
Combinatorial Effect of PLK1 Inhibition with Temozolomide and Radiation in Glioblastoma

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Cell Viability (MTT Assay)
2.3. Irradiation of GBM Cells
2.4. Flow Cytometry
2.5. ROS Assay
2.6. Tumor Sphere (TS) Formation
2.7. Side Population Analysis
2.8. Human Phosphokinase Array
2.9. Immunoblotting
2.10. Neutral Comet Assay
2.11. Clonogenic Cell Survival Analysis
2.12. RT2 Profiler PCR Array
2.13. Xenograft Models and Treatment
2.14. Immunohistochemistry Analysis
2.15. Statistical Analysis
3. Results
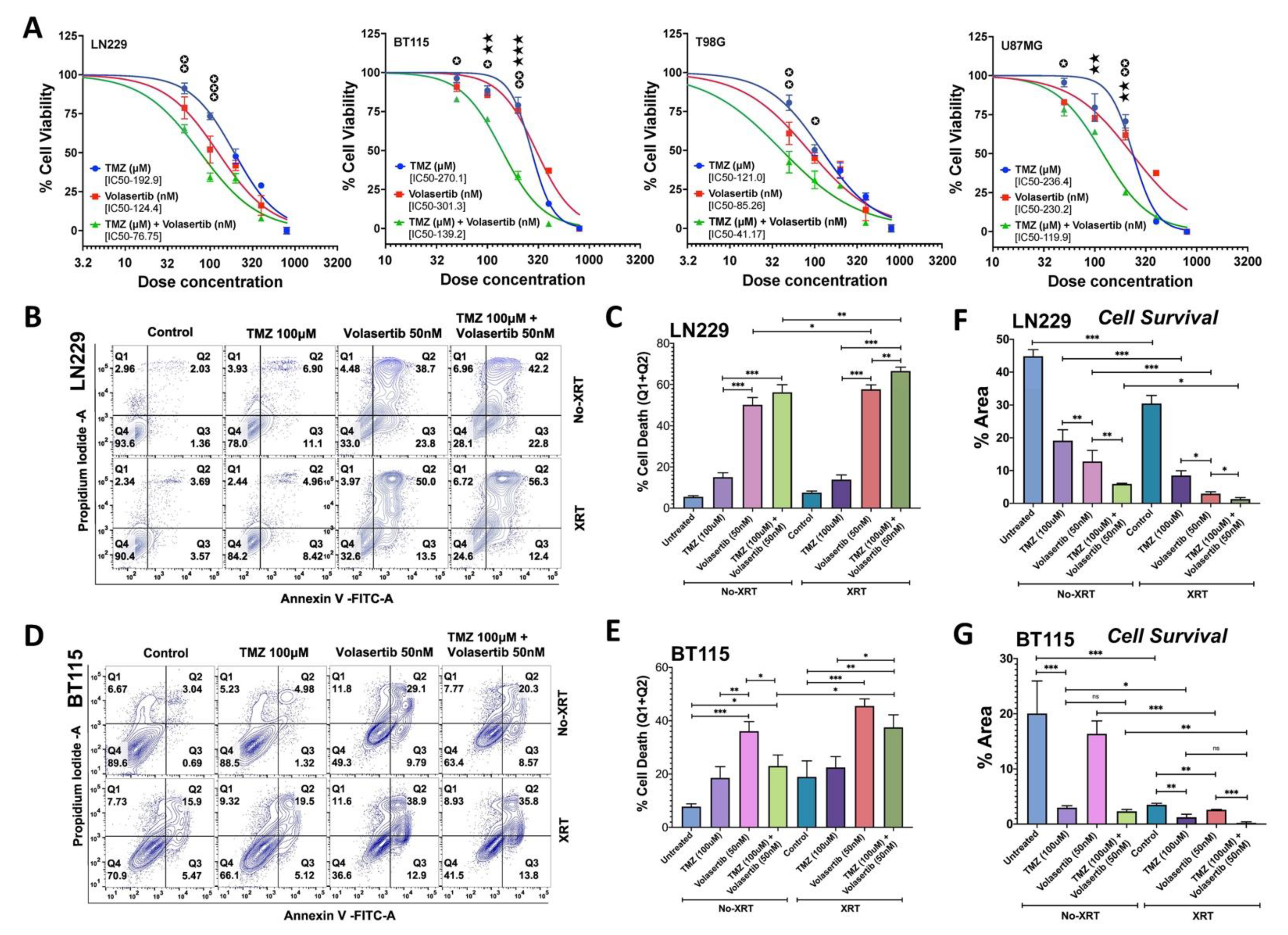
3.1. PLK1 Inhibition Promotes Apoptotic Cell Death and Reduces Cell Survival Alone and in Combination with TMZ and Radiation
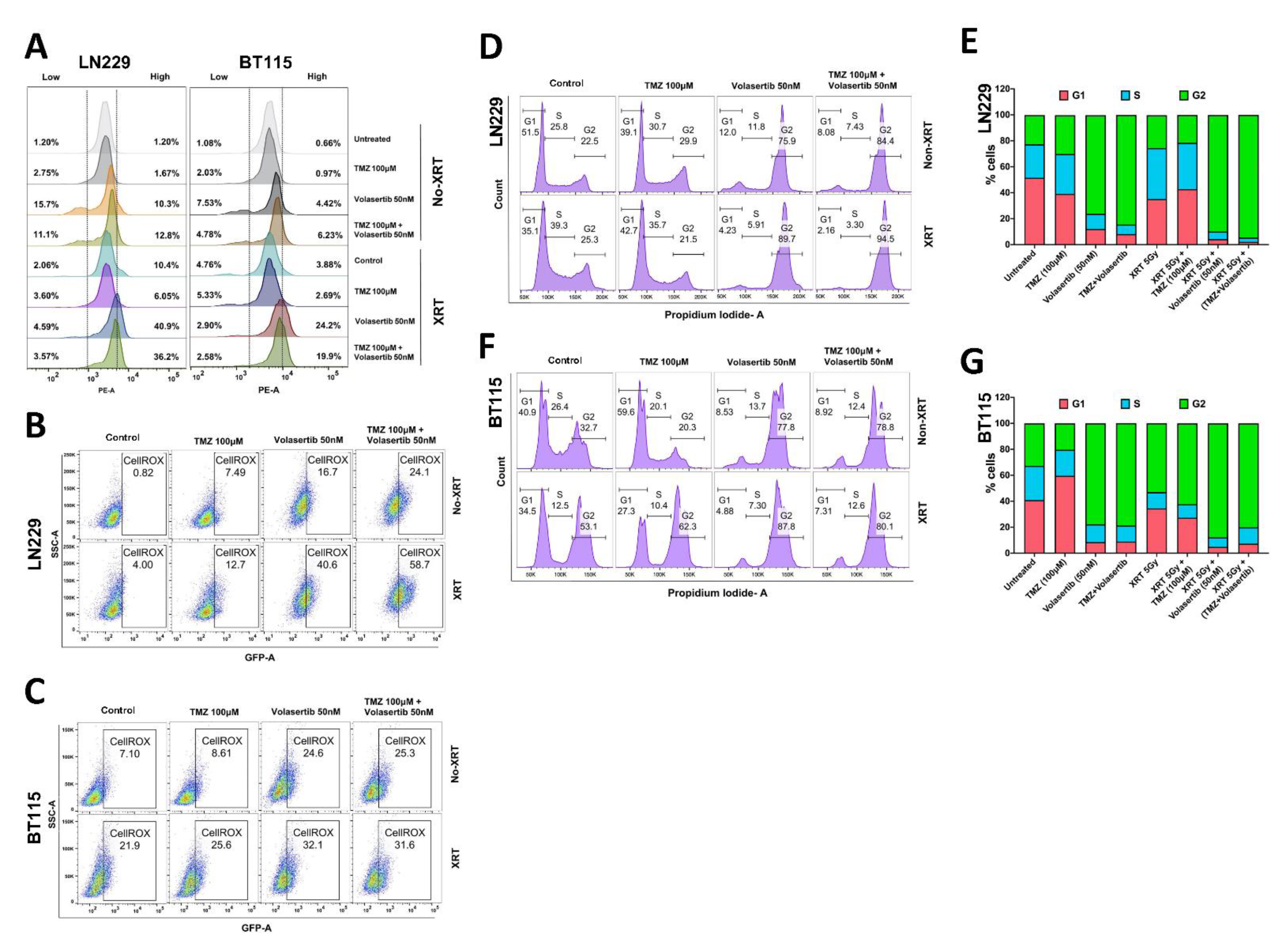
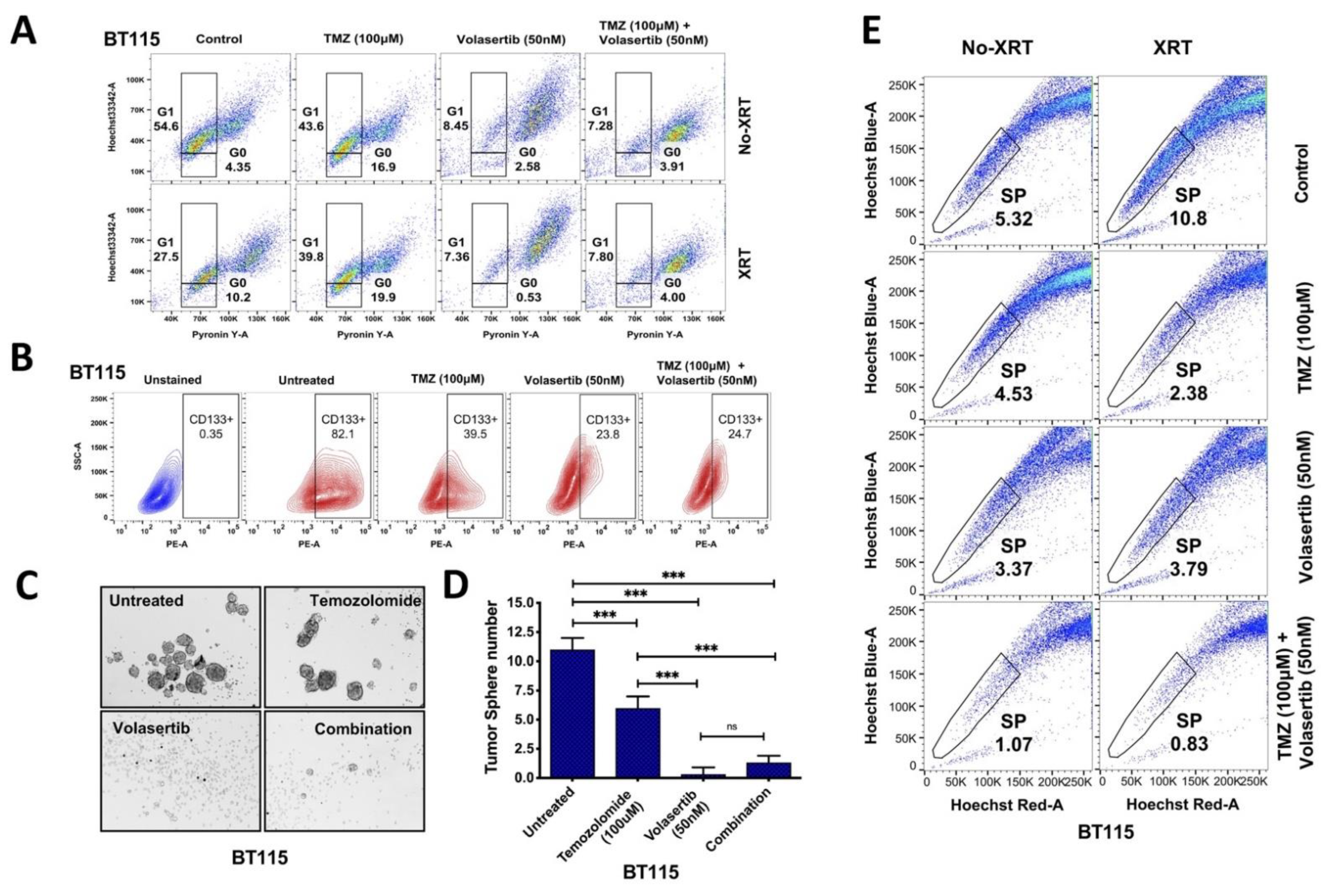
3.2. Volasertib Drives Concerted Disruption of MtMP, Activation of ROS and G2-M Arrest in GBM Cells without the Transition to the Quiescent State
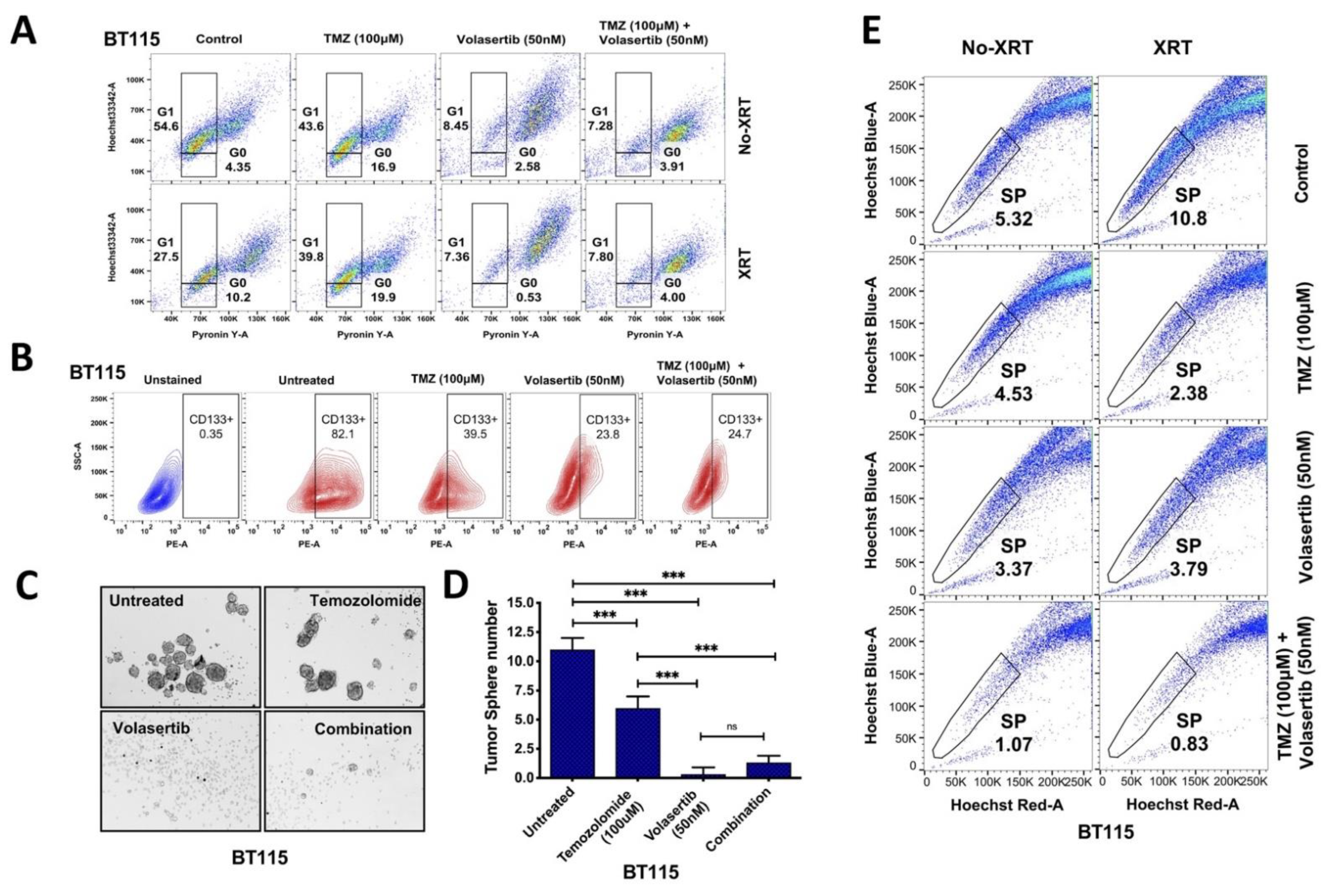
3.3. Inhibition of PLK1 Reduces Cancer Cell Stemness and Abrogates Side Population (SP) in GBM Cells
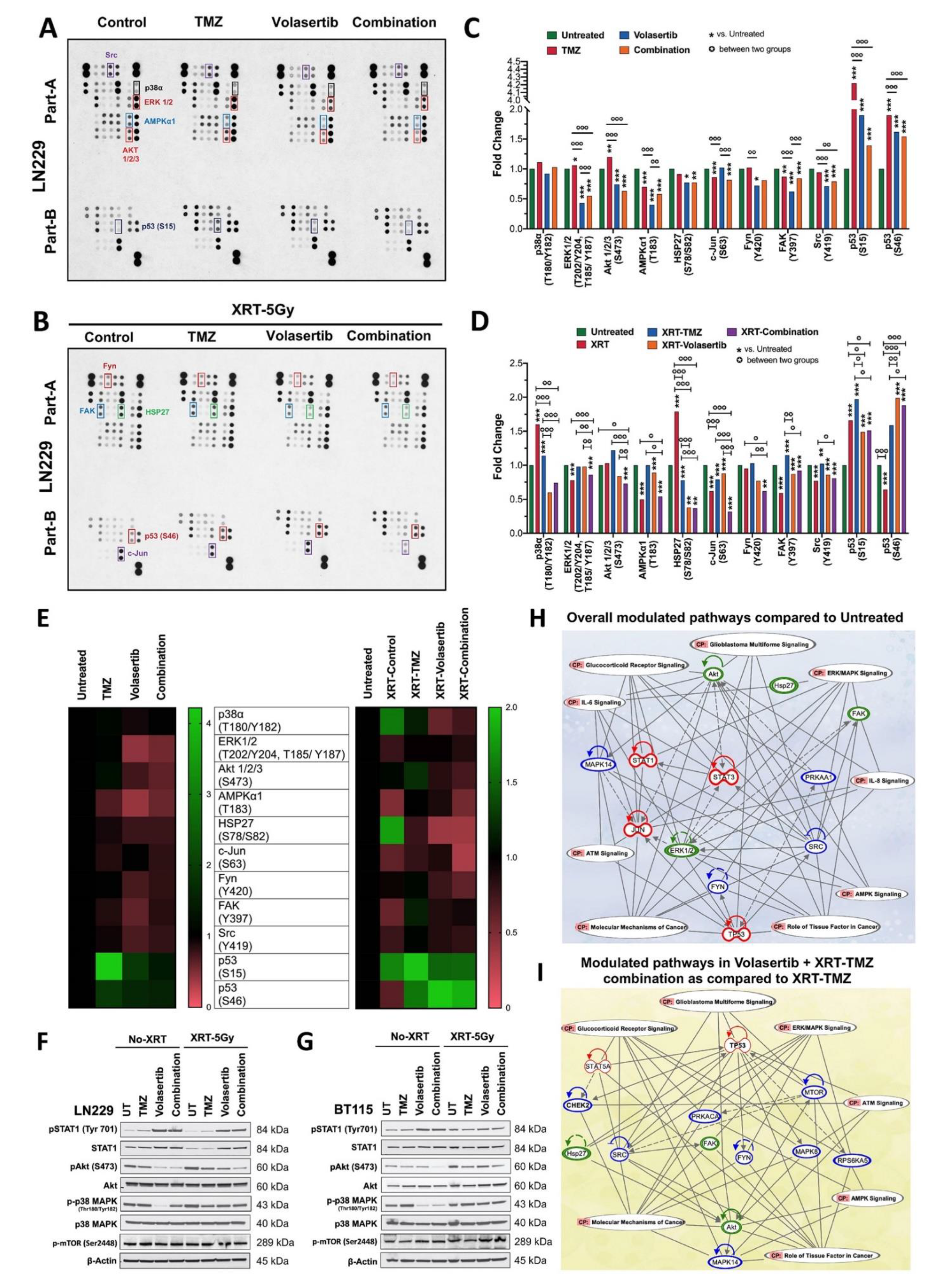
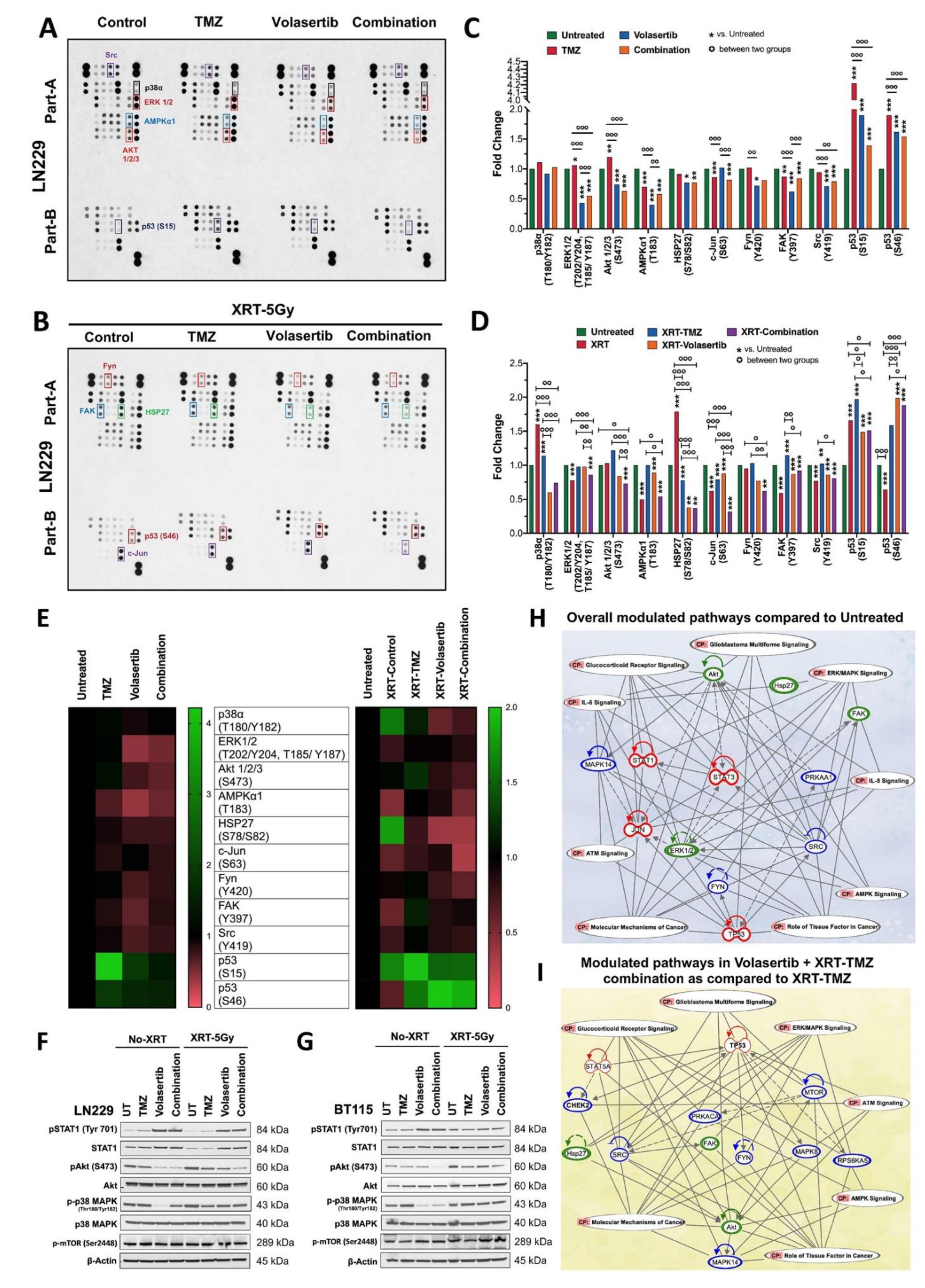
3.4. Volasertib Inhibits Key Tumor-Promoting Signaling Pathways in GBM Cells
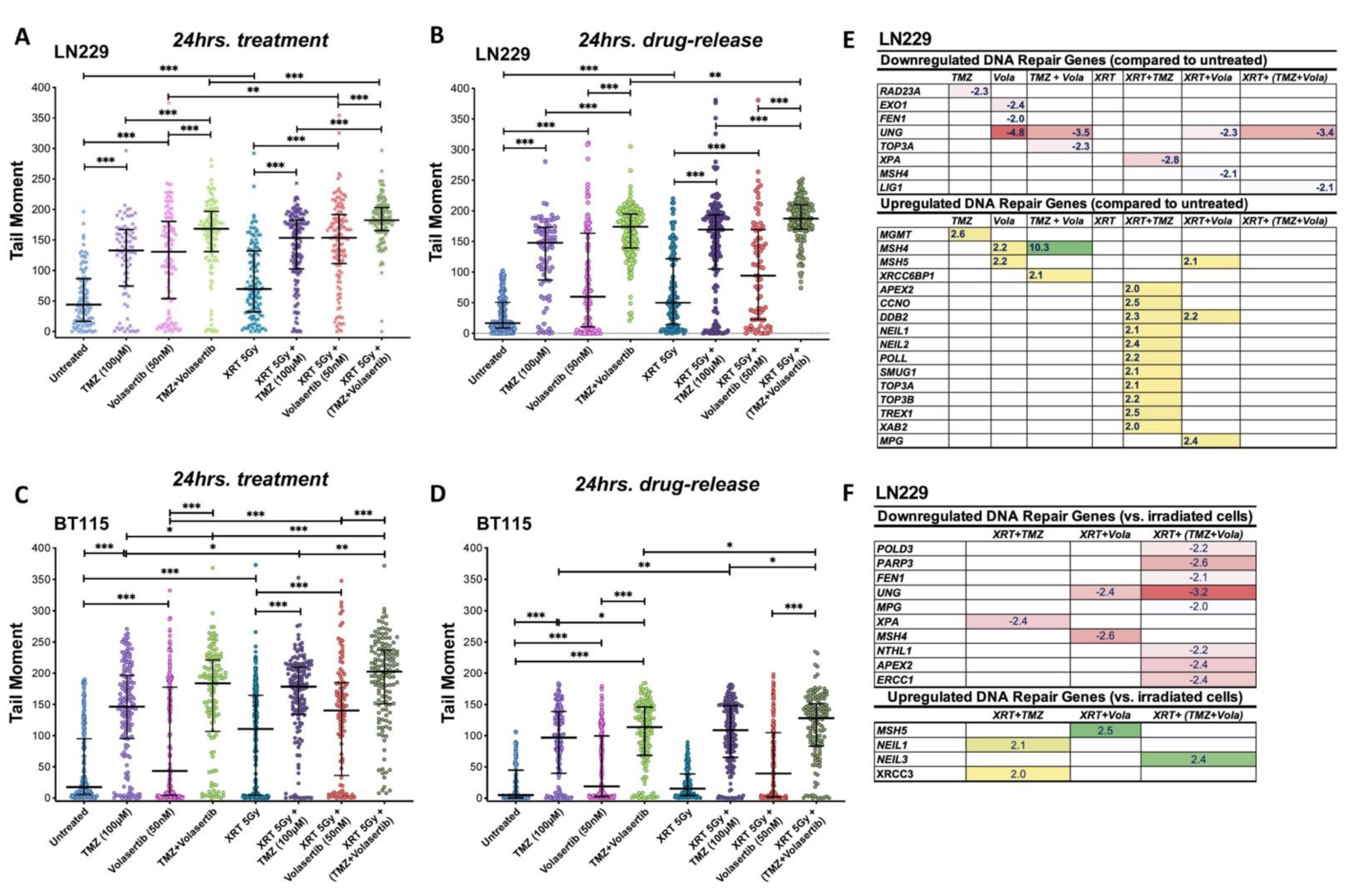
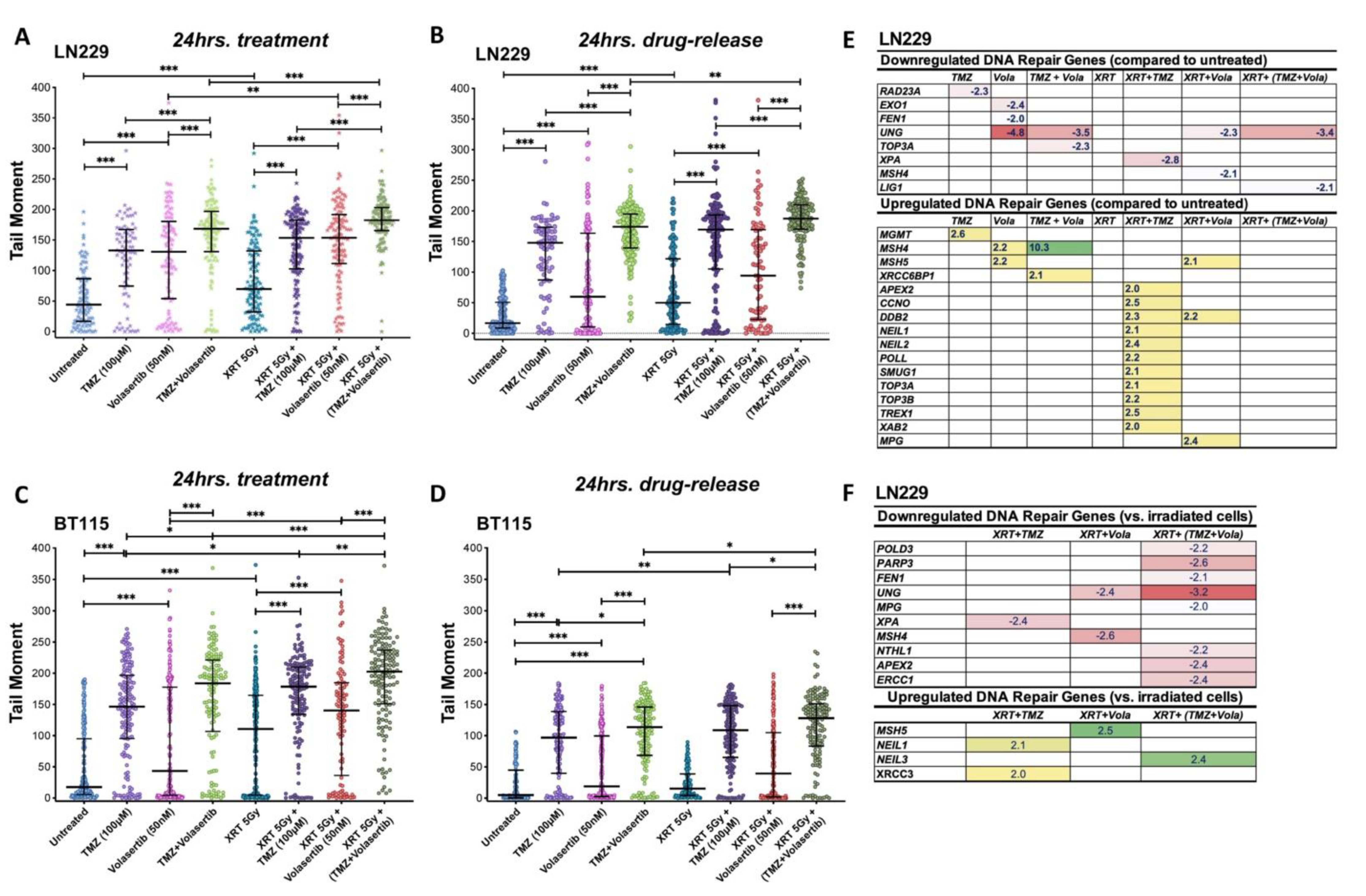
3.5. Volasertib Synergizes with TMZ and Radiation to Induce DNA Damage and Modulate DNA Repair Genes Expression in GBM Cells
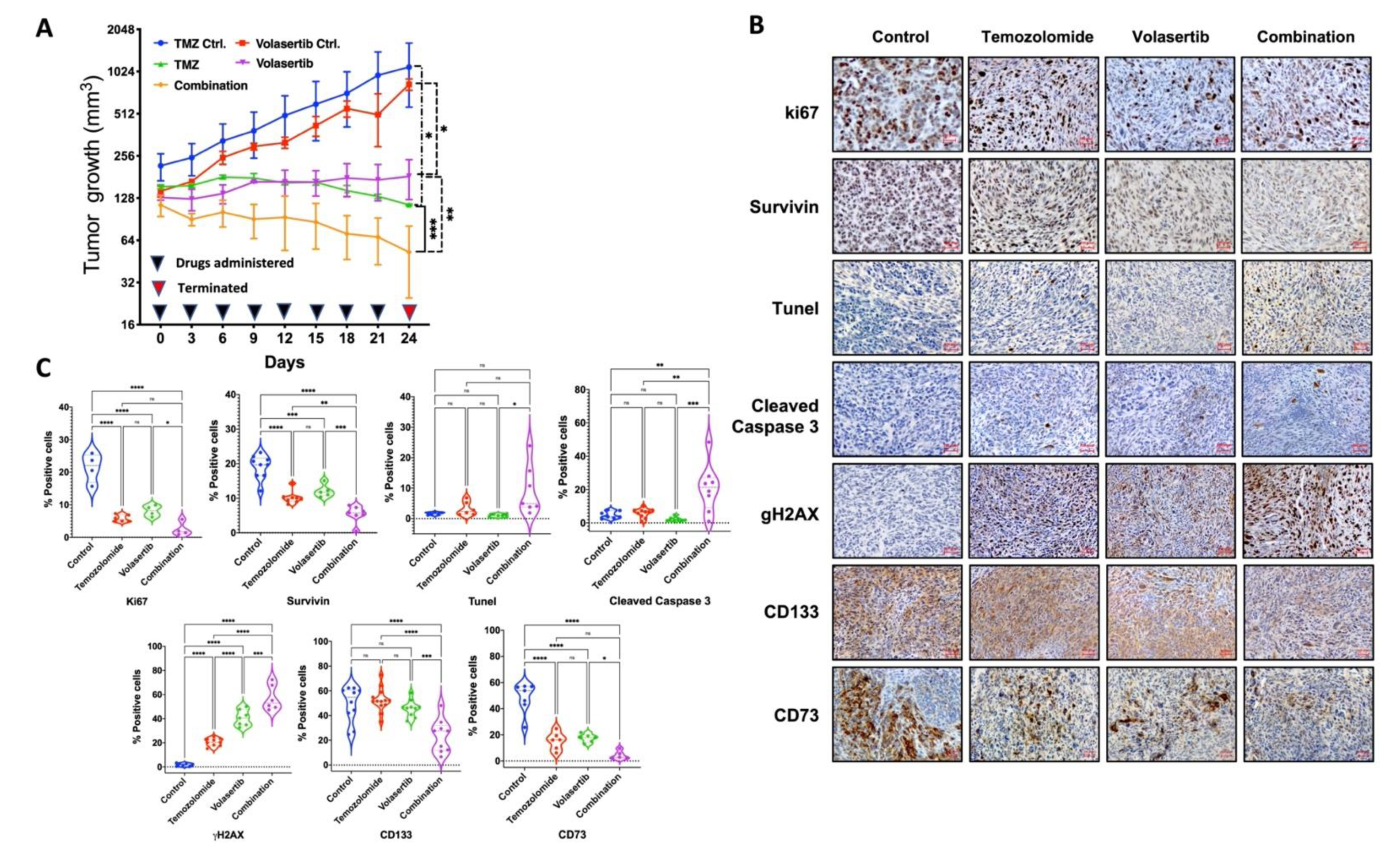
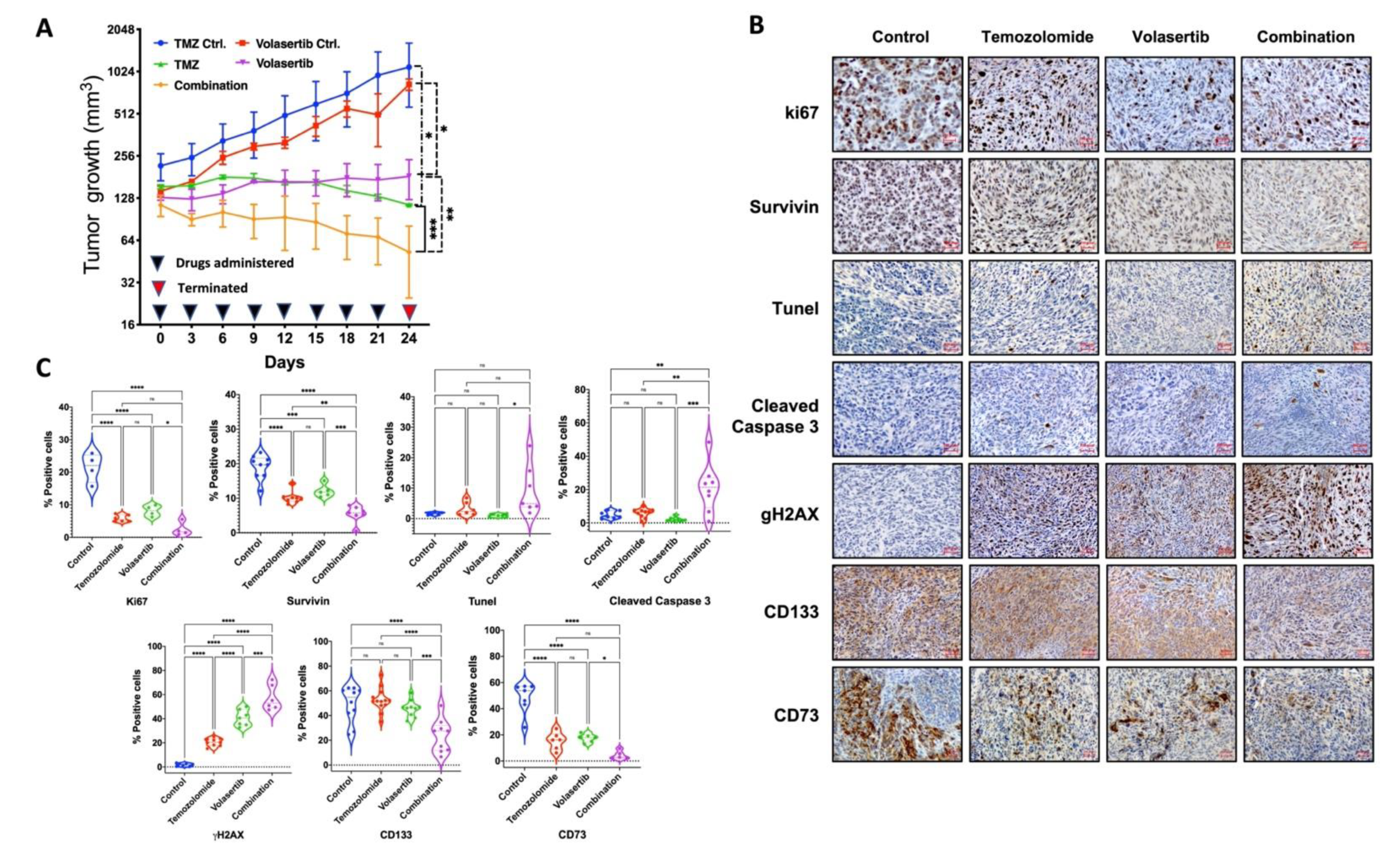
3.6. Combined Treatment of Volasertib and TMZ Reduces Tumor Growth and Stemness and Increases DNA Damage and Apoptotic Cell Death in Glioblastoma Cells In Vivo
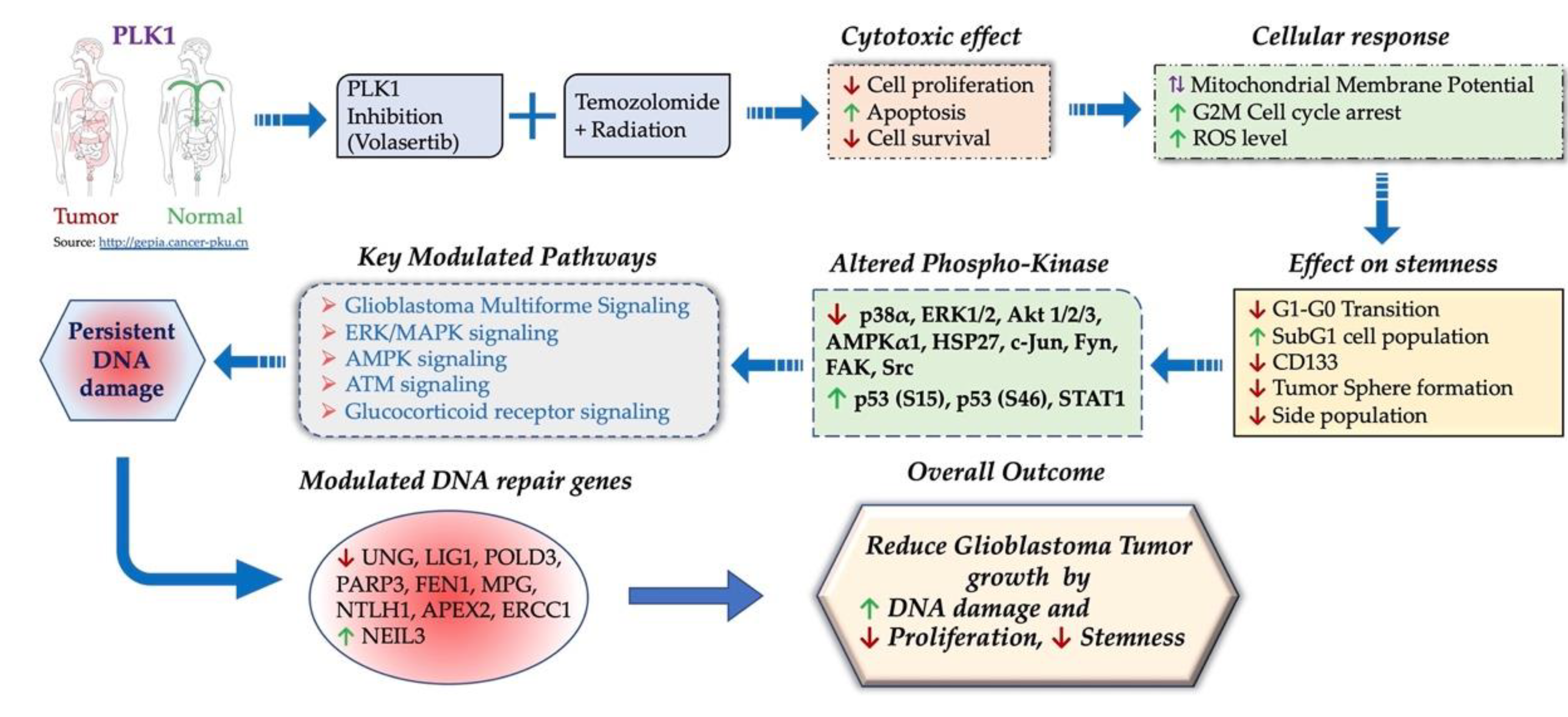
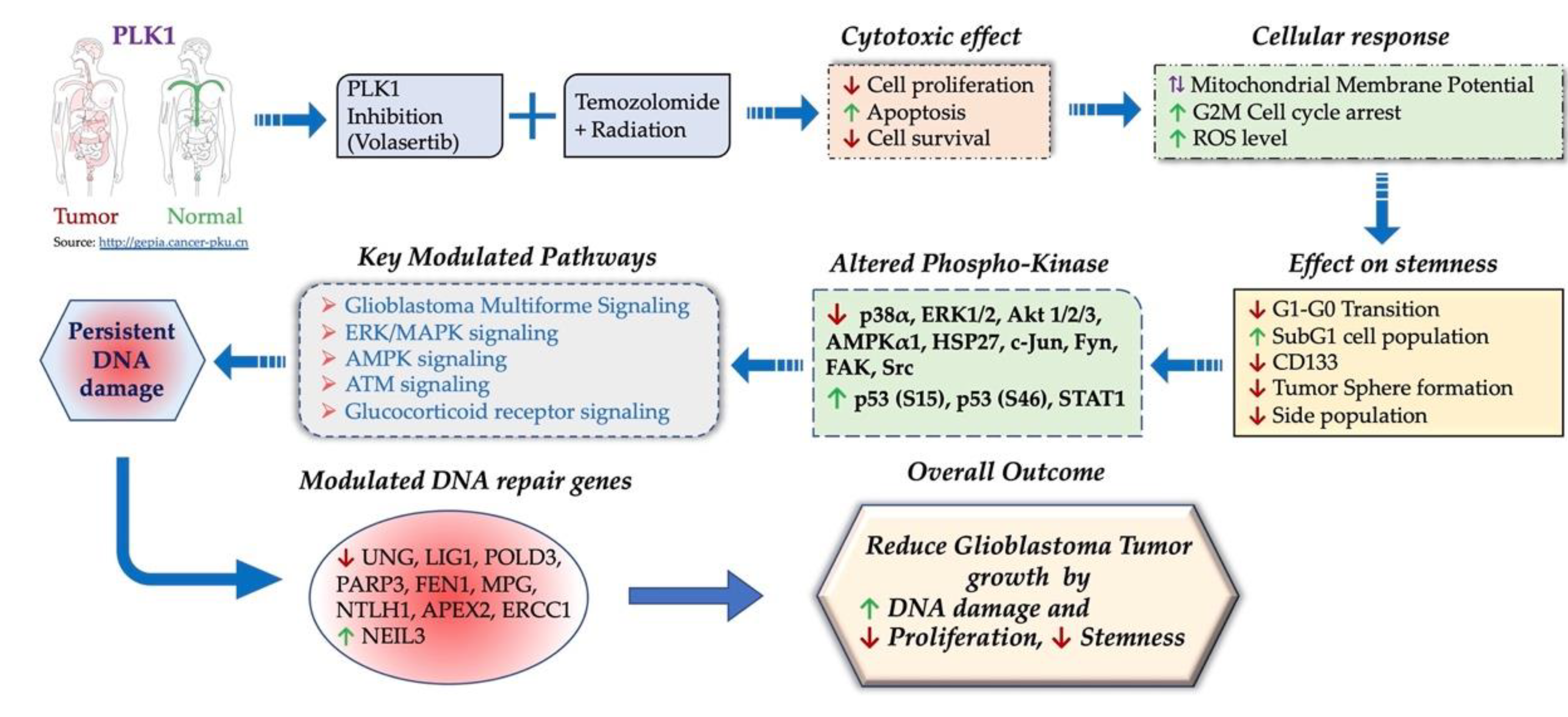
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neurooncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Hu, G.; Wang, H.; Li, Z.; Guo, Z. PLK1 inhibitor facilitates the suppressing effect of temozolomide on human brain glioma stem cells. J. Cell. Mol. Med. 2018, 22, 5300–5310. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, S.A.; Ye, X.; Piantadosi, S.; Desideri, S.; Nabors, L.B.; Rosenfeld, M.; Fisher, J.; Consortium, N.C. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin. Cancer Res. 2010, 16, 2443–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goroshchuk, O.; Kolosenko, I.; Vidarsdottir, L.; Azimi, A.; Palm-Apergi, C. Polo-like kinases and acute leukemia. Oncogene 2019, 38, 1–16. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential target for cancer therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Benada, J.; Burdova, K.; Lidak, T.; von Morgen, P.; Macurek, L. Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 2015, 14, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Pezuk, J.A.; Brassesco, M.S.; Morales, A.G.; de Oliveira, J.C.; de Paula Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; Scrideli, C.A.; Tone, L.G. Polo-like kinase 1 inhibition causes decreased proliferation by cell cycle arrest, leading to cell death in glioblastoma. Cancer Gene Ther. 2013, 20, 499–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, F.; Fink, A.L.; Kiyokawa, J.; Miller, J.J.; Koerner, M.V.A.; Cahill, D.P.; Wakimoto, H. PLK1 inhibition targets myc-activated malignant glioma cells irrespective of mismatch repair deficiency-mediated acquired resistance to temozolomide. Mol. Cancer Ther. 2018, 17, 2551–2563. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Park, S.Y.; Nguyen, N.; Ezhilarasan, R.; Martinez-Ledesma, E.; Wu, S.; Henry, V.; Piao, Y.; Tiao, N.; Brunell, D.; et al. The polo-like kinase 1 inhibitor volasertib synergistically increases radiation efficacy in glioma stem cells. Oncotarget 2018, 9, 10497–10509. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Vishnoi, K.; Mahata, S.; Tripathi, S.C.; Misra, S.P.; Misra, V.; Mehrotra, R.; Dwivedi, M.; Bharti, A.C. Berberine and curcumin target survivin and STAT3 in gastric cancer cells and synergize actions of standard chemotherapeutic 5-fluorouracil. Nutr. Cancer 2015, 67, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Kim, K.H.; Sederstrom, J.M. Assaying cell cycle status using flow cytometry. Curr. Protoc. Mol. Biol. 2015, 111, 28.6.1–28.6.11. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.K.; Goodell, M.A. Purification of hematopoietic stem cells using the side population. Methods Enzymol. 2006, 420, 255–264. [Google Scholar] [CrossRef]
- Vishnoi, K.; Mahata, S.; Tyagi, A.; Pandey, A.; Verma, G.; Jadli, M.; Singh, T.; Singh, S.M.; Bharti, A.C. Cross-talk between human papillomavirus oncoproteins and hedgehog signaling synergistically promotes stemness in cervical cancer cells. Sci. Rep. 2016, 6, 34377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, J.; Shoemake, J.D.; Ma, T.; Rizzo, A.E.; Godley, A.R.; Wu, Q.; Mohammadi, A.M.; Bao, S.; Rich, J.N.; Yu, J.S. Hyperthermia Sensitizes Glioma Stem-like Cells to Radiation by Inhibiting AKT Signaling. Cancer Res. 2015, 75, 1760–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.S.; Hsu, D.; Clement, M.V. OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Guzman, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Esteras Gallego, N.; Wray, S.; Preza, E.; Abramov, A.Y. Higher mitochondrial membrane potential induces ROS production in the familiar form of frontotemporal dementia with MAPT mutations. Biophys. J. 2015, 108. [Google Scholar] [CrossRef] [Green Version]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between mitochondrial membrane potential and ROS formation. Methods Mol. Biol. 2012, 810, 183–205. [Google Scholar] [CrossRef]
- Zheng, W.L.; Wang, B.J.; Wang, L.; Shan, Y.P.; Zou, H.; Song, R.L.; Wang, T.; Gu, J.H.; Yuan, Y.; Liu, X.Z.; et al. ROS-mediated cell cycle arrest and apoptosis induced by zearalenone in mouse sertoli cells via ER stress and the ATP/AMPK pathway. Toxins 2018, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Cho, I.J.; Lui, P.P.; Obajdin, J.; Riccio, F.; Stroukov, W.; Willis, T.L.; Spagnoli, F.; Watt, F.M. Mechanisms, hallmarks, and implications of stem cell quiescence. Stem Cell Rep. 2019, 12, 1190–1200. [Google Scholar] [CrossRef] [Green Version]
- Harris, M.A.; Yang, H.; Low, B.E.; Mukherjee, J.; Guha, A.; Bronson, R.T.; Shultz, L.D.; Israel, M.A.; Yun, K. Cancer stem cells are enriched in the side population cells in a mouse model of glioma. Cancer Res. 2008, 68, 10051–10059. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhou, K.; Gao, L.; Zhang, B.; Li, W.; Yan, W.; Song, X.; Yu, H.; Wang, S.; Yu, N.; et al. Radiation induces the generation of cancer stem cells: A novel mechanism for cancer radioresistance. Oncol. Lett. 2016, 12, 3059–3065. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Finnegan, C.E.; Rogers, K.M.; Purcell, J.W.; Trimble, A.; Johnston, P.G.; Boland, M.P. STAT1: A modulator of chemotherapy-induced apoptosis. Cancer Res. 2004, 64, 8357–8364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, L.; Karsy, M.; Murali, R.; Jhanwar-Uniyal, M. Inhibition of mTOR activates the MAPK pathway in glioblastoma multiforme. Cancer Genom. Proteom. 2009, 6, 255–261. [Google Scholar]
- Ando, K.; Ozaki, T.; Yamamoto, H.; Furuya, K.; Hosoda, M.; Hayashi, S.; Fukuzawa, M.; Nakagawara, A. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem. 2004, 279, 25549–25561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.; Farzan, R.; Ali, S.; Buluwela, L.; Saurin, A.T.; Meek, D.W. The responses of cancer cells to PLK1 inhibitors reveal a novel protective role for p53 in maintaining centrosome separation. Sci. Rep. 2017, 7, 16115. [Google Scholar] [CrossRef] [Green Version]
- Santucci-Darmanin, S.; Neyton, S.; Lespinasse, F.; Saunieres, A.; Gaudray, P.; Paquis-Flucklinger, V. The DNA mismatch-repair MLH3 protein interacts with MSH4 in meiotic cells, supporting a role for this MutL homolog in mammalian meiotic recombination. Hum. Mol. Genet. 2002, 11, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- Weeks, L.D.; Zentner, G.E.; Scacheri, P.C.; Gerson, S.L. Uracil DNA glycosylase (UNG) loss enhances DNA double strand break formation in human cancer cells exposed to pemetrexed. Cell Death Dis. 2014, 5, e1045. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Luo, L.; Zhu, H.; Yang, H.; Zhang, Y.; Wu, H.; Sun, H.; Jiang, F.; Kathera, C.S.; Liu, L.; et al. FEN1 promotes tumor progression and confers cisplatin resistance in non-small-cell lung cancer. Mol. Oncol. 2017, 11, 1302–1303. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Duan, J.; Shu, S.; Wang, X.; Gao, L.; Guo, J.; Zhang, Y. Ligase I and ligase III mediate the DNA double-strand break ligation in alternative end-joining. Proc. Natl. Acad. Sci. USA 2016, 113, 1256–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnihotri, S.; Burrell, K.; Buczkowicz, P.; Remke, M.; Golbourn, B.; Chornenkyy, Y.; Gajadhar, A.; Fernandez, N.A.; Clarke, I.D.; Barszczyk, M.S.; et al. ATM regulates 3-methylpurine-DNA glycosylase and promotes therapeutic resistance to alkylating agents. Cancer Discov. 2014, 4, 1198–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccard, S.G.; Marand, S.V.; Geraci, S.; Pycroft, L.; Berger, F.R.; Pelletier, L.A. Inhibition of DNA-repair genes Ercc1 and Mgmt enhances temozolomide efficacy in gliomas treatment: A pre-clinical study. Oncotarget 2015, 6, 29456–29468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehler, C.; Gauthier, L.R.; Mortusewicz, O.; Biard, D.S.; Saliou, J.M.; Bresson, A.; Sanglier-Cianferani, S.; Smith, S.; Schreiber, V.; Boussin, F.; et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. USA 2011, 108, 2783–2788. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Swenberg, J.; Nakamura, J. Abstract 1281: POLD3 is required for DNA damage response to endogenous and exogenous DNA damage in human cells. Cancer Res. 2014, 73, 1281. [Google Scholar] [CrossRef]
- Cheng, M.W.; Wang, B.C.; Weng, Z.Q.; Zhu, X.W. Clinicopathological significance of Polo-like kinase 1 (PLK1) expression in human malignant glioma. Acta Histochem. 2012, 114, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Fotovati, A.; Triscott, J.; Chen, J.; Venugopal, C.; Singhal, A.; Dunham, C.; Kerr, J.M.; Verreault, M.; Yip, S.; et al. Polo-like kinase 1 inhibition kills glioblastoma multiforme brain tumor cells in part through loss of SOX2 and delays tumor progression in mice. Stem Cells 2012, 30, 1064–1075. [Google Scholar] [CrossRef]
- Amani, V.; Prince, E.W.; Alimova, I.; Balakrishnan, I.; Birks, D.; Donson, A.M.; Harris, P.; Levy, J.M.; Handler, M.; Foreman, N.K.; et al. Polo-like kinase 1 as a potential therapeutic target in Diffuse Intrinsic Pontine Glioma. BMC Cancer 2016, 16, 647. [Google Scholar] [CrossRef] [Green Version]
- Maharjan, S.; Oku, M.; Tsuda, M.; Hoseki, J.; Sakai, Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci. Rep. 2014, 4, 5896. [Google Scholar] [CrossRef] [Green Version]
- Ghisolfi, L.; Keates, A.C.; Hu, X.; Lee, D.K.; Li, C.J. Ionizing radiation induces stemness in cancer cells. PLoS ONE 2012, 7, e43628. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bhatia, R. Stem cell quiescence. Clin. Cancer Res. 2011, 17, 4936–4941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murad, H.; Hawat, M.; Ekhtiar, A.; AlJapawe, A.; Abbas, A.; Darwish, H.; Sbenati, O.; Ghannam, A. Induction of G1-phase cell cycle arrest and apoptosis pathway in MDA-MB-231 human breast cancer cells by sulfated polysaccharide extracted from Laurencia papillosa. Cancer Cell Int. 2016, 16, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostopoulou, O.N.; Mohammad, A.A.; Bartek, J., Jr.; Winter, J.; Jung, M.; Stragliotto, G.; Soderberg-Naucler, C.; Landazuri, N. Glucocorticoids promote a glioma stem cell-like phenotype and resistance to chemotherapy in human glioblastoma primary cells: Biological and prognostic significance. Int. J. Cancer 2018, 142, 1266–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhipa, R.R.; Fan, Q.; Anderson, J.; Muraleedharan, R.; Huang, Y.; Ciraolo, G.; Chen, X.; Waclaw, R.; Chow, L.M.; Khuchua, Z.; et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol. 2018, 20, 823–835. [Google Scholar] [CrossRef]
- Belkacemi, L.; Hebb, M.O. HSP27 knockdown produces synergistic induction of apoptosis by HSP90 and kinase inhibitors in glioblastoma multiforme. Anticancer Res. 2014, 34, 4915–4927. [Google Scholar] [PubMed]
- Blau, L.; Knirsh, R.; Ben-Dror, I.; Oren, S.; Kuphal, S.; Hau, P.; Proescholdt, M.; Bosserhoff, A.K.; Vardimon, L. Aberrant expression of c-Jun in glioblastoma by internal ribosome entry site (IRES)-mediated translational activation. Proc. Natl. Acad. Sci. USA 2012, 109, E2875–E2884. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Guo, S.; Li, Y.; Ran, H.; Huang, H.; Mi, L.; Wu, J.; Wang, X.; Xiao, D.; Chen, L.; et al. Glioma stem-like cells evade interferon suppression through MBD3/NuRD complex-mediated STAT1 downregulation. J. Exp. Med. 2020, 217, e20191340. [Google Scholar] [CrossRef] [PubMed]
- Townsend, P.A.; Scarabelli, T.M.; Davidson, S.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J. Biol. Chem. 2004, 279, 5811–5820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karicheva, O.; Rodriguez-Vargas, J.M.; Wadier, N.; Martin-Hernandez, K.; Vauchelles, R.; Magroun, N.; Tissier, A.; Schreiber, V.; Dantzer, F. PARP3 controls TGFbeta and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget 2016, 7, 64109–64123. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, A.; Tripathi, S.C.; Mai, J.; Hanash, S.M.; Shen, H.; Mitra, S.; Rostomily, R.C. Combinatorial Effect of PLK1 Inhibition with Temozolomide and Radiation in Glioblastoma. Cancers 2021, 13, 5114. https://doi.org/10.3390/cancers13205114
Pandey A, Tripathi SC, Mai J, Hanash SM, Shen H, Mitra S, Rostomily RC. Combinatorial Effect of PLK1 Inhibition with Temozolomide and Radiation in Glioblastoma. Cancers. 2021; 13(20):5114. https://doi.org/10.3390/cancers13205114
Chicago/Turabian StylePandey, Arvind, Satyendra C. Tripathi, Junhua Mai, Samir M. Hanash, Haifa Shen, Sankar Mitra, and Robert C. Rostomily. 2021. "Combinatorial Effect of PLK1 Inhibition with Temozolomide and Radiation in Glioblastoma" Cancers 13, no. 20: 5114. https://doi.org/10.3390/cancers13205114
APA StylePandey, A., Tripathi, S. C., Mai, J., Hanash, S. M., Shen, H., Mitra, S., & Rostomily, R. C. (2021). Combinatorial Effect of PLK1 Inhibition with Temozolomide and Radiation in Glioblastoma. Cancers, 13(20), 5114. https://doi.org/10.3390/cancers13205114