
Genomic Abnormalities as Biomarkers and Therapeutic Targets in Acute Myeloid Leukemia



Abstract
Simple Summary
Abstract
1. Introduction
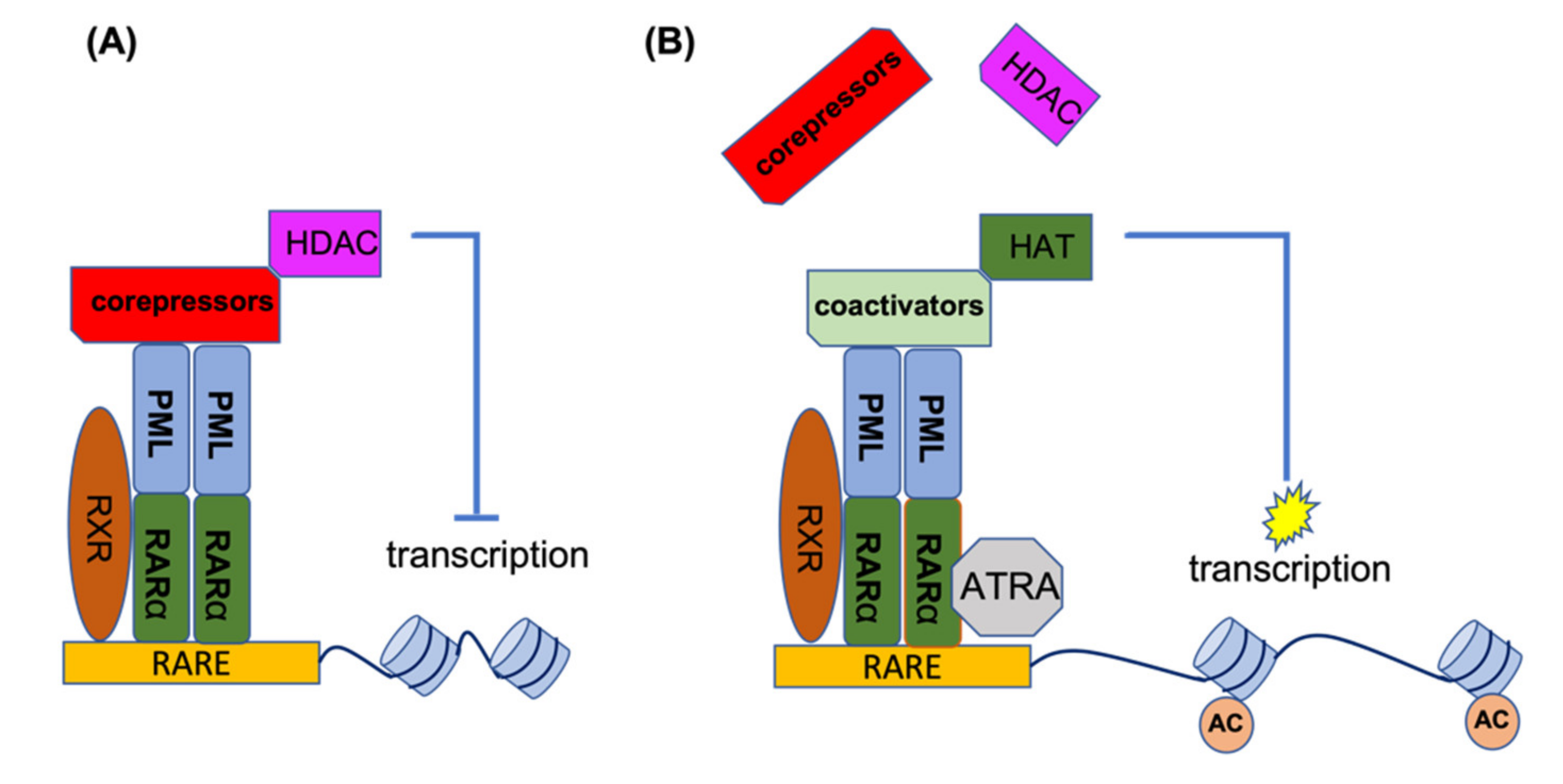
2. PML-RARA
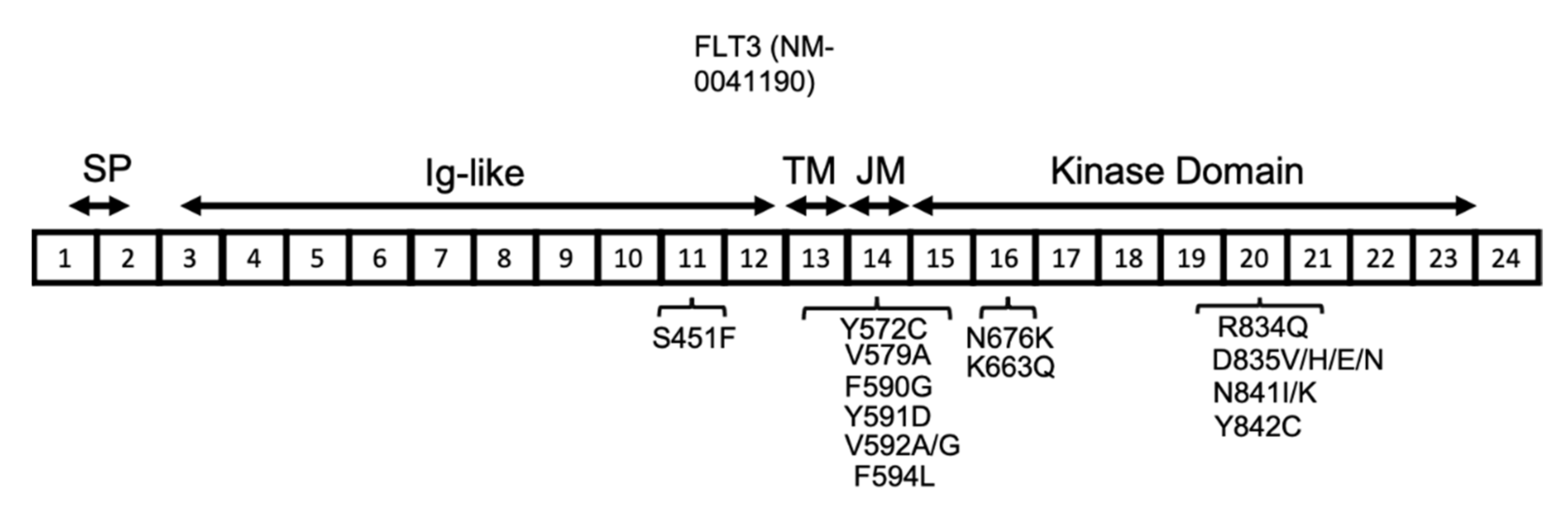
3. FLT3
4. BCL-2
5. IDH1/IDH2
6. Functional Genomics and Target Discovery
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- O’Hare, T.; Zabriskie, M.S.; Eiring, A.M.; Deininger, M.W. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer. 2012, 12, 513–526. [Google Scholar] [CrossRef]
- O’Dwyer, M.E.; Druker, B.J. STI571: An inhibitor of the BCR-ABL tyrosine kinase for the treatment of chronic myelogenous leukaemia. Lancet Oncol. 2000, 1, 207–211. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.-X.; Rea, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Torres-Ayuso, P.; Brognard, J. Combing the Cancer Genome for Novel Kinase Drivers and New Therapeutic Targets. Cancers 2019, 11, 1972. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Noguera, N.; Catalano, G.; Banella, C.; Divona, M.; Faraoni, I.; Ottone, T.; Arcese, W.; Voso, M. Acute Promyelocytic Leukemia: Update on the Mechanisms of Leukemogenesis, Resistance and on Innovative Treatment Strategies. Cancers 2019, 11, 1591. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, C.; Zhu, X. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.; Brown, C.; Iland, H. Retinoic acid and arsenic trioxide in the treatment of acute promyelocytic leukemia: Current perspectives. OncoTargets Ther. 2017, 10, 1585–1601. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Burd, A.; Levine, R.L.; Ruppert, A.S.; Mims, A.S.; Borate, U.; Stein, E.M.; Patel, P.; Baer, M.R.; Stock, W.; Deininger, M.; et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: Feasibility and preliminary efficacy of the Beat AML Master Trial. Nat. Med. 2020, 26, 1852–1858. [Google Scholar] [CrossRef]
- Guglielmi, C.; Martelli, M.P.; Diverio, D.; Fenu, S.; Vegna, M.L.; Cantù-Rajnoldi, A.; Biondi, A.; Cocito, M.G.; Del Vecchio, L.; Tabilio, A.; et al. Immunophenotype of adult and childhood acute promyelocytic leukaemia: Correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br. J. Haematol. 1998, 102, 1035–1041. [Google Scholar] [CrossRef]
- Tomita, A.; Kiyoi, H.; Naoe, T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O3) in acute promyelocytic leukemia. Int. J. Hematol. 2013, 97, 717–725. [Google Scholar] [CrossRef]
- Özpolat, B. Acute promyelocytic leukemia and differentiation therapy: Molecular mechanisms of differentiation, retinoic acid resistance and novel treatments. Turk. J. Hematol. 2009, 26, 47–61. [Google Scholar]
- Sobas, M.; Rodriguez-Veiga, R.; Vellenga, E.; Paluszewska, M.; De La Serna, J.; García-Álvarez, F.; Gil, C.; Brunet, S.; Bergua, J.; González-Campos, J.; et al. Characteristics and outcome of adult patients with acute promyelocytic leukemia and increased body mass index treated with the PETHEMA Protocols. Eur. J. Haematol. 2020, 104, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Catalano, A.; Dawson, M.A.; Somana, K.; Opat, S.; Schwarer, A.; Campbell, L.J.; Iland, H. The PRKAR1A gene is fused to RARA in a new variant acute promyelocytic leukemia. Blood 2007, 110, 4073–4076. [Google Scholar] [CrossRef]
- Strehl, S.; König, M.; Boztug, H.; Cooper, B.W.; Suzukawa, K.; Zhang, S.-J.; Chen, H.-Y.; Attarbaschi, A.; Dworzak, M.N. All-trans retinoic acid and arsenic trioxide resistance of acute promyelocytic leukemia with the variant STAT5B-RARA fusion gene. Leukemia 2013, 27, 1606–1610. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Tsuzuki, S.; Tsuzuki, M.; Handa, K.; Inaguma, Y.; Emi, N. BCOR as a novel fusion partner of retinoic acid receptor alpha in a t(X;17)(p11;q12) variant of acute promyelocytic leukemia. Blood 2010, 116, 4274–4283. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Q.; Zhu, J.; Shi, X.G.; Ni, J.H.; Zhong, H.J.; Si, G.Y.; Jin, X.L.; Tang, W.; Li, X.S.; Xong, S.M.; et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood 1996, 88, 1052–1061. [Google Scholar] [CrossRef]
- Mi, J.-Q.; Li, J.-M.; Shen, Z.-X.; Chen, S.-J.; Chen, Z. How to manage acute promyelocytic leukemia. Leukemia 2012, 26, 1743–1751. [Google Scholar] [CrossRef][Green Version]
- Fox, E.; Razzouk, B.; Widemann, B.C.; Xiao, S.; O’Brien, M.; Goodspeed, W.; Reaman, G.H.; Blaney, S.M.; Murgo, A.J.; Balis, F.M.; et al. Phase 1 trial and pharmacokinetic study of arsenic trioxide in children and adolescents with refractory or relapsed acute leukemia, including acute promyelocytic leukemia or lymphoma. Blood 2008, 111, 566–573. [Google Scholar] [CrossRef]
- Zhang, X.-W.; Yan, X.-J.; Zhou, Z.-R.; Yang, F.-F.; Wu, Z.Y.; Sun, H.-B.; Liang, W.-X.; Song, A.-X.; Lallemand-Breitenbach, V.; Jeanne, M.; et al. Arsenic Trioxide Controls the Fate of the PML-RAR Oncoprotein by Directly Binding PML. Science 2010, 328, 240–243. [Google Scholar] [CrossRef]
- Jeanne, M.; Lallemand, V.; Ferhi, O.; Koken, M.; LE Bras, M.; Duffort, S.; Peres, L.; Berthier, C.; Soilihi, H.; Raught, B.; et al. PML/RARA Oxidation and Arsenic Binding Initiate the Antileukemia Response of As2O3. Cancer Cell 2010, 18, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Maroui, M.A.; Kheddache-Atmane, S.; El Asmi, F.; Dianoux, L.; Aubry, M.; Chelbi-Alix, M.K. Requirement of PML SUMO Interacting Motif for RNF4- or Arsenic Trioxide-Induced Degradation of Nuclear PML Isoforms. PLoS ONE 2012, 7, e44949. [Google Scholar] [CrossRef] [PubMed]
- Lång, E.; Grudic, A.; Pankiv, S.; Bruserud, O.; Simonsen, A.; Bjerkvig, R.; Bjørås, M.; Bøe, S.O. The arsenic-based cure of acute promyelocytic leukemia promotes cytoplasmic sequestration of PML and PML/RARA through inhibition of PML body recycling. Blood 2012, 120, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D. The pathogenesis of acute promyelocytic leukaemia: Evaluation of the role of molecular diagnosis and monitoring in the management of the disease. Br. J. Haematol. 1999, 106, 591–613. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, F.; Fromental-Ramain, C.; Yamamoto, K.; Chambon, P. ATP-Driven Chromatin Remodeling Activity and Histone Acetyltransferases Act Sequentially during Transactivation by RAR/RXR In Vitro. Mol. Cell 2000, 6, 1049–1058. [Google Scholar] [CrossRef]
- Di Croce, L.; Raker, V.A.; Corsaro, M.; Fazi, F.; Fanelli, M.; Faretta, M.; Fuks, F.; Lo Coco, F.; Kouzarides, T.; Nervi, C.; et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 2002, 295, 1079–1082. [Google Scholar] [CrossRef]
- Pandolfi, P.P. In vivo analysis of the molecular genetics of acute promyelocytic leukemia. Oncogene 2001, 20, 5726–5735. [Google Scholar] [CrossRef]
- Lo-Coco, F.; Ammatuna, E. The Biology of Acute Promyelocytic Leukemia and Its Impact on Diagnosis and Treatment. Hematology 2006, 2006, 156–161. [Google Scholar] [CrossRef]
- Saeed, S.; Logie, C.; Stunnenberg, H.G.; Martens, J.H. Genome-wide functions of PML-RARalpha in acute promyelocytic leukaemia. Br. J. Cancer 2011, 104, 554–558. [Google Scholar] [CrossRef]
- Gallagher, R.E. Retinoic acid resistance in acute promyelocytic leukemia. Leukemia 2002, 16, 1940–1958. [Google Scholar] [CrossRef]
- Gallagher, R.E.; Moser, B.K.; Racevskis, J.; Poiré, X.; Bloomfield, C.D.; Carroll, A.J.; Ketterling, R.P.; Roulston, D.; Schachter-Tokarz, E.; Zhou, D.-C.; et al. Treatment-influenced associations of PML-RARα mutations, FLT3 mutations, and additional chromosome abnormalities in relapsed acute promyelocytic leukemia. Blood 2012, 120, 2098–2108. [Google Scholar] [CrossRef]
- Cote, S.; Zhou, D.; Bianchini, A.; Nervi, C.; Gallagher, R.E.; Miller, W.H. Altered ligand binding and transcriptional regulation by mutations in the PML/RARalpha ligand-binding domain arising in retinoic acid-resistant patients with acute promyelocytic leukemia. Blood 2000, 96, 3200–3208. [Google Scholar] [CrossRef]
- Marasca, R.; Zucchini, P.; Galimberti, S.; Leonardi, G.; Vaccari, P.; Donelli, A.; Luppi, M.; Petrini, M.; Torelli, G. Missense mutations in the PML/RARalpha ligand binding domain in ATRA-resistant As(2)O(3) sensitive relapsed acute promyelocytic leukemia. Haematologica 1999, 84, 963–968. [Google Scholar]
- Lehmann-Che, J.; Bally, C.; Letouzé, E.; Berthier, C.; Yuan, H.; Jollivet, F.; Ades, L.; Cassinat, B.; Hirsch, P.; Pigneux, A.; et al. Dual origin of relapses in retinoic-acid resistant acute promyelocytic leukemia. Nat. Commun. 2018, 9, 2047. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Kantarjian, H.; Ravandi, F. Acute promyelocytic leukemia current treatment algorithms. Blood Cancer J. 2021, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Hattori, H.; Ishikawa, Y.; Kawashima, N.; Akashi, A.; Yamaguchi, Y.; Harada, Y.; Hirano, D.; Adachi, Y.; Miyao, K.; Ushijima, Y.; et al. Identification of the novel deletion-type PML-RARA mutation associated with the retinoic acid resistance in acute promyelocytic leukemia. PLoS ONE 2018, 13, e0204850. [Google Scholar] [CrossRef] [PubMed]
- Kazi, J.U.; Rönnstrand, L. FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef] [PubMed]
- Matthews, W.; Jordan, C.; Wiegand, G.W.; Pardoll, D.; Lemischka, I.R. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell 1991, 65, 1143–1152. [Google Scholar] [CrossRef]
- Rappold, I.; Ziegler, B.L.; Köhler, I.; Marchetto, S.; Rosnet, O.; Birnbaum, D.; Simmons, P.J.; Zannettino, A.; Hill, B.; Neu, S.; et al. Functional and phenotypic characterization of cord blood and bone marrow subsets expressing FLT3 (CD135) receptor tyrosine kinase. Blood 1997, 90, 111–125. [Google Scholar] [PubMed]
- Williams, A.; Koch, S.; Christian, T.; Brown, P.; Levis, M.; Small, N. Glycosylation and Surface Localization Are Required for FLT3 Activation but Not for FLT3/ITD. Blood 2009, 114, 2748. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.-E.; Böhmer, A.; Markova, B.; Choudhary, C.; Serve, H.; Böhmer, F.-D. Tyrosine Phosphorylation Regulates Maturation of Receptor Tyrosine Kinases. Mol. Cell. Biol. 2005, 25, 3690–3703. [Google Scholar] [CrossRef] [PubMed]
- Razumovskaya, E.; Masson, K.; Khan, R.; Bengtsson, S.; Rönnstrand, L. Oncogenic Flt3 receptors display different specificity and kinetics of autophosphorylation. Exp. Hematol. 2009, 37, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, M.; Pietschmann, K.; Müller, J.P.; Böhmer, F.D.; Heinzel, T.; Krämer, O. Ubiquitin conjugase UBCH8 targets active FMS-like tyrosine kinase 3 for proteasomal degradation. Leukemia 2010, 24, 1412–1421. [Google Scholar] [CrossRef]
- Heiss, E.; Masson, K.; Sundberg, C.; Pedersen, M.; Sun, J.; Bengtsson, S.; Rönnstrand, L. Identification of Y589 and Y599 in the juxtamembrane domain of Flt3 as ligand-induced autophosphorylation sites involved in binding of Src family kinases and the protein tyrosine phosphatase SHP2. Blood 2006, 108, 1542–1550. [Google Scholar] [CrossRef]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The Structural Basis for Autoinhibition of FLT3 by the Juxtamembrane Domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Breitenbuecher, F.; Schnittger, S.; Grundler, R.; Markova, B.; Carius, B.; Brecht, A.; Duyster, J.; Haferlach, T.; Huber, C.; Fischer, T. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood 2009, 113, 4074–4077. [Google Scholar] [CrossRef]
- Kayser, S.; Schlenk, R.F.; Londono, M.C.; Breitenbuecher, F.; Wittke, K.; Du, J.; Groner, S.; Späth, D.; Krauter, J.; Ganser, A.; et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood 2009, 114, 2386–2392. [Google Scholar] [CrossRef]
- Schlenk, R.F.; Kayser, S.; Bullinger, L.; Kobbe, G.; Casper, J.; Ringhoffer, M.; Held, G.; Brossart, P.; Lübbert, M.; Salih, H.R.; et al. Differential impact of allelic ratio and insertion site in FLT3-ITD–positive AML with respect to allogeneic transplantation. Blood 2014, 124, 3441–3449. [Google Scholar] [CrossRef]
- Liu, S.-B.; Qiu, Q.-C.; Bao, X.-B.; Ma, X.; Li, H.-Z.; Liu, Y.-J.; Chen, S.-N.; Song, Y.-H.; Wu, D.-P.; Xue, S.-L. Pattern and prognostic value of FLT 3–ITD mutations in Chinese de novo adult acute myeloid leukemia. Cancer Sci. 2018, 109, 3981–3992. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Du, L.; Luck, T.J.; Benner, A.; Krzykalla, J.; Gathmann, I.; Voso, M.T.; Amadori, S.; Prior, T.W.; Brandwein, J.M.; et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia 2021. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Schoch, C.; Dugas, M.; Kern, W.; Staib, P.; Wuchter, C.; Löffler, H.; Sauerland, C.M.; Serve, H.; Büchner, T.; et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: Correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002, 100, 59–66. [Google Scholar] [CrossRef]
- Whitman, S.P.; Archer, K.; Feng, L.; Baldus, C.; Becknell, B.; Carlson, B.D.; Carroll, A.J.; Mrózek, K.; Vardiman, J.W.; George, S.L.; et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: A cancer and leukemia group B study. Cancer Res. 2001, 61, 7233–7239. [Google Scholar] [PubMed]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef]
- Gale, R.E.; Green, C.; Allen, C.; Mead, A.J.; Burnett, A.K.; Hills, R.K.; Linch, D.C. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008, 111, 2776–2784. [Google Scholar] [CrossRef]
- Linch, D.C.; Hills, R.K.; Burnett, A.K.; Khwaja, A.; Gale, R.E. Impact of FLT3ITD mutant allele level on relapse risk in intermediate-risk acute myeloid leukemia. Blood 2014, 124, 273–276. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar]
- Schnittger, S.; Bacher, U.; Haferlach, C.; Alpermann, T.; Kern, W.; Haferlach, T. Diversity of the juxtamembrane and TKD1 mutations (Exons 13-15) in the FLT3 gene with regards to mutant load, sequence, length, localization, and correlation with biological data. Genes Chromosom. Cancer 2012, 51, 910–924. [Google Scholar] [CrossRef]
- Young, D.J.; Nguyen, B.; Zhu, R.; Seo, J.; Li, L.; Levis, M.J.; Pratz, K.W.; Duffield, A.S.; Small, D. Deletions in FLT-3 juxtamembrane domain define a new class of pathogenic mutations: Case report and systematic analysis. Blood Adv. 2021, 5, 2285–2293. [Google Scholar] [CrossRef]
- Pratz, K.W.; Sato, T.; Murphy, K.M.; Stine, A.; Rajkhowa, T.; Levis, M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood 2010, 115, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.R.; McCormick, M.J.; Grutkoski, P.S.; Ducker, G.S.; Banerji, N.; Higgins, R.R.; Mendiola, J.R.; Reinartz, J.J. FLT3 mutations at diagnosis and relapse in acute myeloid leukemia: Cytogenetic and pathologic correlations, including cuplike blast morphology. Arch. Pathol. Lab. Med. 2010, 134, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Linch, D.C.; Hills, R.; Wheatley, K.; Burnett, A.K.; Gale, R.E. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood 2007, 110, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Hyrenius-Wittsten, A.; Pilheden, M.; Sturesson, H.; Hansson, J.; Walsh, M.P.; Song, G.; Kazi, J.U.; Liu, J.; Ramakrishan, R.; Garcia-Ruiz, C.; et al. De novo activating mutations drive clonal evolution and enhance clonal fitness in KMT2A-rearranged leukemia. Nat. Commun. 2018, 9, 1770. [Google Scholar] [CrossRef] [PubMed]
- Kindler, T.; Breitenbuecher, F.; Kasper, S.; Estey, E.; Giles, F.; Feldman, E.; Ehninger, G.; Schiller, G.; Klimek, V.; Nimer, S.D.; et al. Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML). Blood 2005, 105, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Paez, J.G.; Lee, J.C.; Bo, R.; Stone, R.M.; DeAngelo, D.J.; Galinsky, I.; Wolpin, B.M.; Jonasova, A.; Herman, P.; et al. Identifying and characterizing a novel activating mutation of the FLT3 tyrosine kinase in AML. Blood 2004, 104, 1855–1858. [Google Scholar] [CrossRef]
- Matsuno, N.; Nanri, T.; Kawakita, T.; Mitsuya, H.; Asou, N. A novel FLT3 activation loop mutation N841K in acute myeloblastic leukemia. Leukemia 2005, 19, 480–481. [Google Scholar] [CrossRef]
- Schittenhelm, M.M.; Yee, K.W.H.; Tyner, J.; McGreevey, L.; Haley, A.D.; Town, A.; Griffith, D.J.; Bainbridge, T.; Braziel, R.M.; O’Farrell, A.-M.; et al. FLT3 K663Q is a novel AML-associated oncogenic kinase: Determination of biochemical properties and sensitivity to Sunitinib (SU11248). Leukemia 2006, 20, 2008–2014. [Google Scholar] [CrossRef]
- Reindl, C.; Bagrintseva, K.; Vempati, S.; Schnittger, S.; Ellwart, J.W.; Wenig, K.; Hopfner, K.-P.; Hiddemann, W.; Spiekermann, K. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood 2006, 107, 3700–3707. [Google Scholar] [CrossRef]
- Fröhling, S.; Scholl, C.; Levine, R.L.; Loriaux, M.; Boggon, T.J.; Bernard, O.; Berger, R.; Döhner, H.; Döhner, K.; Ebert, B.L.; et al. Identification of Driver and Passenger Mutations of FLT3 by High-Throughput DNA Sequence Analysis and Functional Assessment of Candidate Alleles. Cancer Cell 2007, 12, 501–513. [Google Scholar] [CrossRef]
- Hayakawa, F.; Towatari, M.; Kiyoi, H.; Tanimoto, M.; Kitamura, T.; Saito, H.; Naoe, T. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 2000, 19, 624–631. [Google Scholar] [CrossRef]
- Choudhary, C.; Schwäble, J.; Brandts, C.; Tickenbrock, L.; Sargin, B.; Kindler, T.; Fischer, T.; Berdel, W.E.; Müller-Tidow, C.; Serve, H. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood 2005, 106, 265–273. [Google Scholar] [CrossRef]
- Ghiaur, G.; Levis, M. Mechanisms of Resistance to FLT3 Inhibitors and the Role of the Bone Marrow Microenvironment. Hematol. Oncol. Clin. N. Am. 2017, 31, 681–692. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; Melgar, K.; Bolanos, L.; Hueneman, K.; Walker, M.M.; Jiang, J.-K.; Wilson, K.M.; Zhang, X.; Shen, J.; Jiang, F.; et al. Targeting AML-associated FLT3 mutations with a type I kinase inhibitor. J. Clin. Investig. 2020, 130, 2017–2023. [Google Scholar] [CrossRef]
- Smith, C.C.; Lin, K.; Stecula, A.; Sali, A.; Shah, N.P. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015, 29, 2390–2392. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Krämer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.; Miller, C.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.L.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Sträng, E.; Theis, F.; Jahn, N.; Panina, E.; Blätte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.K.; Nechiporuk, T.; Bottomly, D.; Piehowski, P.D.; Reisz, J.A.; Pittsenbarger, J.; Kaempf, A.; Gosline, S.J.; Wang, Y.-T.; Hansen, J.R.; et al. The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance. Cancer Cell 2021, 39, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.-S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, C.; Konopleva, M.; Chen, Y.; Jacamo, R.O.; Borthakur, G.; Cortes, J.; Ravandi, F.; Ramachandran, A.; Andreeff, M. Reversal of Acquired Drug Resistance in FLT3-Mutated Acute Myeloid Leukemia Cells via Distinct Drug Combination Strategies. Clin. Cancer Res. 2014, 20, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.-Y.; Singh, V.K.; Coumar, M.; Hsu, Y.C.; Wang, W.-C.; Song, J.-S.; Chen, C.-H.; Lin, W.-H.; Wu, S.-H.; Hsu, J.T.A.; et al. Homology modeling of DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Sci. Rep. 2015, 5, srep11702. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, Y.; Kantarjian, H.M.; Luthra, R.; Ravandi, F.; Borthakur, G.; Garcia-Manero, G.; Konopleva, M.; Estrov, Z.; Andreeff, M.; Cortes, J.E. Treatment with FLT3 inhibitor in patients withFLT3-mutated acute myeloid leukemia is associated with development of secondaryFLT3-tyrosine kinase domain mutations. Cancer 2014, 120, 2142–2149. [Google Scholar] [CrossRef]
- Alotaibi, A.S.; Yilmaz, M.; Kanagal-Shamanna, R.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Konopleva, M.; Pierce, S.A.; Wang, S.A.; et al. Patterns of Resistance Differ in Patients with Acute Myeloid Leukemia Treated with Type I versus Type II FLT3 Inhibitors. Blood Cancer Discov. 2020, 2, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.-T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef]
- Al-Jamal, H.; Jusoh, S.A.M.; Hassan, R.; Johan, M.F. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer 2015, 15, 869. [Google Scholar] [CrossRef]
- Williams, A.B.; Li, L.; Nguyen, B.; Brown, P.; Levis, M.; Small, D. Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood 2012, 120, 3069–3079. [Google Scholar] [CrossRef]
- Minami, Y.; Kiyoi, H.; Yamamoto, Y.; Ueda, R.; Saito, H.; Naoe, T. Selective apoptosis of tandemly duplicated FLT3-transformed leukemia cells by Hsp90 inhibitors. Leukemia 2002, 16, 1535–1540. [Google Scholar] [CrossRef]
- Chen-Levy, Z.; Nourse, J.; Cleary, M.L. The bcl-2 candidate proto-oncogene product is a 24-kilodalton integral-membrane protein highly expressed in lymphoid cell lines and lymphomas carrying the t(14;18) translocation. Mol. Cell. Biol. 1989, 9, 701–710. [Google Scholar] [CrossRef]
- Hockenbery, D.M.; Zutter, M.; Hickey, W.; Nahm, M.; Korsmeyer, S.J. BCL2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proc. Natl. Acad. Sci. USA 1991, 88, 6961–6965. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Ren, D.; Tu, H.-C.; Kim, H.; Wang, G.X.; Bean, G.R.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. BID, BIM, and PUMA Are Essential for Activation of the BAX- and BAK-Dependent Cell Death Program. Science 2010, 330, 1390–1393. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W. Insights into Programmed Cell Death through Structural Biology. Cell 2000, 103, 273–282. [Google Scholar] [CrossRef]
- Lessene, G.; Czabotar, P.; Colman, P.M. BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef]
- Konopleva, M.; Letai, A. BCL-2 inhibition in AML: An unexpected bonus? Blood 2018, 132, 1007–1012. [Google Scholar] [CrossRef]
- Kuwana, T.; Newmeyer, D.D. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr. Opin. Cell Biol. 2003, 15, 691–699. [Google Scholar] [CrossRef]
- Thomadaki, H.; Scorilas, A. BCL2 Family of Apoptosis-Related Genes: Functions and Clinical Implications in Cancer. Crit. Rev. Clin. Lab. Sci. 2006, 43, 1–67. [Google Scholar] [CrossRef]
- Reed, J.C. Proapoptotic multidomain Bcl-2/Bax-family proteins: Mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006, 13, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.G. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat. Rev. Cancer 2008, 8, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.-X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Delia, D.; Martinez, E.; Fontanella, E.; Aiello, A. Two- and three-color immunofluorescence using aminocoumarin, fluorescein, and phycoerythrin-labelled antibodies and single laser flow cytometry. Cytometry 1991, 12, 537–544. [Google Scholar] [CrossRef]
- Karakas, T.; Maurer, U.; Weidmann, E.; Miething, C.C.; Hoelzer, D.; Bergmann, L. High expression of bcl-2 mRNA as a determinant of poor prognosis in acute myeloid leukemia. Ann. Oncol. 1998, 9, 159–165. [Google Scholar] [CrossRef]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef]
- Vo, T.-T.; Ryan, J.; Carrasco, R.; Neuberg, D.; Rossi, D.J.; Stone, R.M.; DeAngelo, D.J.; Frattini, M.G.; Letai, A. Relative Mitochondrial Priming of Myeloblasts and Normal HSCs Determines Chemotherapeutic Success in AML. Cell 2012, 151, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.-I.; Akashi, K.; Korsmeyer, S.J. Obligate Role of Anti-Apoptotic MCL-1 in the Survival of Hematopoietic Stem Cells. Science 2005, 307, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; DeBose, L.; Mu, H.; et al. Selective BCL-2 Inhibition by ABT-199 Causes On-Target Cell Death in Acute Myeloid Leukemia. Cancer Discov. 2013, 4, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Weber, K.; Dinsdale, D.; Schmitz, I.; Schulze-Osthoff, K.; Dyer, M.; Cohen, G.M. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009, 16, 1030–1039. [Google Scholar] [CrossRef]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Rev. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- Bhatt, S.; Pioso, M.S.; Olesinski, E.A.; Yilma, B.; Ryan, J.A.; Mashaka, T.; Leutz, B.; Adamia, S.; Zhu, H.; Kuang, Y.; et al. Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 872–890. [Google Scholar] [CrossRef]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; D’Almeida, A.; Joshi, S.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764. [Google Scholar] [CrossRef]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Fathi, A.T.; Dinardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Dinardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Tallman, M.S. Emerging therapeutic drugs for AML. Blood 2016, 127, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; DiNardo, C.D. The role of IDH mutations in acute myeloid leukemia. Future Oncol. 2018, 14, 979–993. [Google Scholar] [CrossRef]
- Aref, S.; Areida, E.S.K.; Aaal, M.F.A.; Adam, O.M.; El-Ghonemy, M.S.; Elbaiomy, M.; Zeid, T.A. Prevalence and Clinical Effect of IDH1 and IDH2 Mutations Among Cytogenetically Normal Acute Myeloid Leukemia Patients. Clin. Lymphoma Myeloma Leuk. 2015, 15, 550–555. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Ravandi, F.; Agresta, S.; Konopleva, M.; Takahashi, K.; Kadia, T.; Routbort, M.; Patel, K.P.; Brandt, M.; Pierce, S.; et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am. J. Hematol. 2015, 90, 732–736. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Dohner, H.; Campbell, P.J. Genomic Classification in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 375, 900–901. [Google Scholar] [CrossRef]
- Elnahass, Y.; Badawy, R.H.; Elrefaey, F.A.; Nooh, H.A.; Ibrahiem, D.; Nader, H.A.; Mahmoud, H.K.; ElMetnawy, W.H. IDH Mutations in AML Patients; A higher Association with Intermediate Risk Cytogenetics. Asian Pac. J. Cancer Prev. 2020, 21, 721–725. [Google Scholar] [CrossRef]
- Debarri, H.; Lebon, D.; Roumier, C.; Cheok, M.; Marceau-Renaut, A.; Nibourel, O.; Geffroy, S.; Helevaut, N.; Rousselot, P.; Gruson, B.; et al. IDH1/2 but not DNMT3A mutations are suitable targets for minimal residual disease monitoring in acute myeloid leukemia patients: A study by the Acute Leukemia French Association. Oncotarget 2015, 6, 42345–42353. [Google Scholar] [CrossRef]
- Chou, W.-C.; Lei, W.-C.; Ko, B.-S.; Hou, H.-A.; Chen, C.-Y.; Tang, J.-L.; Yao, M.; Tsay, W.; Wu, S.-J.; Huang, S.-Y.; et al. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia 2011, 25, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.; Simpson, M.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Kim, E.S. Enasidenib: First Global Approval. Drugs 2017, 77, 1705–1711. [Google Scholar] [CrossRef]
- Stein, E.M.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Roboz, G.J.; Collins, R.; Sekeres, M.A.; Stone, R.M.; Attar, E.C.; Frattini, M.G.; et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: A phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Haematol. 2020, 7, e309–e319. [Google Scholar] [CrossRef]
- Stein, E.M.; Dinardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.; Levine, R.L.; Flinn, I.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Dinardo, C.D.; Stein, E.M.; DE Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib inIDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; Dinardo, C.D.; Stein, E.M.; De Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.; Marteyn, B.; Farnoud, N.R.; DE Botton, S.; Bernard, O.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef]
- Choe, S.; Wang, H.; Dinardo, C.D.; Stein, E.M.; De Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef]
- Intlekofer, A.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef]
- Issa, G.C.; DiNardo, C.D. Acute myeloid leukemia with IDH1 and IDH2 mutations: 2021 treatment algorithm. Blood Cancer J. 2021, 11, 107. [Google Scholar] [CrossRef]
- Harding, J.J.; Lowery, M.A.; Shih, A.H.; Schvartzman, J.M.; Hou, S.; Famulare, C.; Patel, M.; Roshal, M.; Do, R.K.; Zehir, A.; et al. Isoform Switching as a Mechanism of Acquired Resistance to Mutant Isocitrate Dehydrogenase Inhibition. Cancer Discov. 2018, 8, 1540–1547. [Google Scholar] [CrossRef]
- Gbyli, R.; Song, Y.; Liu, W.; Gao, Y.; Chandhok, N.S.; Fu, X.; Wang, X.; Patel, A.; Sundaram, R.; Tebaldi, T.; et al. PARP Inhibitors Are Effective in IDH1/2 Mutant MDS and AML Resistant to Targeted IDH Inhibitors. Blood 2019, 134, 4222. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef]
- Chandhok, N.S.; Wei, W.; Bindra, R.; Halene, S.; Shyr, Y.; Li, J.; Berens, M.; Karlovich, C.; Ivy, S.P.; Prebet, T. The PRIME Trial: PARP Inhibition in IDH Mutant Effectiveness Trial. A Phase II Study of Olaparib in Isocitrate Dehydrogenase (IDH) Mutant Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2019, 134, 3909. [Google Scholar] [CrossRef]
- Jayavelu, A.K.; Schnöder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing factor YBX1 mediates persistence of JAK2-mutated neoplasms. Nature 2020, 588, 157–163. [Google Scholar] [CrossRef]
- Kokkaliaris, K.D.; Scadden, D.T. Cell interactions in the bone marrow microenvironment affecting myeloid malignancies. Blood Adv. 2020, 4, 3795–3803. [Google Scholar] [CrossRef]
- Cucchi, D.; Groen, R.; Janssen, J.; Cloos, J. Ex vivo cultures and drug testing of primary acute myeloid leukemia samples: Current techniques and implications for experimental design and outcome. Drug Resist. Updat. 2020, 53, 100730. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Yang, W.F.; Bankhead, A.; Fan, G.; Fletcher, L.B.; Bryant, J.; Glover, J.M.; Chang, B.H.; Spurgeon, S.E.; Fleming, W.H.; et al. Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res. 2013, 73, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Khorashad, J.S.; Eiring, A.M.; Mason, C.C.; Gantz, K.C.; Bowler, A.D.; Redwine, H.M.; Yu, F.; Kraft, I.L.; Pomicter, A.D.; Reynolds, K.R.; et al. shRNA library screening identifies nucleocytoplasmic transport as a mediator of BCR-ABL1 kinase-independent resistance. Blood 2015, 125, 1772–1781. [Google Scholar] [CrossRef]
- Mason, C.C.; Fiol, C.R.; Baker, M.J.; Nadal-Melsio, E.; Yebra-Fernandez, E.; Bicalho, L.; Chowdhury, A.; Albert, M.; Reid, A.G.; Claudiani, S.; et al. Identification of genetic targets in acute myeloid leukaemia for designing targeted therapy. Br. J. Haematol. 2021, 192, 137–145. [Google Scholar] [CrossRef]
- Yamauchi, T.; Masuda, T.; Canver, M.C.; Seiler, M.; Semba, Y.; Shboul, M.; Al-Raqad, M.; Maeda, M.; Schoonenberg, V.A.C.; Cole, M.A.; et al. Genome-wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-mRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 2018, 33, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Pomicter, A.; Tantravahi, S.; Mason, C.C.; Senina, A.V.; Ahmann, J.M.; Wang, Q.; Than, H.; Patel, A.B.; Heaton, W.L.; et al. Nuclear–Cytoplasmic Transport Is a Therapeutic Target in Myelofibrosis. Clin. Cancer Res. 2019, 25, 2323–2335. [Google Scholar] [CrossRef]
- Bhavanasi, D.; Gwenn, D.-D.; Liu, X.; Vergez, F.; Danet-Desnoyers, G.; Carroll, M.; Huang, J.; Klein, P.S. Signaling mechanisms that regulate ex vivo survival of human acute myeloid leukemia initiating cells. Blood Cancer J. 2017, 7, 636. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yan, D.; Franzini, A.; Pomicter, A.D.; Halverson, B.J.; Antelope, O.; Mason, C.C.; Ahmann, J.M.; Senina, A.V.; Vellore, N.A.; Jones, C.L.; et al. SIRT5 Is a Druggable Metabolic Vulnerability in Acute Myeloid Leukemia. Blood Cancer Discov. 2021, 2, 266–287. [Google Scholar] [CrossRef]
- Borella, G.; Da Ros, A.; Borile, G.; Porcù, E.; Tregnago, C.; Benetton, M.; Marchetti, A.; Bisio, V.; Montini, B.; Michielotto, B.; et al. Targeting mesenchymal stromal cells plasticity to reroute acute myeloid leukemia course. Blood 2021, 138, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Bray, L.J.; Binner, M.; Körner, Y.; Von Bonin, M.; Bornhäuser, M.; Werner, C. A three-dimensional ex vivo tri-culture model mimics cell-cell interactions between acute myeloid leukemia and the vascular niche. Haematologica 2017, 102, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Cartledge Wolf, D.M.; Langhans, S.A. Moving Myeloid Leukemia Drug Discovery into the Third Dimension. Front. Pediatr. 2019, 7, 314. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef]
- Tian, X.; Gu, T.; Patel, S.; Bode, A.M.; Lee, M.-H.; Dong, Z. CRISPR/Cas9—An evolving biological tool kit for cancer biology and oncology. NPJ Precis. Oncol. 2019, 3, 8. [Google Scholar] [CrossRef]
- Hunker, A.C.; Soden, M.E.; Krayushkina, D.; Heymann, G.; Awatramani, R.; Zweifel, L.S. Conditional Single Vector CRISPR/SaCas9 Viruses for Efficient Mutagenesis in the Adult Mouse Nervous System. Cell Rep. 2020, 30, 4303–4316. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Fusion | Translocation | Frequency | Sensitivity | ||
|---|---|---|---|---|---|
| ATRA | AS2O3 | ||||
| PML-RARα | t(15;17)(q24;q12) | bcr1 | ~55% | + | + |
| bcr2 | ~5% | + | + | ||
| bcr3 | ~40% | + | + | ||
| rare variant | rare | ||||
| ZBTB16-RARα (also known as PLZF-RARα) | t(11,17) (q23, q21) | Up to 0.8% | − | − | |
| NUMA-RARα | t(11;17) (q13;q21) | rare | + | + | |
| NPM-RARα | t(5;17) (q32;q12) | ~0.5% | + | + | |
| BCOR-RARα | t(X;17) (p11;q21) | rare | + | − | |
| FIP1L1-RARα | t(4;17) (q12;q21) | rare | + | NR | |
| STAT5-RARα | t(17;17) (q11;q21) | rare | − | − | |
| PRKAR1A-RARα | t(17;17) (q12;q21) | rare | + | + | |
| Gene | Mutation |
|---|---|
| RARA | Y208N, K227-T233del, K207-Y208del, L224P, K238E, Y208N, R272Q, I273F, R276W, R276Q, T285A, S287L, S287W, G289R, G289E, L290V, G391E, R394W, 412-414del, M413T |
| WT1 | R242fs, R462W |
| CDK12 | D877N |
| KMT2C | R209W |
| KRAS | G12R, Q61K |
| MED12 | R356P, L2162F |
| MYB | I135KfsTer77 |
| NRAS | G12R, Q61K |
| NSD1 | N1664K, E1948K |
| NT5C2 | K404N, R367Q, R246W |
| SALL4 | V191M, A149V |
| TET2 | K148NfsTer3 |
| TFE3 | K68T |
| Molecular Marker | Genomic Alterations Associated with Resistance |
|---|---|
| FLT3 | FLT3-ITD, FLT3-TKD(D835H), N676K |
| KRAS | G12D, G13D, Q61H |
| NRAS | G12D, G13R, G12A, Q61H, Q61K, Q61R |
| PTPN11 | F71L, A72T |
| TP53 | TP53 loss, R248W, M246K, V272M, A161T, G154V, R342 *, P278H, E336fs |
| BAX | downregulation |
| MCL-1 | upregulation |
| Molecular Marker | Genomic Alterations Associated with Response |
| NPM1 | NPM1 mutation |
| IDH1 | R132C, R132H, R132L, R132Q, R132S |
| IDH2 | R140Q, R172G, R172K, R172M, R172S |
| SRSF2 | P95L, P95_R102del |
| ZRSR2 | K203fs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, S.; Eiring, A.M.; Khorashad, J.S. Genomic Abnormalities as Biomarkers and Therapeutic Targets in Acute Myeloid Leukemia. Cancers 2021, 13, 5055. https://doi.org/10.3390/cancers13205055
Ribeiro S, Eiring AM, Khorashad JS. Genomic Abnormalities as Biomarkers and Therapeutic Targets in Acute Myeloid Leukemia. Cancers. 2021; 13(20):5055. https://doi.org/10.3390/cancers13205055
Chicago/Turabian StyleRibeiro, Sara, Anna M. Eiring, and Jamshid S. Khorashad. 2021. "Genomic Abnormalities as Biomarkers and Therapeutic Targets in Acute Myeloid Leukemia" Cancers 13, no. 20: 5055. https://doi.org/10.3390/cancers13205055
APA StyleRibeiro, S., Eiring, A. M., & Khorashad, J. S. (2021). Genomic Abnormalities as Biomarkers and Therapeutic Targets in Acute Myeloid Leukemia. Cancers, 13(20), 5055. https://doi.org/10.3390/cancers13205055