.; Álvarez-Mon, M.; Buján, J.; Teus, M.A. Update on uveal melanoma: Translational research from biology to clinical practice (Review). Int. J. Oncol. 2020, 57, 1262–1279. [Google Scholar] [CrossRef]
- De Lange, M.J.; Razzaq, L.; Versluis, M.; Verlinde, S.; Dogrusoz, M.; Bohringer, S.; Marinkovic, M.; Luyten, G.P.; de Keizer, R.J.; de Gruijl, F.R.; et al. Distribution of GNAQ and GNA11 Mutation Signatures in Uveal Melanoma Points to a Light Dependent Mutation Mechanism. PLoS ONE 2015, 10, e0138002. [Google Scholar] [CrossRef][Green Version]
- Versluis, M.; de Lange, M.J.; van Pelt, S.I.; Ruivenkamp, C.A.; Kroes, W.G.; Cao, J.; Jager, M.J.; Luyten, G.P.; van der Velden, P.A. Digital PCR validates 8q dosage as prognostic tool in uveal melanoma. PLoS ONE 2015, 10, e0116371. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Smit, K.N.; Jager, M.J.; de Klein, A.; Kiliҫ, E. Uveal melanoma: Towards a molecular understanding. Prog. Retin. Eye Res. 2020, 75, 100800. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. New Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef]
- Vaque, J.P.; Dorsam, R.T.; Feng, X.; Iglesias-Bartolome, R.; Forsthoefel, D.J.; Chen, Q.; Debant, A.; Seeger, M.A.; Ksander, B.R.; Teramoto, H.; et al. A genome-wide RNAi screen reveals a Trio-regulated Rho GTPase circuitry transducing mitogenic signals initiated by G protein-coupled receptors. Mol. Cell 2013, 49, 94–108. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Kazmi, M.A.; Taylor, B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; van Bodegom, A.; Paridaens, D.; Kiliç, E.; de Klein, A. Uveal Melanomas with SF3B1 Mutations: A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef]
- Martin, M.; Maßhöfer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef]
- Saraiva, V.S.; Caissie, A.L.; Segal, L.; Edelstein, C.; Burnier, M.N., Jr. Immunohistochemical expression of phospho-Akt in uveal melanoma. Melanoma Res. 2005, 15, 245–250. [Google Scholar] [CrossRef]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef]
- Väisänen, A.; Kallioinen, M.; von Dickhoff, K.; Laatikainen, L.; Höyhtyä, M.; Turpeenniemi-Hujanen, T. Matrix metalloproteinase-2 (MMP-2) immunoreactive protein--a new prognostic marker in uveal melanoma? J. Pathol. 1999, 188, 56–62. [Google Scholar] [CrossRef]
- Lai, K.; Conway, R.M.; Crouch, R.; Jager, M.J.; Madigan, M.C. Expression and distribution of MMPs and TIMPs in human uveal melanoma. Exp. Eye Res. 2008, 86, 936–941. [Google Scholar] [CrossRef]
- Béliveau, A.; Bérubé, M.; Rousseau, A.; Pelletier, G.; Guérin, S.L. Expression of integrin alpha5beta1 and MMPs associated with epithelioid morphology and malignancy of uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2363–2372. [Google Scholar]
- Babchia, N.; Landreville, S.; Clément, B.; Coulouarn, C.; Mouriaux, F. The bidirectional crosstalk between metastatic uveal melanoma cells and hepatic stellate cells engenders an inflammatory microenvironment. Exp. Eye Res. 2019, 181, 213–222. [Google Scholar] [CrossRef]
- Barisione, G.; Fabbi, M.; Gino, A.; Queirolo, P.; Orgiano, L.; Spano, L.; Picasso, V.; Pfeffer, U.; Mosci, C.; Jager, M.J.; et al. Potential Role of Soluble c-Met as a New Candidate Biomarker of Metastatic Uveal Melanoma. JAMA Ophthalmol. 2015, 133, 1013–1021. [Google Scholar] [CrossRef]
- Vivet-Noguer, R.; Tarin, M.; Roman-Roman, S.; Alsafadi, S. Emerging Therapeutic Opportunities Based on Current Knowledge of Uveal Melanoma Biology. Cancers 2019, 11, 1019. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Kirschmann, D.A.; Gardner, L.M.; Boldt, H.C.; Meyer, M.; Pe’er, J.; Folberg, R. Regulation of uveal melanoma interconverted phenotype by hepatocyte growth factor/scatter factor (HGF/SF). Am. J. Pathol. 1998, 152, 855–863. [Google Scholar]
- Yoshida, M.; Selvan, S.; McCue, P.A.; DeAngelis, T.; Baserga, R.; Fujii, A.; Rui, H.; Mastrangelo, M.J.; Sato, T. Expression of insulin-like growth factor-1 receptor in metastatic uveal melanoma and implications for potential autocrine and paracrine tumor cell growth. Pigment. Cell Melanoma Res. 2014, 27, 297–308. [Google Scholar] [CrossRef]
- Barak, V.; Pe’er, J.; Kalickman, I.; Frenkel, S. VEGF as a biomarker for metastatic uveal melanoma in humans. Curr. Eye Res. 2011, 36, 386–390. [Google Scholar] [CrossRef]
- Koch, K.R.; Refaian, N.; Hos, D.; Schlereth, S.L.; Bosch, J.J.; Cursiefen, C.; Heindl, L.M. Autocrine impact of VEGF-A on uveal melanoma cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2697–2704. [Google Scholar] [CrossRef]
- Sharma, A.; Stei, M.M.; Fröhlich, H.; Holz, F.G.; Loeffler, K.U.; Herwig-Carl, M.C. Genetic and epigenetic insights into uveal melanoma. Clin. Genet. 2018, 93, 952–961. [Google Scholar] [CrossRef]
- Field, M.G.; Durante, M.A.; Anbunathan, H.; Cai, L.Z.; Decatur, C.L.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Punctuated evolution of canonical genomic aberrations in uveal melanoma. Nat. Commun. 2018, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Maat, W.; van der Velden, P.A.; Out-Luiting, C.; Plug, M.; Dirks-Mulder, A.; Jager, M.J.; Gruis, N.A. Epigenetic inactivation of RASSF1a in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 486–490. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Venza, M.; Visalli, M.; Biondo, C.; Lentini, M.; Catalano, T.; Teti, D.; Venza, I. Epigenetic regulation of p14ARF and p16INK4A expression in cutaneous and uveal melanoma. Biochim. Biophys. Acta 2015, 1849, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Venza, M.; Visalli, M.; Catalano, T.; Fortunato, C.; Oteri, R.; Teti, D.; Venza, I. Impact of DNA methyltransferases on the epigenetic regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in malignant melanoma. Biochem. Biophys. Res. Commun. 2013, 441, 743–750. [Google Scholar] [CrossRef]
- van der Velden, P.A.; Metzelaar-Blok, J.A.; Bergman, W.; Monique, H.; Hurks, H.; Frants, R.R.; Gruis, N.A.; Jager, M.J. Promoter hypermethylation: A common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res. 2001, 61, 5303–5306. [Google Scholar]
- Abdel-Rahman, M.H.; Yang, Y.; Zhou, X.P.; Craig, E.L.; Davidorf, F.H.; Eng, C. High frequency of submicroscopic hemizygous deletion is a major mechanism of loss of expression of PTEN in uveal melanoma. J. Clin. Oncol. 2006, 24, 288–295. [Google Scholar] [CrossRef]
- Park, J.J.; Diefenbach, R.J.; Joshua, A.M.; Kefford, R.F.; Carlino, M.S.; Rizos, H. Oncogenic signaling in uveal melanoma. Pigment. Cell Melanoma Res. 2018, 31, 661–672. [Google Scholar] [CrossRef]
- Chua, V.; Lapadula, D.; Randolph, C.; Benovic, J.L.; Wedegaertner, P.B.; Aplin, A.E. Dysregulated GPCR Signaling and Therapeutic Options in Uveal Melanoma. Mol. Cancer Res. MCR 2017, 15, 501–506. [Google Scholar] [CrossRef]
- Croce, M.; Ferrini, S.; Pfeffer, U.; Gangemi, R. Targeted Therapy of Uveal Melanoma: Recent Failures and New Perspectives. Cancers 2019, 11, 846. [Google Scholar] [CrossRef]
- Li, Y.; Shi, J.; Yang, J.; Ge, S.; Zhang, J.; Jia, R.; Fan, X. Uveal melanoma: Progress in molecular biology and therapeutics. Ther Adv. Med. Oncol. 2020, 12, 1758835920965852. [Google Scholar] [CrossRef]
- Ambrosini, G.; Pratilas, C.A.; Qin, L.X.; Tadi, M.; Surriga, O.; Carvajal, R.D.; Schwartz, G.K. Identification of unique MEK-dependent genes in GNAQ mutant uveal melanoma involved in cell growth, tumor cell invasion, and MEK resistance. Clin. Cancer Res. 2012, 18, 3552–3561. [Google Scholar] [CrossRef]
- Steeb, T.; Wessely, A.; Ruzicka, T.; Heppt, M.V.; Berking, C. How to MEK the best of uveal melanoma: A systematic review on the efficacy and safety of MEK inhibitors in metastatic or unresectable uveal melanoma. Eur. J. Cancer 2018, 103, 41–51. [Google Scholar] [CrossRef]
- Daud, A.; Kluger, H.M.; Kurzrock, R.; Schimmoller, F.; Weitzman, A.L.; Samuel, T.A.; Moussa, A.H.; Gordon, M.S.; Shapiro, G.I. Phase II randomised discontinuation trial of the MET/VEGF receptor inhibitor cabozantinib in metastatic melanoma. Br. J. Cancer 2017, 116, 432–440. [Google Scholar] [CrossRef]
- Mahipal, A.; Tijani, L.; Chan, K.; Laudadio, M.; Mastrangelo, M.J.; Sato, T. A pilot study of sunitinib malate in patients with metastatic uveal melanoma. Melanoma Res. 2012, 22, 440–446. [Google Scholar] [CrossRef]
- Valsecchi, M.E.; Orloff, M.; Sato, R.; Chervoneva, I.; Shields, C.L.; Shields, J.A.; Mastrangelo, M.J.; Sato, T. Adjuvant Sunitinib in High-Risk Patients with Uveal Melanoma: Comparison with Institutional Controls. Ophthalmology 2018, 125, 210–217. [Google Scholar] [CrossRef]
- Yang, J.; Manson, D.K.; Marr, B.P.; Carvajal, R.D. Treatment of uveal melanoma: Where are we now? Ther. Adv. Med. Oncol. 2018, 10, 1758834018757175. [Google Scholar] [CrossRef]
- Yang, C.; Wang, Y.; Hardy, P. Emerging roles of microRNAs and their implications in uveal melanoma. Cell. Mol. Life Sci. CMLS 2020. [Google Scholar] [CrossRef]
- Peng, D.; Dong, J.; Zhao, Y.; Peng, X.; Tang, J.; Chen, X.; Wang, L.; Hu, D.N.; Reinach, P.S.; Qu, J.; et al. miR-142-3p suppresses uveal melanoma by targeting CDC25C, TGFbetaR1, GNAQ, WASL, and RAC1. Cancer Manag. Res. 2019, 11, 4729–4742. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Q.; Gao, X.; Shi, D.; Mi, S.; Han, Q. MicroRNA-454 functions as an oncogene by regulating PTEN in uveal melanoma. FEBS Lett. 2015, 589, 2791–2796. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef]
- Aughton, K.; Kalirai, H.; Coupland, S.E. MicroRNAs and Uveal Melanoma: Understanding the Diverse Role of These Small Molecular Regulators. Int. J. Mol. Sci. 2020, 21, 5648. [Google Scholar] [CrossRef]
- Wang, Y.C.; Yang, X.; Wei, W.B.; Xu, X.L. Role of microRNA-21 in uveal melanoma cell invasion and metastasis by regulating p53 and its downstream protein. Int. J. Ophthalmol. 2018, 11, 1258–1268. [Google Scholar] [PubMed]
- Zhou, Y.; Zhang, L.; Fan, J.; Jia, R.; Song, X.; Xu, X.; Dai, L.; Zhuang, A.; Ge, S.; Fan, X. Let-7b overexpression leads to increased radiosensitivity of uveal melanoma cells. Melanoma Res. 2015, 25, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Chen, H.; Han, N.; Zhang, C.; Yan, H. Long Noncoding RNA PVT1 Silencing Prevents the Development of Uveal Melanoma by Impairing MicroRNA-17-3p-Dependent MDM2 Upregulation. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4904–4914. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Wang, J.Y. Targeting the RB-pathway in cancer therapy. Clin. Cancer Res. 2010, 16, 1094–1099. [Google Scholar] [CrossRef]
- Chen, X.; He, D.; Dong, X.D.; Dong, F.; Wang, J.; Wang, L.; Tang, J.; Hu, D.N.; Yan, D.; Tu, L. MicroRNA-124a is epigenetically regulated and acts as a tumor suppressor by controlling multiple targets in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2248–2256. [Google Scholar] [CrossRef]
- Moreno, C.S. SOX4: The unappreciated oncogene. Semin. Cancer Biol. 2020, 67, 57–64. [Google Scholar] [CrossRef]
- Zhao, G.; Yin, Y.; Zhao, B. miR-140-5p is negatively correlated with proliferation, invasion, and tumorigenesis in malignant melanoma by targeting SOX4 via the Wnt/β-catenin and NF-κB cascades. J. Cell. Physiol. 2020, 235, 2161–2170. [Google Scholar] [CrossRef]
- Li, Y.; Huang, Q.; Shi, X.; Jin, X.; Shen, L.; Xu, X.; Wei, W. MicroRNA 145 may play an important role in uveal melanoma cell growth by potentially targeting insulin receptor substrate-1. Chin. Med. J. 2014, 127, 1410–1416. [Google Scholar]
- Yang, J.Y.; Li, Y.; Wang, Q.; Zhou, W.J.; Yan, Y.N.; Wei, W.B. MicroRNA-145 suppresses uveal melanoma angiogenesis and growth by targeting neuroblastoma RAS viral oncogene homolog and vascular endothelial growth factor. Chin. Med. J. 2020, 133, 1922–1929. [Google Scholar] [CrossRef]
- Yan, D.; Dong, X.D.; Chen, X.; Yao, S.; Wang, L.; Wang, J.; Wang, C.; Hu, D.N.; Qu, J.; Tu, L. Role of microRNA-182 in posterior uveal melanoma: Regulation of tumor development through MITF, BCL2 and cyclin D2. PLoS ONE 2012, 7, e40967. [Google Scholar] [CrossRef]
- McGill, G.G.; Haq, R.; Nishimura, E.K.; Fisher, D.E. c-Met expression is regulated by Mitf in the melanocyte lineage. J. Biol. Chem. 2006, 281, 10365–10373. [Google Scholar] [CrossRef]
- Bakalian, S.; Marshall, J.C.; Logan, P.; Faingold, D.; Maloney, S.; Di Cesare, S.; Martins, C.; Fernandes, B.F.; Burnier, M.N., Jr. Molecular pathways mediating liver metastasis in patients with uveal melanoma. Clin. Cancer Res. 2008, 14, 951–956. [Google Scholar] [CrossRef]
- Tabatabaei, S.N.; Derbali, R.M.; Yang, C.; Superstein, R.; Hamel, P.; Chain, J.L.; Hardy, P. Co-delivery of miR-181a and melphalan by lipid nanoparticles for treatment of seeded retinoblastoma. J. Control. Release 2019, 298, 177–185. [Google Scholar] [CrossRef]
- Bader, A.G.; Brown, D.; Winkler, M. The promise of microRNA replacement therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar] [CrossRef]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef]
- Aquino-Jarquin, G. Emerging Role of CRISPR/Cas9 Technology for MicroRNAs Editing in Cancer Research. Cancer Res. 2017, 77, 6812–6817. [Google Scholar] [CrossRef]
- Guzman-Villanueva, D.; El-Sherbiny, I.M.; Herrera-Ruiz, D.; Vlassov, A.V.; Smyth, H.D. Formulation approaches to short interfering RNA and MicroRNA: Challenges and implications. J. Pharm. Sci. 2012, 101, 4046–4066. [Google Scholar] [CrossRef]
- Reda El Sayed, S.; Cristante, J.; Guyon, L.; Denis, J.; Chabre, O.; Cherradi, N. MicroRNA Therapeutics in Cancer: Current Advances and Challenges. Cancers 2021, 13, 2680. [Google Scholar] [CrossRef]
- Gou, Y.; Miao, D.; Zhou, M.; Wang, L.; Zhou, H.; Su, G. Bio-Inspired Protein-Based Nanoformulations for Cancer Theranostics. Front. Pharmacol. 2018, 9, 421. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, J.; Huang, Z. Recent progress in microRNA-based delivery systems for the treatment of human disease. ExRNA 2019, 1, 1–14. [Google Scholar] [CrossRef]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef]
- Tyagi, N.; Arora, S.; Deshmukh, S.K.; Singh, S.; Marimuthu, S.; Singh, A.P. Exploiting Nanotechnology for the Development of MicroRNA-Based Cancer Therapeutics. J. Biomed. Nanotechnol. 2016, 12, 28–42. [Google Scholar] [CrossRef]
- Bayda, S.; Hadla, M.; Palazzolo, S.; Riello, P.; Corona, G.; Toffoli, G.; Rizzolio, F. Inorganic Nanoparticles for Cancer Therapy: A Transition from Lab to Clinic. Curr. Med. Chem. 2018, 25, 4269–4303. [Google Scholar] [CrossRef]
- Ghosh, R.; Singh, L.C.; Shohet, J.M.; Gunaratne, P.H. A gold nanoparticle platform for the delivery of functional microRNAs into cancer cells. Biomaterials 2013, 34, 807–816. [Google Scholar] [CrossRef]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold nanoparticles for nucleic acid delivery. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Tivnan, A.; Orr, W.S.; Gubala, V.; Nooney, R.; Williams, D.E.; McDonagh, C.; Prenter, S.; Harvey, H.; Domingo-Fernández, R.; Bray, I.M.; et al. Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles. PLoS ONE 2012, 7, e38129. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Cilurzo, F.; Maiuolo, J.; Federico, C.; Di Martino, M.T.; Cristiano, M.C.; Tassone, P.; Fresta, M.; Paolino, D. Delivery of miR-34a by chitosan/PLGA nanoplexes for the anticancer treatment of multiple myeloma. Sci. Rep. 2015, 5, 17579. [Google Scholar] [CrossRef] [PubMed]
- Na, J.H.; Koo, H.; Lee, S.; Min, K.H.; Park, K.; Yoo, H.; Lee, S.H.; Park, J.H.; Kwon, I.C.; Jeong, S.Y.; et al. Real-time and non-invasive optical imaging of tumor-targeting glycol chitosan nanoparticles in various tumor models. Biomaterials 2011, 32, 5252–5261. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.W.; Son, S.; Jang, J.; Youn, H.; Lee, S.; Lee, D.; Lee, Y.S.; Jeong, J.M.; Kim, W.J.; Lee, D.S. A brain-targeted rabies virus glycoprotein-disulfide linked PEI nanocarrier for delivery of neurogenic microRNA. Biomaterials 2011, 32, 4968–4975. [Google Scholar] [CrossRef]
- Yang, Y.P.; Chien, Y.; Chiou, G.Y.; Cherng, J.Y.; Wang, M.L.; Lo, W.L.; Chang, Y.L.; Huang, P.I.; Chen, Y.W.; Shih, Y.H.; et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials 2012, 33, 1462–1476. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Prow, T.W.; Bhutto, I.; Kim, S.Y.; Grebe, R.; Merges, C.; McLeod, D.S.; Uno, K.; Mennon, M.; Rodriguez, L.; Leong, K.; et al. Ocular nanoparticle toxicity and transfection of the retina and retinal pigment epithelium. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 340–349. [Google Scholar] [CrossRef]
- Yang, C.; Jiang, L.; Bu, S.; Zhang, L.; Xie, X.; Zeng, Q.; Zhu, D.; Zheng, Y. Intravitreal administration of dexamethasone-loaded PLGA-TPGS nanoparticles for the treatment of posterior segment diseases. J. Biomed. Nanotechnol. 2013, 9, 1617–1623. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, J.; Hu, Z.; Nair, A.; Tang, L. Nanoparticles for Uveal Melanoma Treatment. Proc IEEE Conf. Nanotechnol. 2008, 2008, 822–825. [Google Scholar]
- Gray, W.D.; Wu, R.J.; Yin, X.; Zhou, J.; Davis, M.E.; Luo, Y. Dendrimeric bowties featuring hemispheric-selective decoration of ligands for microRNA-based therapy. Biomacromolecules 2013, 14, 101–109. [Google Scholar] [CrossRef]
- Liu, X.; Li, G.; Su, Z.; Jiang, Z.; Chen, L.; Wang, J.; Yu, S.; Liu, Z. Poly(amido amine) is an ideal carrier of miR-7 for enhancing gene silencing effects on the EGFR pathway in U251 glioma cells. Oncol. Rep. 2013, 29, 1387–1394. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef]
- Wu, Y.; Crawford, M.; Yu, B.; Mao, Y.; Nana-Sinkam, S.P.; Lee, L.J. MicroRNA delivery by cationic lipoplexes for lung cancer therapy. Mol. Pharm. 2011, 8, 1381–1389. [Google Scholar] [CrossRef]
- Piao, L.; Zhang, M.; Datta, J.; Xie, X.; Su, T.; Li, H.; Teknos, T.N.; Pan, Q. Lipid-based nanoparticle delivery of Pre-miR-107 inhibits the tumorigenicity of head and neck squamous cell carcinoma. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1261–1269. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef]
- Mittal, A.; Chitkara, D.; Behrman, S.W.; Mahato, R.I. Efficacy of gemcitabine conjugated and miRNA-205 complexed micelles for treatment of advanced pancreatic cancer. Biomaterials 2014, 35, 7077–7087. [Google Scholar] [CrossRef]
- Cavalli, R.; Caputo, O.; Gasco, M.R. Solid lipospheres of doxorubicin and idarubicin. Int. J. Pharm. 1993, 89, R9–R12. [Google Scholar] [CrossRef]
- Scheideler, M.; Vidakovic, I.; Prassl, R. Lipid nanocarriers for microRNA delivery. Chem. Phys. Lipids 2020, 226, 104837. [Google Scholar] [CrossRef]
- MacDiarmid, J.A.; Madrid-Weiss, J.; Amaro-Mugridge, N.B.; Phillips, L.; Brahmbhatt, H. Bacterially-derived nanocells for tumor-targeted delivery of chemotherapeutics and cell cycle inhibitors. Cell Cycle 2007, 6, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.B.; Ruggieri, R.; Jamil, E.; Tran, N.L.; Gonzalez, C.; Mugridge, N.; Gao, S.; MacDiarmid, J.; Brahmbhatt, H.; Sarkaria, J.N.; et al. Nanocell-mediated delivery of miR-34a counteracts temozolomide resistance in glioblastoma. Mol. Med. 2021, 27, 28. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Ríos, A.J.; Molina-Crespo, Á.; Bouzo, B.L.; López-López, R.; Moreno-Bueno, G.; de la Fuente, M. Exosome-mimetic nanoplatforms for targeted cancer drug delivery. J. Nanobiotechnol. 2019, 17, 85. [Google Scholar] [CrossRef] [PubMed]
- Vandghanooni, S.; Eskandani, M.; Barar, J.; Omidi, Y. AS1411 aptamer-decorated cisplatin-loaded poly(lactic-co-glycolic acid) nanoparticles for targeted therapy of miR-21-inhibited ovarian cancer cells. Nanomedicine 2018, 13, 2729–2758. [Google Scholar] [CrossRef]
- Wu, C.; Tian, Y.; Zhang, Y.; Xu, J.; Wang, Y.; Guan, X.; Li, T.; Yang, H.; Li, S.; Qin, X.; et al. Acid-Triggered Charge-Convertible Graphene-Based All-in-One Nanocomplex for Enhanced Genetic Phototherapy of Triple-Negative Breast Cancer. Adv. Healthc. Mater. 2020, 9, e1901187. [Google Scholar] [CrossRef]
- Li, J.; Huang, J.; Yang, X.; Yang, Y.; Quan, K.; Xie, N.; Wu, Y.; Ma, C.; Wang, K. Gold nanoparticle-based 2’-O-methyl modified DNA probes for breast cancerous theranostics. Talanta 2018, 183, 11–17. [Google Scholar] [CrossRef]
- Costa, P.M.; Cardoso, A.L.; Custódia, C.; Cunha, P.; Pereira de Almeida, L.; Pedroso de Lima, M.C. MiRNA-21 silencing mediated by tumor-targeted nanoparticles combined with sunitinib: A new multimodal gene therapy approach for glioblastoma. J. Control Release 2015, 207, 31–39. [Google Scholar] [CrossRef]
- Lee, T.J.; Yoo, J.Y.; Shu, D.; Li, H.; Zhang, J.; Yu, J.G.; Jaime-Ramirez, A.C.; Acunzo, M.; Romano, G.; Cui, R.; et al. RNA Nanoparticle-Based Targeted Therapy for Glioblastoma through Inhibition of Oncogenic miR-21. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1544–1555. [Google Scholar] [CrossRef]
- Maghsoudnia, N.; Eftekhari, R.B.; Sohi, A.N.; Dorkoosh, F.A. Chloroquine Assisted Delivery of microRNA Mimic Let-7b to NSCLC Cell Line by PAMAM (G5)—HA Nano-Carrier. Curr. Drug Deliv. 2021, 18, 31–43. [Google Scholar] [CrossRef]
- Di Paolo, D.; Pastorino, F.; Brignole, C.; Corrias, M.V.; Emionite, L.; Cilli, M.; Tamma, R.; Priddy, L.; Amaro, A.; Ferrari, D.; et al. Combined Replenishment of miR-34a and let-7b by Targeted Nanoparticles Inhibits Tumor Growth in Neuroblastoma Preclinical Models. Small 2020, 16, e1906426. [Google Scholar] [CrossRef]
- Vencken, S.; Foged, C.; Ramsey, J.M.; Sweeney, L.; Cryan, S.A.; MacLoughlin, R.J.; Greene, C.M. Nebulised lipid-polymer hybrid nanoparticles for the delivery of a therapeutic anti-inflammatory microRNA to bronchial epithelial cells. ERJ Open Res. 2019, 5. [Google Scholar] [CrossRef]
- Zilio, S.; Vella, J.L.; De la Fuente, A.C.; Daftarian, P.M.; Weed, D.T.; Kaifer, A.; Marigo, I.; Leone, K.; Bronte, V.; Serafini, P. 4PD Functionalized Dendrimers: A Flexible Tool for In Vivo Gene Silencing of Tumor-Educated Myeloid Cells. J. Immunol. 2017, 198, 4166–4177. [Google Scholar] [CrossRef]
- Chin, D.D.; Poon, C.; Wang, J.; Joo, J.; Ong, V.; Jiang, Z.; Cheng, K.; Plotkin, A.; Magee, G.A.; Chung, E.J. miR-145 micelles mitigate atherosclerosis by modulating vascular smooth muscle cell phenotype. Biomaterials 2021, 273, 120810. [Google Scholar] [CrossRef]
- Zhai, J.; Zhu, Y.; Liu, J.; An, J.; Yu, Y.; Li, Y.; Fan, Y. Enhanced Suppression of Disulfide Cross-Linking Micelles Nanocarriers Loaded miR-145 Delivering System via Down-Regulation of MYC and FSCN1 in Colon Cancer Cells. J. Biomed. Nanotechnol. 2020, 16, 1183–1195. [Google Scholar] [CrossRef]
- Reimondez-Troitiño, S.; González-Aramundiz, J.V.; Ruiz-Bañobre, J.; López-López, R.; Alonso, M.J.; Csaba, N.; de la Fuente, M. Versatile protamine nanocapsules to restore miR-145 levels and interfere tumor growth in colorectal cancer cells. Eur. J. Pharm. Biopharm. 2019, 142, 449–459. [Google Scholar] [CrossRef]
- Nishio, H.; Masumoto, H.; Sakamoto, K.; Yamazaki, K.; Ikeda, T.; Minatoya, K. MicroRNA-145-loaded poly(lactic-co-glycolic acid) nanoparticles attenuate venous intimal hyperplasia in a rabbit model. J. Thorac. Cardiovasc. Surg. 2019, 157, 2242–2251. [Google Scholar] [CrossRef]
- Tekie, F.S.M.; Soleimani, M.; Zakerian, A.; Dinarvand, M.; Amini, M.; Dinarvand, R.; Arefian, E.; Atyabi, F. Glutathione responsive chitosan-thiolated dextran conjugated miR-145 nanoparticles targeted with AS1411 aptamer for cancer treatment. Carbohydr. Polym. 2018, 201, 131–140. [Google Scholar] [CrossRef]
- Tekie, F.S.; Kiani, M.; Zakerian, A.; Pilevarian, F.; Assali, A.; Soleimani, M.; Dinarvand, R.; Arefian, E.; Atashi, A.; Amini, M.; et al. Nano polyelectrolyte complexes of carboxymethyl dextran and chitosan to improve chitosan-mediated delivery of miR-145. Carbohydr. Polym. 2017, 159, 66–75. [Google Scholar] [CrossRef]
- Setua, S.; Khan, S.; Yallapu, M.M.; Behrman, S.W.; Sikander, M.; Khan, S.S.; Jaggi, M.; Chauhan, S.C. Restitution of Tumor Suppressor MicroRNA-145 Using Magnetic Nanoformulation for Pancreatic Cancer Therapy. J. Gastrointest. Surg. 2017, 21, 94–105. [Google Scholar] [CrossRef]
- Zhang, T.; Xue, X.; He, D.; Hsieh, J.T. A prostate cancer-targeted polyarginine-disulfide linked PEI nanocarrier for delivery of microRNA. Cancer Lett. 2015, 365, 156–165. [Google Scholar] [CrossRef]
- Ekin, A.; Karatas, O.F.; Culha, M.; Ozen, M. Designing a gold nanoparticle-based nanocarrier for microRNA transfection into the prostate and breast cancer cells. J. Gene Med. 2014, 16, 331–335. [Google Scholar] [CrossRef]
- Liang, G.; Zhu, Y.; Jing, A.; Wang, J.; Hu, F.; Feng, W.; Xiao, Z.; Chen, B. Cationic microRNA-delivering nanocarriers for efficient treatment of colon carcinoma in xenograft model. Gene Ther. 2016, 23, 829–838. [Google Scholar] [CrossRef]
- Tekie, F.S.; Atyabi, F.; Soleimani, M.; Arefian, E.; Atashi, A.; Kiani, M.; Khoshayand, M.R.; Amini, M.; Dinarvand, R. Chitosan polyplex nanoparticle vector for miR-145 expression in MCF-7: Optimization by design of experiment. Int. J. Biol. Macromol. 2015, 81, 828–837. [Google Scholar] [CrossRef]
- Gilam, A.; Conde, J.; Weissglas-Volkov, D.; Oliva, N.; Friedman, E.; Artzi, N.; Shomron, N. Local microRNA delivery targets Palladin and prevents metastatic breast cancer. Nat. Commun. 2016, 7, 12868. [Google Scholar] [CrossRef]
- Kouri, F.M.; Hurley, L.A.; Daniel, W.L.; Day, E.S.; Hua, Y.; Hao, L.; Peng, C.Y.; Merkel, T.J.; Queisser, M.A.; Ritner, C.; et al. miR-182 integrates apoptosis, growth, and differentiation programs in glioblastoma. Genes Dev. 2015, 29, 732–745. [Google Scholar] [CrossRef]
- Milan Rois, P.; Latorre, A.; Rodriguez Diaz, C.; Del Moral, A.; Somoza, A. Reprogramming Cells for Synergistic Combination Therapy with Nanotherapeutics against Uveal Melanoma. Biomimetics 2018, 3, 28. [Google Scholar] [CrossRef]
- Lee, T.J.; Haque, F.; Vieweger, M.; Yoo, J.Y.; Kaur, B.; Guo, P.; Croce, C.M. Functional assays for specific targeting and delivery of RNA nanoparticles to brain tumor. Methods Mol. Biol. 2015, 1297, 137–152. [Google Scholar]
- Vu, V.P.; Gifford, G.B.; Chen, F.; Benasutti, H.; Wang, G.; Groman, E.V.; Scheinman, R.; Saba, L.; Moghimi, S.M.; Simberg, D. Immunoglobulin deposition on biomolecule corona determines complement opsonization efficiency of preclinical and clinical nanoparticles. Nat. Nanotechnol. 2019, 14, 260–268. [Google Scholar] [CrossRef]
- Lainé, A.L.; Gravier, J.; Henry, M.; Sancey, L.; Béjaud, J.; Pancani, E.; Wiber, M.; Texier, I.; Coll, J.L.; Benoit, J.P.; et al. Conventional versus stealth lipid nanoparticles: Formulation and in vivo fate prediction through FRET monitoring. J. Control. Release 2014, 188, 1–8. [Google Scholar] [CrossRef]
- Idlas, P.; Lepeltier, E.; Jaouen, G.; Passirani, C. Ferrocifen Loaded Lipid Nanocapsules: A Promising Anticancer Medication against Multidrug Resistant Tumors. Cancers 2021, 13, 2291. [Google Scholar] [CrossRef]
- Pasut, G.; Veronese, F.M. State of the art in PEGylation: The great versatility achieved after forty years of research. J. Control. Release 2012, 161, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Laine, A.L.; Huynh, N.T.; Clavreul, A.; Balzeau, J.; Béjaud, J.; Vessieres, A.; Benoit, J.P.; Eyer, J.; Passirani, C. Brain tumour targeting strategies via coated ferrociphenol lipid nanocapsules. Eur. J. Pharm. Biopharm. 2012, 81, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, W.M.; de Waard-Siebinga, I.; Danen, E.H.J.; Luyten, G.P.M.; van Muijen, G.N.P.; Jager, M.J. Cytokine-mediated modulation of integrin, ICAM-1 and CD44 expression on human uveal melanoma cells in vitro. Melanoma Res. 1995, 5, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Huang, J.; Wang, L.; Jia, D.; Yang, J.; Dillon, D.A.; Zurakowski, D.; Mao, H.; Moses, M.A.; Auguste, D.T. ICAM-1 as a molecular target for triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14710–14715. [Google Scholar] [CrossRef]
- Khan, M.M.; Madni, A.; Filipczak, N.; Pan, J.; Rehman, M.; Rai, N.; Attia, S.A.; Torchilin, V.P. Folate targeted lipid chitosan hybrid nanoparticles for enhanced anti-tumor efficacy. Nanomed. Nanotechnol. Biol. Med. 2020, 28, 102228. [Google Scholar] [CrossRef]
- Li, J.; Li, M.; Tian, L.; Qiu, Y.; Yu, Q.; Wang, X.; Guo, R.; He, Q. Facile strategy by hyaluronic acid functional carbon dot-doxorubicin nanoparticles for CD44 targeted drug delivery and enhanced breast cancer therapy. Int. J. Pharm. 2020, 578, 119122. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Y.; Kumar, A.; Tan, A.; Jin, S.; Mozhi, A.; Liang, X.J. pH-sensitive nano-systems for drug delivery in cancer therapy. Biotechnol. Adv. 2014, 32, 693–710. [Google Scholar] [CrossRef]
- Aughton, K.; Shahidipour, H.; Djirackor, L.; Coupland, S.E.; Kalirai, H. Characterization of Uveal Melanoma Cell Lines and Primary Tumor Samples in 3D Culture. Transl. Vis. Sci. Technol. 2020, 9, 39. [Google Scholar] [CrossRef]
- You, S.; Luo, J.; Grossniklaus, H.E.; Gou, M.L.; Meng, K.; Zhang, Q. Nanomedicine in the application of uveal melanoma. Int. J. Ophthalmol. 2016, 9, 1215–1225. [Google Scholar]
- Byon, I.S.; Jeon, H.S.; Kim, H.W.; Lee, S.J.; Lee, J.E.; Oum, B.S. The effect of a systemic angiotensin receptor blocker on vascular endothelial growth factor in the vitreous of patients with proliferative diabetic retinopathy. Curr. Eye Res. 2013, 38, 774–780. [Google Scholar] [CrossRef]
- Mack, W.P. Complications in periocular rejuvenation. Facial. Plast. Surg. Clin. N. Am. 2010, 18, 435–456. [Google Scholar] [CrossRef]
- Ranta, V.P.; Mannermaa, E.; Lummepuro, K.; Subrizi, A.; Laukkanen, A.; Antopolsky, M.; Murtomäki, L.; Hornof, M.; Urtti, A. Barrier analysis of periocular drug delivery to the posterior segment. J. Control. Release 2010, 148, 42–48. [Google Scholar] [CrossRef]
- Ma, J.; Roelofs, K.A.; Russell, L.; Weis, E.; Chen, S.H. Rapid growth of primary uveal melanoma following intravitreal bevacizumab injection: A case report and review of the literature. Digit. J. Ophthalmol. 2021, 26, 27–30. [Google Scholar] [CrossRef]
- Wan, C.R.; Muya, L.; Kansara, V.; Ciulla, T.A. Suprachoroidal Delivery of Small Molecules, Nanoparticles, Gene and Cell Therapies for Ocular Diseases. Pharmaceutics 2021, 13, 288. [Google Scholar] [CrossRef]
- Kansara, V.S.; Cooper, M.; Sesenoglu-Laird, O.; Muya, L.; Moen, R.; Ciulla, T.A. Suprachoroidally Delivered DNA Nanoparticles Transfect Retina and Retinal Pigment Epithelium/Choroid in Rabbits. Transl. Vis. Sci. Technol. 2020, 9, 21. [Google Scholar] [CrossRef]
- Shen, J.; Kim, J.; Tzeng, S.Y.; Ding, K.; Hafiz, Z.; Long, D.; Wang, J.; Green, J.J.; Campochiaro, P.A. Suprachoroidal gene transfer with nonviral nanoparticles. Sci. Adv. 2020, 6, eaba1606. [Google Scholar] [CrossRef]
- Habot-Wilner, Z.; Noronha, G.; Wykoff, C.C. Suprachoroidally injected pharmacological agents for the treatment of chorio-retinal diseases: A targeted approach. Acta Ophthalmol. 2019, 97, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Varela-Fernández, R.; Díaz-Tomé, V.; Luaces-Rodríguez, A.; Conde-Penedo, A.; García-Otero, X.; Luzardo-Álvarez, A.; Fernández-Ferreiro, A.; Otero-Espinar, F.J. Drug Delivery to the Posterior Segment of the Eye: Biopharmaceutic and Pharmacokinetic Considerations. Pharmaceutics 2020, 12, 269. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Wang, P.Y.; Lin, I.C.; Huang, H.; Liu, G.S.; Tseng, C.L. Ocular Drug Delivery: Role of Degradable Polymeric Nanocarriers for Ophthalmic Application. Int. J. Mol. Sci. 2018, 19, 2830. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yeo, Y. Controlled Drug Release from Pharmaceutical Nanocarriers. Chem. Eng. Sci. 2015, 125, 75–84. [Google Scholar] [CrossRef]
- Kageyama, K.; Ohara, M.; Saito, K.; Ozaki, S.; Terai, M.; Mastrangelo, M.J.; Fortina, P.; Aplin, A.E.; Sato, T. Establishment of an orthotopic patient-derived xenograft mouse model using uveal melanoma hepatic metastasis. J. Transl. Med. 2017, 15, 145. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| miRNA | Preclinical Studies | Function | Target(s) | Ref. |
|---|---|---|---|---|
| miR-21-3p (oncomiR) | OCM-1 cells stably transfected with miR-21-3p inhibition vector s.c. injected into the right side of the axilla in nude mice | Reduces in vivo UM tumor growth | p53 and LASP1 | [54] |
| Let-7b | OCM-1 cells stably overexpressing let-7b s.c. injected into the right flank in thymic nude mice | Increases radiosensitivity of UM cells | Cyclin D1 | [55] |
| miR-17-3p | OCM-1A cells transfected with miR-17-3p agomir s.c. injected into the left axilla in nude mice | Suppresses tumorigenesis and metastasis of UM | MDM2 | [56] |
| miR-124a | M23 cells or SP6.5 cells expressing miR-124a s.c. inoculated into the flank in nude mice | Suppresses UM tumor growth in vivo and inhibits UM cell invasion | CDK4/6, cyclin D2 and EZH2 | [59] |
| miR-142-3p | miR-142-3p-transfected SP6.5 or M17 cells inoculated into the suprachoroidal space in nude mice | Inhibits cell proliferation, migration and invasion | CDC25C, TGFβR1, GNAQ, WASL and RAC1 | [48] |
| miR-145 | Lentivirus-miR-145-transduced OCM-1 cells s.c. injected in the right side of the axilla in nude mice | Reduces xenograft tumor growth and angiogenesis | IRS-1, N-RAS and VEGF | [63] |
| miR-182 | M23 or SP6.5 cells expressing miR-182 s.c. inoculated into the flanks of nude mice | Suppresses in vivo UM growth | MITF, BCL2 and cyclin D2 | [64] |
| Potential Therapeutic miRNA for UM | Nanocarriers/NPs | Targeting Cells | Ref. |
|---|---|---|---|
| Anti-miR-21 oligonucleotide | Aptamer-decorated PEGylated PLGA NPs | Ovarian cancers | [104] |
| Acid-triggered charge-reversible graphene-based multilayer polymers | Triple-negative breast cancer | [105] | |
| GNPs | Breast cancer cells | [106] | |
| Chlorotoxin-coupled stable nucleic acid lipid NPs | Glioblastoma | [107] | |
| 3WJ-based RNA NPs | Glioblastoma | [108] | |
| Let-7b | HA-G5 PAMAM dendrimer | CD44+ non-small-cell lung cancer cells | [109] |
| Cationic liposomes | Neuroblastoma | [110] | |
| miR-17 | DOTAP-modified PLGA lipid–polymer hybrid NPs | Bronchial epithelial cells | [111] |
| miR-124a | Disulfide-linked PEI NPs | Neuron cells | [86] |
| miR-142-3p | G5 PAMAM dendrimers | Myeloid cells | [112] |
| miR-145 | Micelles | Vascular smooth muscle cells | [113] |
| Disulfide cross-linked micelles | Colon cancer cells | [114] | |
| Nanocapsules | Colorectal cancer cells | [115] | |
| PLGA NPs | Vascular smooth muscle cells | [116] | |
| Chitosan-thiolated dextran NPs | Cancer cells | [117,118] | |
| Magnetic nanoformulation | Pancreatic cancer | [119] | |
| Polyarginine-disulfide-linked PEI NPs | Prostate cancer | [120] | |
| GNPs | Prostate and breast cancer cells | [121] | |
| HA-PLGA/PEI with miR-145 expression plasmid | Colon carcinoma | [122] | |
| Chitosan polyplex NPs with miR-145 expression plasmid | MCF-7 | [123] | |
| miR-182 | PEGylated GNP nanogel | Metastatic breast cancer | [124] |
| PEGylated GNPs | Glioblastoma | [125] | |
| miR-34a, miR-137, miR-144 and miR-182 | GNPs | UM cells | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Wang, R.; Hardy, P. Potential of miRNA-Based Nanotherapeutics for Uveal Melanoma. Cancers 2021, 13, 5192. https://doi.org/10.3390/cancers13205192
Yang C, Wang R, Hardy P. Potential of miRNA-Based Nanotherapeutics for Uveal Melanoma. Cancers. 2021; 13(20):5192. https://doi.org/10.3390/cancers13205192
Chicago/Turabian StyleYang, Chun, Rui Wang, and Pierre Hardy. 2021. "Potential of miRNA-Based Nanotherapeutics for Uveal Melanoma" Cancers 13, no. 20: 5192. https://doi.org/10.3390/cancers13205192
APA StyleYang, C., Wang, R., & Hardy, P. (2021). Potential of miRNA-Based Nanotherapeutics for Uveal Melanoma. Cancers, 13(20), 5192. https://doi.org/10.3390/cancers13205192