
Relapsed/Refractory Multiple Myeloma in 2020/2021 and Beyond



Abstract
:Simple Summary
Abstract
1. Introduction
2. Recent Registration Trials in the Context of Current Treatment Standards
2.1. Treatment Options for MM Patients in First Relapse
2.2. Treatment Options for MM Patients in Second or Higher Relapse
2.3. Evolution of Immunotherapies for RRMM Patients
2.3.1. Antibody Drug Conjugates (ADCs)
2.3.2. Bispecific Antibodies (BiTEs/T Cell Engagers)
2.3.3. Chimeric Antigen Receptor T Cells (CAR T Cells)
3. The Therapeutic Future of RRMM
3.1. Molecularly-Based Innovative Approaches of Treatment Stratification and Personalization
3.1.1. Recent Insights into MM Genomics and the Impact of the MM Microenvironment in RRMM
3.1.2. Assessment of MRD for Clinical Decision-Making and Personalized Targeted Therapies in RRMM
3.2. Innovative Approaches to Treat MM-Specific Vulnerabilities
3.2.1. Novel Strategies to Target the UPR
3.2.2. Protein Degradation: A Novel Approach to Target Tumor-Specific Molecules
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group Consensus Criteria for Response and Minimal Residual Disease Assessment in Multiple Myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Nooka, A.K.; Kastritis, E.; Dimopoulos, M.A.; Lonial, S. Treatment Options for Relapsed and Refractory Multiple Myeloma. Blood 2015, 125, 3085–3099. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Harousseau, J.-L.; Durie, B.; Anderson, K.C.; Dimopoulos, M.; Kyle, R.; Blade, J.; Richardson, P.; Orlowski, R.; Siegel, D.; et al. Consensus Recommendations for the Uniform Reporting of Clinical Trials: Report of the International Myeloma Workshop Consensus Panel 1. Blood 2011, 117, 4691–4695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Kumar, S.K.; San Miguel, J.; Davies, F.; Zamagni, E.; Bahlis, N.; Ludwig, H.; Mikhael, J.; Terpos, E.; Schjesvold, F.; et al. Treatment of Relapsed and Refractory Multiple Myeloma: Recommendations from the International Myeloma Working Group. Lancet Oncol. 2021, 22, e105–e118. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Dimopoulos, M.A.; White, D.J.; Benboubker, L.; Cook, G.; Leiba, M.; Ho, P.J.; Kim, K.; Takezako, N.; Moreau, P.; et al. Daratumumab plus Lenalidomide and Dexamethasone in Relapsed/Refractory Multiple Myeloma: Extended Follow-up of POLLUX, a Randomized, Open-Label, Phase 3 Study. Leukemia 2020, 34, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.; Quach, H.; Mateos, M.-V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Yang, H.; Klippel, Z.; Zahlten-Kumeli, A.; et al. Carfilzomib, Dexamethasone, and Daratumumab versus Carfilzomib and Dexamethasone for Patients with Relapsed or Refractory Multiple Myeloma (CANDOR): Results from a Randomised, Multicentre, Open-Label, Phase 3 Study. Lancet 2020, 396, 186–197. [Google Scholar] [CrossRef]
- Leleu, X.; Beksac, M.; Chou, T.; Dimopoulos, M.; Yoon, S.-S.; Prince, H.M.; Pour, L.; Shelekhova, T.; Chari, A.; Khurana, M.; et al. Efficacy and Safety of Weekly Carfilzomib (70 Mg/M2), Dexamethasone, and Daratumumab (KdD70) Is Comparable to Twice-Weekly KdD56 While Being a More Convenient Dosing Option: A Cross-Study Comparison of the CANDOR and EQUULEUS Studies. Leuk. Lymphoma 2021, 62, 358–367. [Google Scholar] [CrossRef]
- Richardson, P.G.; Oriol, A.; Beksac, M.; Liberati, A.M.; Galli, M.; Schjesvold, F.; Lindsay, J.; Weisel, K.; White, D.; Facon, T.; et al. Pomalidomide, Bortezomib, and Dexamethasone for Patients with Relapsed or Refractory Multiple Myeloma Previously Treated with Lenalidomide (OPTIMISMM): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 781–794. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Weisel, K.; Moreau, P.; Anderson, L.D.; White, D.; San-Miguel, J.; Sonneveld, P.; Engelhardt, M.; Jenner, M.; Corso, A.; et al. Pomalidomide, Bortezomib, and Dexamethasone for Multiple Myeloma Previously Treated with Lenalidomide (OPTIMISMM): Outcomes by Prior Treatment at First Relapse. Leukemia 2021, 35, 1722–1731. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Leleu, X.; Moreau, P.; Richardson, P.G.; Liberati, A.M.; Harrison, S.J.; Miles Prince, H.; Ocio, E.M.; Assadourian, S.; Campana, F.; et al. Isatuximab plus Pomalidomide and Dexamethasone in Relapsed/Refractory Multiple Myeloma Patients with Renal Impairment: ICARIA-MM Subgroup Analysis. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Suvannasankha, A.; Fay, J.W.; Arnulf, B.; Kaufman, J.L.; Ifthikharuddin, J.J.; Weiss, B.M.; Krishnan, A.; Lentzsch, S.; Comenzo, R.; et al. Daratumumab plus Pomalidomide and Dexamethasone in Relapsed and/or Refractory Multiple Myeloma. Blood 2017, 130, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus Pomalidomide and Low-Dose Dexamethasone versus Pomalidomide and Low-Dose Dexamethasone in Patients with Relapsed and Refractory Multiple Myeloma (ICARIA-MM): A Randomised, Multicentre, Open-Label, Phase 3 Study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- Richardson, P.G.; Perrot, A.F.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; Huang, J.S.; et al. Updates from ICARIA-MM, a Phase 3 Study of Isatuximab (Isa) plus Pomalidomide and Low-Dose Dexamethasone (Pd) versus Pd in Relapsed and Refractory Multiple Myeloma (RRMM). J. Clin. Oncol. 2021, 39, 8017. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Terpos, E.; Boccadoro, M.; Delimpasi, S.; Beksac, M.; Katodritou, E.; Moreau, P.; Baldini, L.; Symeonidis, A.; Bila, J.; et al. Daratumumab plus Pomalidomide and Dexamethasone versus Pomalidomide and Dexamethasone Alone in Previously Treated Multiple Myeloma (APOLLO): An Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2021, 22, 801–812. [Google Scholar] [CrossRef]
- Moreau, P.; Dimopoulos, M.-A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Špička, I.; Baker, R.; Kim, K.; et al. Isatuximab, Carfilzomib, and Dexamethasone in Relapsed Multiple Myeloma (IKEMA): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet 2021, 397, 2361–2371. [Google Scholar] [CrossRef]
- Spicka, I.; Moreau, P.; Martin, T.G.; Facon, T.; Martinez, G.; Oriol, A.; Koh, Y.; Lim, A.; Mikala, G.; Rosiñol, L.; et al. Isatuximab plus Carfilzomib and Dexamethasone in Relapsed Multiple Myeloma Patients with High-Risk Cytogenetics: IKEMA Subgroup Analysis. J. Clin. Oncol. 2021, 39, 8042. [Google Scholar] [CrossRef]
- Facon, T.; Moreau, P.; Martin, T.G.; Spicka, I.; Oriol, A.; Koh, Y.; Lim, A.; Mikala, G.; Rosiñol, L.; Yağci, M.; et al. Isatuximab plus Carfilzomib and Dexamethasone versus Carfilzomib and Dexamethasone in Elderly Patients with Relapsed Multiple Myeloma: IKEMA Subgroup Analysis. J. Clin. Oncol. 2021, 39, 8026. [Google Scholar] [CrossRef]
- Hajek, R.; Moreau, P.; Augustson, B.; Castro, N.; Pika, T.; Delimpasi, S.; de La Rubia, J.; Maiolino, A.; Reiman, T.J.; Kryuchkova, I.; et al. Isatuximab plus Carfilzomib and Dexamethasone in Patients with Relapsed Multiple Myeloma According to Prior Lines of Treatment and Refractory Status: IKEMA Subgroup Analysis. J. Clin. Oncol. 2021, 39, 8034. [Google Scholar] [CrossRef]
- Costa, L.J.; Hari, P.; Kumar, S.K.; Tang, S.; Gandhi, U.H.; Shah, J.J.; Jagannath, S.; Chari, A.; Lakshman, A.; Shacham, S.; et al. Overall Survival of Triple Class Refractory, Penta-Exposed Multiple Myeloma (MM) Patients Treated with Selinexor plus Dexamethasone or Conventional Care: A Combined Analysis of the STORM and Mammoth Studies. Blood 2019, 134, 3125. [Google Scholar] [CrossRef]
- Podar, K.; Shah, J.; Chari, A.; Richardson, P.G.; Jagannath, S. Selinexor for the Treatment of Multiple Myeloma. Expert Opin. Pharmacother. 2020, 21, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Grosicki, S.; Simonova, M.; Spicka, I.; Pour, L.; Kriachok, I.; Gavriatopoulou, M.; Pylypenko, H.; Auner, H.W.; Leleu, X.; Doronin, V.; et al. Once-per-Week Selinexor, Bortezomib, and Dexamethasone versus Twice-per-Week Bortezomib and Dexamethasone in Patients with Multiple Myeloma (BOSTON): A Randomised, Open-Label, Phase 3 Trial. Lancet 2020, 396, 1563–1573. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Kotb, R.; Sebag, M.; Sutherland, H.J.; LeBlanc, R.; White, D.; Venner, C.P.; Kouroukis, T.; Bergstrom, D.; McCurdy, A.; et al. Selinexor in Combination with Bortezomib and Dexamethasone (SdB) Demonstrates Significant Activity in Patients with Refractory Multiple Myeloma (MM) Including Proteasome-Inhibitor Refractory Patients: Results of the Phase I Stomp Trial. Blood 2016, 128, 977. [Google Scholar] [CrossRef]
- Gasparetto, C.; Lipe, B.; Tuchman, S.; Callander, N.S.; Lentzsch, S.; Baljevic, M.; Rossi, A.C.; Bahlis, N.J.; White, D.; Chen, C.; et al. Once Weekly Selinexor, Carfilzomib, and Dexamethasone (SKd) in Patients with Relapsed/Refractory Multiple Myeloma (MM). J. Clin. Oncol. 2020, 38, 8530. [Google Scholar] [CrossRef]
- Chen, C.; Bahlis, N.; Gasparetto, C.; Tuchman, S.A.; Lipe, B.; Baljevic, M.; Kotb, R.; Sutherland, H.J.; Bensinger, W.I.; Sebag, M.; et al. Selinexor in Combination with Pomalidomide and Dexamethasone (SPd) for Treatment of Patients with Relapsed Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 18–19. [Google Scholar] [CrossRef]
- Gasparetto, C.; Lentzsch, S.; Schiller, G.J.; Callander, N.S.; Tuchman, S.; Bahlis, N.J.; White, D.; Chen, C.; Baljevic, M.; Sutherland, H.J.; et al. Selinexor, Daratumumab, and Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma (MM). J. Clin. Oncol. 2020, 38, 8510. [Google Scholar] [CrossRef]
- Richardson, P.G.; Oriol, A.; Larocca, A.; Bladé, J.; Cavo, M.; Rodriguez-Otero, P.; Leleu, X.; Nadeem, O.; Hiemenz, J.W.; Hassoun, H.; et al. Melflufen and Dexamethasone in Heavily Pretreated Relapsed and Refractory Multiple Myeloma. J. Clin. Oncol. 2021, 39, 757–767. [Google Scholar] [CrossRef]
- Dhakal, B.; D’Souza, A.; Kleman, A.; Chhabra, S.; Mohan, M.; Hari, P. Salvage Second Transplantation in Relapsed Multiple Myeloma. Leukemia 2021, 35, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Garderet, L.; Iacobelli, S.; Koster, L.; Goldschmidt, H.; Johansson, J.-E.; Bourhis, J.H.; Krejci, M.; Leleu, X.; Potter, M.; Blaise, D.; et al. Outcome of a Salvage Third Autologous Stem Cell Transplantation in Multiple Myeloma. Biol. Blood Marrow Transpl. 2018, 24, 1372–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, G.; Ashcroft, A.J.; Cairns, D.A.; Williams, C.D.; Brown, J.M.; Cavenagh, J.D.; Snowden, J.A.; Parrish, C.; Yong, K.; Cavet, J.; et al. The Effect of Salvage Autologous Stem-Cell Transplantation on Overall Survival in Patients with Relapsed Multiple Myeloma (Final Results from BSBMT/UKMF Myeloma X Relapse [Intensive]): A Randomised, Open-Label, Phase 3 Trial. Lancet Haematol. 2016, 3, e340–e351. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.-F.; Lin, L.; Xing, L.; Li, Y.; Yu, T.; Anderson, K.C.; Tai, Y.-T. BCMA-Targeting Therapy: Driving a New Era of Immunotherapy in Multiple Myeloma. Cancers 2020, 12, 1473. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.; Dardac, A.; Madduri, D.; Richard, S.; Richter, J. B-Cell Maturation Antigen (BCMA) in Multiple Myeloma: The New Frontier of Targeted Therapies. Ther. Adv. Hematol. 2021, 12, 2040620721989585. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab Mafodotin for Relapsed or Refractory Multiple Myeloma (DREAMM-2): A Two-Arm, Randomised, Open-Label, Phase 2 Study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Bruins, W.S.C.; Zheng, W.; Higgins, J.P.; Willert, E.K.; Newcomb, J.; Dash, A.B.; Van De Donk, N.W.C.J.; Zweegman, S.; Mutis, T. TAK-169, a Novel Recombinant Immunotoxin Specific for CD38, Induces Powerful Preclinical Activity against Patient-Derived Multiple Myeloma Cells. Blood 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Vogl, D.T.; Kaufman, J.L.; Holstein, S.A.; Nadeem, O.; O’Donnell, E.; Suryanarayan, K.; Collins, S.; Parot, X.; Chaudhry, M. TAK-573, an Anti-CD38/Attenuated Ifnα Fusion Protein, Has Clinical Activity and Modulates the Ifnα Receptor (IFNAR) Pathway in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 37–38. [Google Scholar] [CrossRef]
- Figueroa-Vazquez, V.; Ko, J.; Breunig, C.; Baumann, A.; Giesen, N.; Pálfi, A.; Müller, C.; Lutz, C.; Hechler, T.; Kulke, M.; et al. HDP-101, an Anti-BCMA Antibody-Drug Conjugate, Safely Delivers Amanitin to Induce Cell Death in Proliferating and Resting Multiple Myeloma Cells. Mol. Cancer Ther. 2021, 20, 367–378. [Google Scholar] [CrossRef]
- Xing, L.; Wang, S.; Liu, J.; Yu, T.; Chen, H.; Wen, K.; Li, Y.; Lin, L.; Hsieh, P.A.; Cho, S.-F.; et al. BCMA-Specific ADC MEDI2228 and Daratumumab Induce Synergistic Myeloma Cytotoxicity via IFN-Driven Immune Responses and Enhanced CD38 Expression. Clin. Cancer Res. 2021, 27, 5376–5388. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel Anti-B-Cell Maturation Antigen Antibody-Drug Conjugate (GSK2857916) Selectively Induces Killing of Multiple Myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Lee, H.C.; Raje, N.S.; Landgren, O.; Upreti, V.V.; Wang, J.; Avilion, A.A.; Hu, X.; Rasmussen, E.; Ngarmchamnanrith, G.; Fujii, H.; et al. Phase 1 Study of the Anti-BCMA Antibody-Drug Conjugate AMG 224 in Patients with Relapsed/Refractory Multiple Myeloma. Leukemia 2021, 35, 255–258. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef]
- Harrison, S.J.; Minnema, M.C.; Lee, H.C.; Spencer, A.; Kapoor, P.; Madduri, D.; Larsen, J.; Ailawadhi, S.; Kaufman, J.L.; Raab, M.S.; et al. A Phase 1 First in Human (FIH) Study of AMG 701, an Anti-B-Cell Maturation Antigen (BCMA) Half-Life Extended (HLE) BiTE (Bispecific T-Cell Engager) Molecule, in Relapsed/Refractory (RR) Multiple Myeloma (MM). Blood 2020, 136, 28–29. [Google Scholar] [CrossRef]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, L.J.; Wong, S.W.; Bermúdez, A.; de la Rubia, J.; Mateos, M.V.; Ocio, E.M.; Rodríguez-Otero, P.; San Miguel, J.; Li, S.; Sarmiento, R.; et al. Interim Results from the First Phase 1 Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients with Relapsed/Refractory Multiple Myeloma. Available online: https://library.ehaweb.org/eha/2020/eha25th/295025/luciano.j.costa.interim.results.from.the.first.phase.1.clinical.study.of.the (accessed on 12 October 2020).
- Usmani, S.Z.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Rosinol, L.; Chari, A.; Bhutani, M.; Karlin, L.; et al. Teclistamab, a B-Cell Maturation Antigen × CD3 Bispecific Antibody, in Patients with Relapsed or Refractory Multiple Myeloma (MajesTEC-1): A Multicentre, Open-Label, Single-Arm, Phase 1 Study. Lancet 2021, 398, 665–674. [Google Scholar] [CrossRef]
- Madduri, D.; Rosko, A.; Brayer, J.; Zonder, J.; Bensinger, W.I.; Li, J.; Xu, L.; Adriaens, L.; Chokshi, D.; Zhang, W.; et al. REGN5458, a BCMA × CD3 Bispecific Monoclonal Antibody, Induces Deep and Durable Responses in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 41–42. [Google Scholar] [CrossRef]
- Rodriguez, C.; D’Souza, A.; Shah, N.; Voorhees, P.M.; Buelow, B.; Vij, R.; Kumar, S.K. Initial Results of a Phase I Study of TNB-383B, a BCMA × CD3 Bispecific T-Cell Redirecting Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 43–44. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Raje, N.S.; Costello, C.; Dholaria, B.R.; Solh, M.M.; Levy, M.Y.; Tomasson, M.H.; Dube, H.; Liu, F.; Liao, K.H.; et al. Efficacy and Safety of Elranatamab (PF-06863135), a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients with Relapsed or Refractory Multiple Myeloma (MM). J. Clin. Oncol. 2021, 39, 8006. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Krishnan, A.Y.; Oriol, A.; van de Donk, N.W.C.J.; Rodríguez-Otero, P.; Askari, E.; Mateos, M.-V.; Minnema, M.C.; Costa, L.J.; Verona, R.; et al. Updated Results of a Phase 1, First-in-Human Study of Talquetamab, a G Protein-Coupled Receptor Family C Group 5 Member D (GPRC5D) × CD3 Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma (MM). J. Clin. Oncol. 2021, 39, 8008. [Google Scholar] [CrossRef]
- Sumiyoshi, T.; Nakamura, R.; Lear, S.; Wilson, D.; Choeurng, V.; Vaze, A.; Trudel, S.; Spencer, A.; Cohen, A.D.; Fonseca, R.; et al. FCRH5 Target Expression in Patients with Relapsed Refractory Multiple Myeloma (RRMM) Treated with Cevostamab in an Ongoing Phase I Dose Escalation Study. EHA Libr. 2021, 325723, EP965. [Google Scholar]
- Bae, J.; Parayath, N.; Ma, W.; Amiji, M.; Munshi, N.; Anderson, K.C. BCMA Peptide-Engineered Nanoparticles Enhance Induction and Function of Antigen-Specific CD8+ Cytotoxic T Lymphocytes against Multiple Myeloma: Clinical Applications. Leukemia 2020, 34, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.; Canale, F.A.; Alati, C.; Vincelli, I.D.; Moscato, T.; Porto, G.; Loteta, B.; Naso, V.; Mazza, M.; Nicolini, F.; et al. CART-Cell Therapy: Recent Advances and New Evidence in Multiple Myeloma. Cancers 2021, 13, 2639. [Google Scholar] [CrossRef] [PubMed]
- Mohyuddin, G.R.; Rooney, A.; Balmaceda, N.; Aziz, M.; Sborov, D.W.; McClune, B.; Kumar, S.K. Chimeric Antigen Receptor T-Cell Therapy in Multiple Myeloma: A Systematic Review and Meta-Analysis of 950 Patients. Blood Adv. 2021, 5, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Agha, M.E.; Cohen, A.D.; Madduri, D.; Cohen, Y.C.; Delforge, M.; Hillengass, J.; Goldschmidt, H.; Weisel, K.; Raab, M.-S.; Scheid, C.; et al. CARTITUDE-2: Efficacy and Safety of Ciltacabtagene Autoleucel (Cilta-Cel), a BCMA-Directed CAR T-Cell Therapy, in Patients with Progressive Multiple Myeloma (MM) after One to Three Prior Lines of Therapy. J. Clin. Oncol. 2021, 39, 8013. [Google Scholar] [CrossRef]
- Mailankody, S.; Jakubowiak, A.J.; Htut, M.; Costa, L.J.; Lee, K.; Ganguly, S.; Kaufman, J.L.; Siegel, D.S.D.; Bensinger, W.; Cota, M.; et al. Orvacabtagene Autoleucel (Orva-Cel), a B-Cell Maturation Antigen (BCMA)-Directed CAR T Cell Therapy for Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Update of the Phase 1/2 EVOLVE Study (NCT03430011). J. Clin. Oncol. 2020, 38, 8504. [Google Scholar] [CrossRef]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-Cell Therapy in Multiple Myeloma: Can We Do Better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical Lessons Learned from the First Leg of the CAR T Cell Journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.-T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic Loss of BCMA as a Resistance Mechanism to CAR T Cell Therapy in a Patient with Multiple Myeloma. Nat. Commun. 2021, 12, 868. [Google Scholar] [CrossRef] [PubMed]
- Da Vià, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Heidemeier, A.; Zhou, X.; Danhof, S.; Kraus, S.; Chatterjee, M.; et al. Homozygous BCMA Gene Deletion in Response to Anti-BCMA CAR T Cells in a Patient with Multiple Myeloma. Nat. Med. 2021, 27, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Frerichs, K.A.; Broekmans, M.E.C.; Marin Soto, J.A.; van Kessel, B.; Heymans, M.W.; Holthof, L.C.; Verkleij, C.P.M.; Boominathan, R.; Vaidya, B.; Sendecki, J.; et al. Preclinical Activity of JNJ-7957, a Novel BCMA × CD3 Bispecific Antibody for the Treatment of Multiple Myeloma, Is Potentiated by Daratumumab. Clin. Cancer Res. 2020, 26, 2203–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Donk, N.W.C.J.; Themeli, M.; Usmani, S.Z. Determinants of Response and Mechanisms of Resistance of CAR T-Cell Therapy in Multiple Myeloma. Blood Cancer Discov. 2021, 2, 302–318. [Google Scholar] [CrossRef]
- Yan, Z.; Cao, J.; Cheng, H.; Qiao, J.; Zhang, H.; Wang, Y.; Shi, M.; Lan, J.; Fei, X.; Jin, L.; et al. A Combination of Humanised Anti-CD19 and Anti-BCMA CAR T Cells in Patients with Relapsed or Refractory Multiple Myeloma: A Single-Arm, Phase 2 Trial. Lancet Haematol. 2019, 6, e521–e529. [Google Scholar] [CrossRef]
- Li, C.; Mei, H.; Hu, Y.; Guo, T.; Liu, L.; Jiang, H.; Tang, L.; Wu, Y.; Ai, L.; Deng, J.; et al. Improved Efficacy and Safety of a Dual-Target CAR-T Cell Therapy Targeting BCMA and CD38 for Relapsed/Refractory Multiple Myeloma from a Phase I Study. EHA Libr. 2019, 267409, S826. [Google Scholar] [CrossRef]
- Rodríguez-Lobato, L.G.; Ganzetti, M.; Fernández de Larrea, C.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1243. [Google Scholar] [CrossRef]
- Mailankody, S.; Matous, J.V.; Liedtke, M.; Sidana, S.; Malik, S.; Nath, R.; Oluwole, O.O.; Karski, E.E.; Lovelace, W.; Zhou, X.; et al. Universal: An Allogeneic First-in-Human Study of the Anti-BCMA ALLO-715 and the Anti-CD52 ALLO-647 in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 24–25. [Google Scholar] [CrossRef]
- Marofi, F.; Saleh, M.M.; Rahman, H.S.; Suksatan, W.; Al-Gazally, M.E.; Abdelbasset, W.K.; Thangavelu, L.; Yumashev, A.V.; Hassanzadeh, A.; Yazdanifar, M.; et al. CAR-Engineered NK Cells; a Promising Therapeutic Option for Treatment of Hematological Malignancies. Stem Cell Res. Ther. 2021, 12, 374. [Google Scholar] [CrossRef]
- Miller, J.S.; Rooney, C.M.; Curtsinger, J.; McElmurry, R.; McCullar, V.; Verneris, M.R.; Lapteva, N.; McKenna, D.; Wagner, J.E.; Blazar, B.R.; et al. Expansion and Homing of Adoptively Transferred Human Natural Killer Cells in Immunodeficient Mice Varies with Product Preparation and in Vivo Cytokine Administration: Implications for Clinical Therapy. Biol. Blood Marrow Transpl. 2014, 20, 1252–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Zhao, X.; Li, Z.; Hu, Y.; Wang, H. From CAR-T Cells to CAR-NK Cells: A Developing Immunotherapy Method for Hematological Malignancies. Front. Oncol. 2021, 11, 720501. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Cho, S.-F.; Xing, L.; Wen, K.; Li, Y.; Yu, T.; Hsieh, P.A.; Chen, H.; Kurtoglu, M.; Zhang, Y.; et al. Preclinical Evaluation of CD8+ Anti-BCMA MRNA CAR T Cells for Treatment of Multiple Myeloma. Leukemia 2021, 35, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.N.; Sherbenou, D.W. Emerging Therapeutic Strategies to Overcome Drug Resistance in Multiple Myeloma. Cancers 2021, 13, 1686. [Google Scholar] [CrossRef]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.-T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. The JAK-STAT Pathway Regulates CD38 on Myeloma Cells in the Bone Marrow Microenvironment: Therapeutic Implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef] [PubMed]
- Croft, J.; Ellis, S.; Sherborne, A.L.; Sharp, K.; Price, A.; Jenner, M.W.; Drayson, M.T.; Owen, R.G.; Chown, S.; Lindsay, J.; et al. Copy Number Evolution and Its Relationship with Patient Outcome-an Analysis of 178 Matched Presentation-Relapse Tumor Pairs from the Myeloma XI Trial. Leukemia 2021, 35, 2043–2053. [Google Scholar] [CrossRef]
- Fonseca, R.; Debes-Marun, C.S.; Picken, E.B.; Dewald, G.W.; Bryant, S.C.; Winkler, J.M.; Blood, E.; Oken, M.M.; Santana-Dávila, R.; González-Paz, N.; et al. The Recurrent IgH Translocations Are Highly Associated with Nonhyperdiploid Variant Multiple Myeloma. Blood 2003, 102, 2562–2567. [Google Scholar] [CrossRef]
- Smadja, N.V.; Bastard, C.; Brigaudeau, C.; Leroux, D.; Fruchart, C.; Groupe Français de Cytogénétique Hématologique. Hypodiploidy Is a Major Prognostic Factor in Multiple Myeloma. Blood 2001, 98, 2229–2238. [Google Scholar] [CrossRef] [Green Version]
- Smadja, N.V.; Leroux, D.; Soulier, J.; Dumont, S.; Arnould, C.; Taviaux, S.; Taillemite, J.L.; Bastard, C. Further Cytogenetic Characterization of Multiple Myeloma Confirms That 14q32 Translocations Are a Very Rare Event in Hyperdiploid Cases. Genes Chromosom. Cancer 2003, 38, 234–239. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, S.V. The Multiple Myelomas—Current Concepts in Cytogenetic Classification and Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 409–421. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic Complexity of Multiple Myeloma and Its Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Hoang, P.H.; Cornish, A.J.; Sherborne, A.L.; Chubb, D.; Kimber, S.; Jackson, G.; Morgan, G.J.; Cook, G.; Kinnersley, B.; Kaiser, M.; et al. An Enhanced Genetic Model of Relapsed IGH-Translocated Multiple Myeloma Evolutionary Dynamics. Blood Cancer J. 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of Novel Mutational Drivers Reveals Oncogene Dependencies in Multiple Myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Caprio, C.; Sacco, A.; Giustini, V.; Roccaro, A.M. Epigenetic Aberrations in Multiple Myeloma. Cancers 2020, 12, 2996. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Vallet, S.; Sacco, A.; Roccaro, A.; Lentzsch, S.; Podar, K. Targeting Transcription Factors in Multiple Myeloma: Evolving Therapeutic Strategies. Expert Opin. Investig. Drugs 2019, 28, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.S.Y.; Chooi, J.Y.; Lim, J.S.L.; Toh, S.H.M.; Tan, T.Z.; Chng, W.-J. SMARCA2 Is a Novel Interactor of NSD2 and Regulates Prometastatic PTP4A3 through Chromatin Remodeling in t(4;14) Multiple Myeloma. Cancer Res. 2021, 81, 2332–2344. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jia, Y.; Tan, T.K.; Chung, T.-H.; Sanda, T.; Chng, W.J. Super-Enhancer Profiling Identifies Novel Oncogenes and Therapeutic Targets in Multiple Myeloma. Blood 2019, 134, 362. [Google Scholar] [CrossRef]
- Gupta, V.A.; Barwick, B.G.; Matulis, S.M.; Shirasaki, R.; Jaye, D.L.; Keats, J.J.; Oberlton, B.; Joseph, N.S.; Hofmeister, C.C.; Heffner, L.T.; et al. Venetoclax Sensitivity in Multiple Myeloma Is Associated with B-Cell Gene Expression. Blood 2021, 137, 3604–3615. [Google Scholar] [CrossRef]
- Kumar, S.K.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Venetoclax or Placebo in Combination with Bortezomib and Dexamethasone in Patients with Relapsed or Refractory Multiple Myeloma (BELLINI): A Randomised, Double-Blind, Multicentre, Phase 3 Trial. Lancet Oncol. 2020, 21, 1630–1642. [Google Scholar] [CrossRef]
- Costa, L.J.; Davies, F.E.; Monohan, G.P.; Kovacsovics, T.J.; Burwick, N.; Jakubowiak, A.J.; Kaufman, J.L.; Hong, W.-J.; Dail, M.; Salem, A.H.; et al. Phase 2 Study of Venetoclax plus Carfilzomib and Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma. Blood Adv. 2021, 5, 3748–3759. [Google Scholar] [CrossRef]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of Action of Proteasome Inhibitors and Deacetylase Inhibitors and the Biological Basis of Synergy in Multiple Myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef] [Green Version]
- San-Miguel, J.F.; Hungria, V.T.M.; Yoon, S.-S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Overall Survival of Patients with Relapsed Multiple Myeloma Treated with Panobinostat or Placebo plus Bortezomib and Dexamethasone (the PANORAMA 1 Trial): A Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Haematol. 2016, 3, e506–e515. [Google Scholar] [CrossRef]
- Laubach, J.P.; Schjesvold, F.; Mariz, M.; Dimopoulos, M.A.; Lech-Maranda, E.; Spicka, I.; Hungria, V.T.; Shelekhova, T.; Abdo, A.; Jacobasch, L.; et al. Efficacy and Safety of Oral Panobinostat plus Subcutaneous Bortezomib and Oral Dexamethasone in Patients with Relapsed or Relapsed and Refractory Multiple Myeloma (PANORAMA 3): An Open-Label, Randomised, Phase 2 Study. Lancet Oncol. 2021, 22, 142–154. [Google Scholar] [CrossRef]
- Moschetta, M.; Kawano, Y.; Podar, K. Targeting the Bone Marrow Microenvironment. Cancer Treat. Res. 2016, 169, 63–102. [Google Scholar] [CrossRef]
- Lomas, O.C.; Tahri, S.; Ghobrial, I.M. The Microenvironment in Myeloma. Curr. Opin. Oncol. 2020, 32, 170–175. [Google Scholar] [CrossRef]
- Lind, J.; Czernilofsky, F.; Vallet, S.; Podar, K. Emerging Protein Kinase Inhibitors for the Treatment of Multiple Myeloma. Expert Opin. Emerg. Drugs 2019, 24, 133–152. [Google Scholar] [CrossRef]
- Chong, P.S.Y.; Chng, W.-J.; de Mel, S. STAT3: A Promising Therapeutic Target in Multiple Myeloma. Cancers 2019, 11, 731. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.H.-H.; Shin, E.M.; Tergaonkar, V.; Chng, W.-J. Targeting NF-ΚB Signaling for Multiple Myeloma. Cancers 2020, 12, 2203. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Gonzalez-Paz, N.; Price-Troska, T.; Jacobus, S.; Rajkumar, S.V.; Oken, M.M.; Kyle, R.A.; Henderson, K.J.; Van Wier, S.; Greipp, P.; et al. Clinical and Biological Significance of RAS Mutations in Multiple Myeloma. Leukemia 2008, 22, 2280–2284. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Pfarr, N.; Endris, V.; Mai, E.K.; Md Hanafiah, N.H.; Lehners, N.; Penzel, R.; Weichert, W.; Ho, A.D.; Schirmacher, P.; et al. Molecular Signaling in Multiple Myeloma: Association of RAS/RAF Mutations and MEK/ERK Pathway Activation. Oncogenesis 2017, 6, e337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortüm, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.-X.; Zhu, Y.-X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted Sequencing of Refractory Myeloma Reveals a High Incidence of Mutations in CRBN and Ras Pathway Genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The Genetic Architecture of Multiple Myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of Genomic Evolution and Mutational Profiles in Multiple Myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Fu, J.; Yang, J.; Ma, H.; Bhutani, D.; Mapara, M.Y.; Marcireau, C.; Lentzsch, S. Targeting the GCK Pathway: A Novel and Selective Therapeutic Strategy against RAS-Mutated Multiple Myeloma. Blood 2021, 137, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Rajagopalan, A.; Flietner, E.D.; Yun, G.; Chesi, M.; Furumo, Q.; Burns, R.T.; Papadas, A.; Ranheim, E.A.; Pagenkopf, A.C.; et al. Expression of NrasQ61R and MYC Transgene in Germinal Center B Cells Induces a Highly Malignant Multiple Myeloma in Mice. Blood 2021, 137, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Andrulis, M.; Lehners, N.; Capper, D.; Penzel, R.; Heining, C.; Huellein, J.; Zenz, T.; von Deimling, A.; Schirmacher, P.; Ho, A.D.; et al. Targeting the BRAF V600E Mutation in Multiple Myeloma. Cancer Discov. 2013, 3, 862–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mey, U.J.M.; Renner, C.; von Moos, R. Vemurafenib in Combination with Cobimetinib in Relapsed and Refractory Extramedullary Multiple Myeloma Harboring the BRAF V600E Mutation. Hematol. Oncol. 2017, 35, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Sharman, J.P.; Chmielecki, J.; Morosini, D.; Palmer, G.A.; Ross, J.S.; Stephens, P.J.; Stafl, J.; Miller, V.A.; Ali, S.M. Vemurafenib Response in 2 Patients with Posttransplant Refractory BRAF V600E-Mutated Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2014, 14, e161–e163. [Google Scholar] [CrossRef]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial Genomic Heterogeneity in Multiple Myeloma Revealed by Multi-Region Sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef]
- Szalat, R.; Avet-Loiseau, H.; Munshi, N.C. Gene Expression Profiles in Myeloma: Ready for the Real World? Clin. Cancer Res. 2016, 22, 5434–5442. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.J.; Mishima, Y.; Shi, J.; Sklavenitis-Pistofidis, R.; Redd, R.A.; Moschetta, M.; Manier, S.; Roccaro, A.M.; Sacco, A.; Tai, Y.-T.; et al. Progression Signature Underlies Clonal Evolution and Dissemination of Multiple Myeloma. Blood 2021, 137, 2360–2372. [Google Scholar] [CrossRef]
- Bustoros, M.; Sklavenitis-Pistofidis, R.; Park, J.; Redd, R.; Zhitomirsky, B.; Dunford, A.J.; Salem, K.; Tai, Y.-T.; Anand, S.; Mouhieddine, T.H.; et al. Genomic Profiling of Smoldering Multiple Myeloma Identifies Patients at a High Risk of Disease Progression. J. Clin. Oncol. 2020, 38, 2380–2389. [Google Scholar] [CrossRef] [PubMed]
- Huhn, S.; Weinhold, N.; Nickel, J.; Pritsch, M.; Hielscher, T.; Hummel, M.; Bertsch, U.; Huegle-Doerr, B.; Vogel, M.; Angermund, R.; et al. Circulating Tumor Cells as a Biomarker for Response to Therapy in Multiple Myeloma Patients Treated within the GMMG-MM5 Trial. Bone Marrow Transpl. 2017, 52, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Circulating Tumour DNA Analysis Demonstrates Spatial Mutational Heterogeneity That Coincides with Disease Relapse in Myeloma. Leukemia 2017, 31, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Garcés, J.-J.; Bretones, G.; Burgos, L.; Valdes-Mas, R.; Puig, N.; Cedena, M.-T.; Alignani, D.; Rodriguez, I.; Puente, D.Á.; Álvarez, M.-G.; et al. Circulating Tumor Cells for Comprehensive and Multiregional Non-Invasive Genetic Characterization of Multiple Myeloma. Leukemia 2020, 34, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Yasui, H.; Kobayashi, M.; Sato, K.; Kondoh, K.; Ishida, T.; Kaito, Y.; Tamura, H.; Handa, H.; Tsukune, Y.; Sasaki, M.; et al. Circulating Cell-Free DNA in the Peripheral Blood Plasma of Patients Is an Informative Biomarker for Multiple Myeloma Relapse. Int. J. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Park, S.-S.; Lim, J.-Y.; Kim, T.W.; Ko, Y.H.; Jeon, W.-J.; Lee, S.-Y.; Lee, J.-H.; Min, C.-K. Predictive Impact of Circulating MicroRNA-193a-5p on Early Relapse after Autologous Stem Cell Transplantation in Patients with Multiple Myeloma. Br. J. Haematol. 2020, 189, 518–523. [Google Scholar] [CrossRef]
- Derman, B.A.; Stefka, A.T.; Jiang, K.; McIver, A.; Kubicki, T.; Jasielec, J.K.; Jakubowiak, A.J. Measurable Residual Disease Assessed by Mass Spectrometry in Peripheral Blood in Multiple Myeloma in a Phase II Trial of Carfilzomib, Lenalidomide, Dexamethasone and Autologous Stem Cell Transplantation. Blood Cancer J. 2021, 11, 19. [Google Scholar] [CrossRef]
- Eveillard, M.; Rustad, E.; Roshal, M.; Zhang, Y.; Ciardiello, A.; Korde, N.; Hultcrantz, M.; Lu, S.; Shah, U.; Hassoun, H.; et al. Comparison of MALDI-TOF Mass Spectrometry Analysis of Peripheral Blood and Bone Marrow-Based Flow Cytometry for Tracking Measurable Residual Disease in Patients with Multiple Myeloma. Br. J. Haematol. 2020, 189, 904–907. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Krishnan, A.Y.; Arendt, B.; Dasari, S.; Efebera, Y.A.; Geller, N.; Giralt, S.; Hahn, T.; Kohlhagen, M.C.; Landau, H.J.; et al. MASS-FIX versus Standard Methods to Predict for PFS and OS among Multiple Myeloma Patients Participating on the STAMINA Trial. J. Clin. Oncol. 2021, 39, 8009. [Google Scholar] [CrossRef]
- Puig, N.; Mateos, M.-V.; Contreras, T.; Paiva, B.; Cedena, M.T.; Pérez, J.J.; Aires, I.; Agullo, C.; Martinez-Lopez, J.; Otero, P.R.; et al. Qip-Mass Spectrometry in High Risk Smoldering Multiple Myeloma Patients Included in the GEM-CESAR Trial: Comparison with Conventional and Minimal Residual Disease IMWG Response Assessment. Blood 2019, 134, 581. [Google Scholar] [CrossRef]
- Cenci, S.; Mezghrani, A.; Cascio, P.; Bianchi, G.; Cerruti, F.; Fra, A.; Lelouard, H.; Masciarelli, S.; Mattioli, L.; Oliva, L.; et al. Progressively Impaired Proteasomal Capacity during Terminal Plasma Cell Differentiation. EMBO J. 2006, 25, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Lee, K.P.; Boise, L.H. Proteasome Inhibitors Induce a Terminal Unfolded Protein Response in Multiple Myeloma Cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Ray, A.; Li, S.; Das, D.S.; Tai, Y.T.; Carrasco, R.D.; Chauhan, D.; Anderson, K.C. Targeting Proteasome Ubiquitin Receptor Rpn13 in Multiple Myeloma. Leukemia 2016, 30, 1877–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Park, P.M.C.; Wu, L.; Ray, A.; Picaud, S.; Li, D.; Wimalasena, V.K.; Du, T.; Filippakopoulos, P.; Anderson, K.C.; et al. Development and Preclinical Validation of a Novel Covalent Ubiquitin Receptor Rpn13 Degrader in Multiple Myeloma. Leukemia 2019, 33, 2685–2694. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.G.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A Small Molecule Inhibitor of Ubiquitin-Specific Protease-7 Induces Apoptosis in Multiple Myeloma Cells and Overcomes Bortezomib Resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Tian, Z.; D’Arcy, P.; Wang, X.; Ray, A.; Tai, Y.-T.; Hu, Y.; Carrasco, R.D.; Richardson, P.; Linder, S.; Chauhan, D.; et al. A Novel Small Molecule Inhibitor of Deubiquitylating Enzyme USP14 and UCHL5 Induces Apoptosis in Multiple Myeloma and Overcomes Bortezomib Resistance. Blood 2014, 123, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Gulla, A.; Morelli, E.; Samur, M.K.; Botta, C.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Malvestiti, S.; Prabhala, R.H.; Talluri, S.; et al. Bortezomib Induces Anti–Multiple Myeloma Immune Response Mediated by CGAS/STING Pathway Activation. Blood Cancer Discov. 2021, 2, 468–483. [Google Scholar] [CrossRef]
- Ray, A.; Song, Y.; Chauhan, D.; Anderson, K.C. Blockade of Ubiquitin Receptor Rpn13 in Plasmacytoid Dendritic Cells Triggers Anti-Myeloma Immunity. Blood Cancer J. 2019, 9, 64. [Google Scholar] [CrossRef] [Green Version]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, C.C.; Kang, J.; Amatangelo, M.; Polonskaia, A.; Katz, M.; Chiu, H.; Couto, S.; Wang, M.; Ren, Y.; Ortiz, M.; et al. Iberdomide (CC-220) Is a Potent Cereblon E3 Ligase Modulator with Antitumor and Immunostimulatory Activities in Lenalidomide- and Pomalidomide-Resistant Multiple Myeloma Cells with Dysregulated CRBN. Leukemia 2020, 34, 1197–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonial, S.; Richardson, P.G.; Popat, R.; Stadtmauer, E.; Larsen, J.; Oriol, A.; Knop, S.; Jagannath, S.; Cook, G.; Badros, A.Z.; et al. Iberdomide (Iber) in Combination with Dexamethasone (Des) and Daratumumab (Dara), Bortezomib (Boot), or Carfilzomib (Cfz) in Patients (Its) with Relapsed/Refractory Multiple Myeloma (RRMM). EHA Libr. 2021, 324595, S187. [Google Scholar]
- Richardson, P.G.; Vangsted, A.J.; Ramasamy, K.; Trudel, S.; Martínez, J.; Mateos, M.-V.; Rodríguez Otero, P.; Lonial, S.; Popat, R.; Oriol, A.; et al. First-in-Human Phase I Study of the Novel CELMoD Agent CC-92480 Combined with Dexamethasone (DEX) in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM). J. Clin. Oncol. 2020, 38, 8500. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, J.A.; Kirby, R.J.; Perino, S.; Agafonov, R.V.; Chaturvedi, P.; Class, B.; Cocozziello, D.; Eron, S.J.; Good, A.; Hart, A.A.; et al. Abstract LB007: CFT7455: A Novel, IKZF1/3 Degrader That Demonstrates Potent Tumor Regression in IMiD-Resistant Multiple Myeloma (MM) Xenograft Models. In Proceedings of the AACR (American Association for Cancer Research) Annual Meeting 2021, Philadelphia, PA, USA, 10–15 April 2021; Volume 81. [Google Scholar]
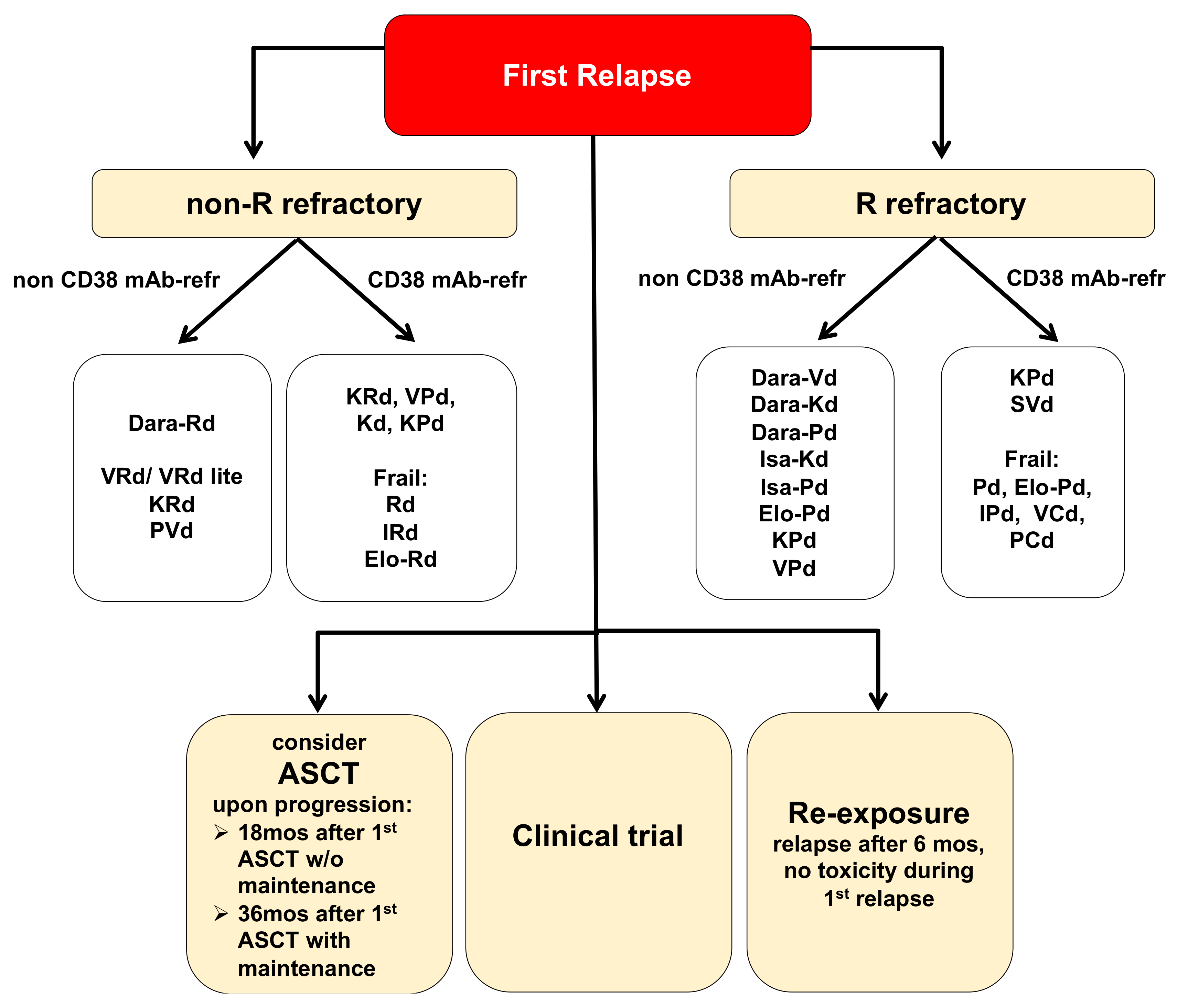


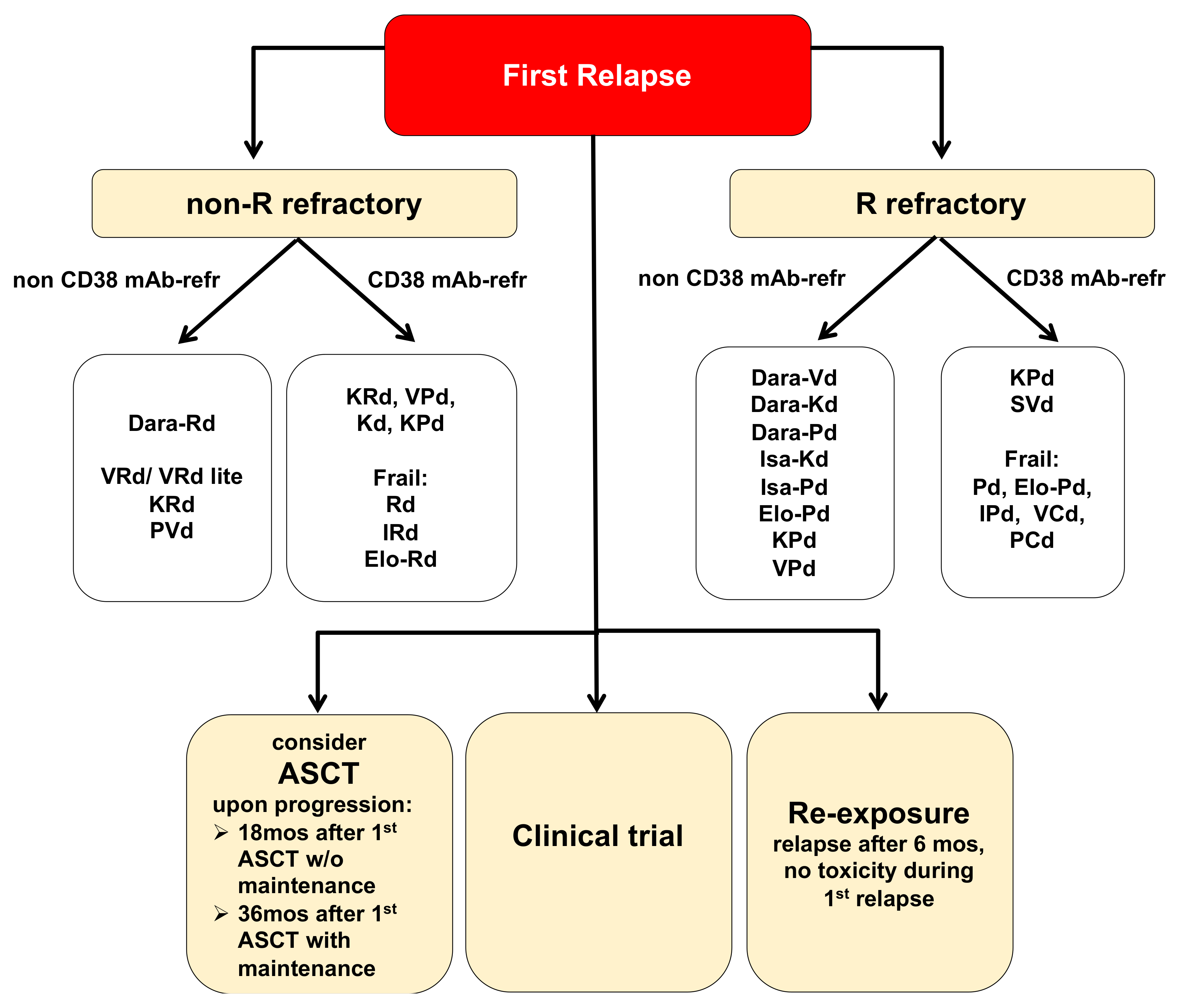{kind=link}
{kind=link}
| FDA/EMA | Regimen | Study | Phase | Prior Lines | N | ORR, % | PFS, mos | OS | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| FDA: 12 July 2021 EMA: 22 June 2021 | Dara-Pd vs Pd | APOLLO | III | ≥1 (Φ 2) | 304 | 69 vs. 46 | 12.4 vs. 6.9 [HR: 0.69] | NR vs. NR | [15] |
| FDA: 31 March 2021 EMA: 19 April 2021 | Isa-Kd vs Kd | IKEMA | III | 1–3 | 302 | 86.6 vs. 82.9 | NR vs. 19.15 [HR: 0.53] | NR vs. NR | [16] |
| FDA 21 December 2020 | S-Vd | BOSTON | III | 1–3 (Φ 2) | 402 | 79 vs. 66 | 13.9 vs. 9.5 [HR: 0.70] | NR | [23] |
| 20, 2020 EMA: 18 December 2020 | Dara-Kd vs Kd | CANDOR | III | 1–3 | 466 | 84.3 vs. 74.7 | NR vs. 15.8 [HR: 0.59] | NR vs. NR | [6] |
| EMA: 16 May 2019 | V-Pd vs Vd | OPTIMISSM | III | 1–3 Φ 2 | 559 | 82.2 vs. 50 (ITT) 90 vs. 54.8 (after 1 prior tx) | HR (ITT): 0.61 [HR: after 1 prior line of Tx 0.54] | NR | [8] |
| FDA: 26 February 2021 | Melflufen-Pd | HORIZON | II | ≥2 | 157 | 31 | 5.7 | 20 | [28] |
| FDA: 2 March 2020 EMA: 2 June 2020 | Isa-Pd | ICARIA | III | ≥2 | 307 | 60.4 vs. 35.3 | 12.7 vs. 7.9 | 1 yr OS: 72 vs. 63 | [13] |
| FDA: 7 November 2020 EMA: 28 August 2019 | Elo-Pd vs Pd | ELOQUENT-3 | III | ≥2 Φ 1–4 | 646 | 79 vs. 66 | 1 yr PFS 68 vs. 57 2 yrs PFS 41 vs. 27 | NR | [10] |
| FDA: 5 August 2020 EMA: 26 August 2020 | Belamaf | DREAMM-2 | II | ≥3 Φ 7 | 97 99 | 32 (2.5 mg/kg) 35 (3.4 mg/kg) | 2.8 (2.5 mg/kg) 3.9 (3.4 mg/kg) | mOS estimate: 13.7 (2.5 mg/kg) 13.8 (3.4 mg/kg) | [34] |
| FDA: 27 March 2021 EMA: 19 August 2021 | Ide-Cel | KarMMa | II | ≥4 Φ 6 | 128 | 73 | 8.8 12.1 at 450 × 106 18 mos PFS 66% | 19.4 | [41] |
| FDA: 3 July 2019 EMA: 29 March 2021 | Sd | STORM | I/II | Φ 7 | 79 122 | 26.2 | 3.7 | 8.6 | [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podar, K.; Leleu, X. Relapsed/Refractory Multiple Myeloma in 2020/2021 and Beyond. Cancers 2021, 13, 5154. https://doi.org/10.3390/cancers13205154
Podar K, Leleu X. Relapsed/Refractory Multiple Myeloma in 2020/2021 and Beyond. Cancers. 2021; 13(20):5154. https://doi.org/10.3390/cancers13205154
Chicago/Turabian StylePodar, Klaus, and Xavier Leleu. 2021. "Relapsed/Refractory Multiple Myeloma in 2020/2021 and Beyond" Cancers 13, no. 20: 5154. https://doi.org/10.3390/cancers13205154
APA StylePodar, K., & Leleu, X. (2021). Relapsed/Refractory Multiple Myeloma in 2020/2021 and Beyond. Cancers, 13(20), 5154. https://doi.org/10.3390/cancers13205154