Immune Functions of Signaling Lymphocytic Activation Molecule Family Molecules in Multiple Myeloma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
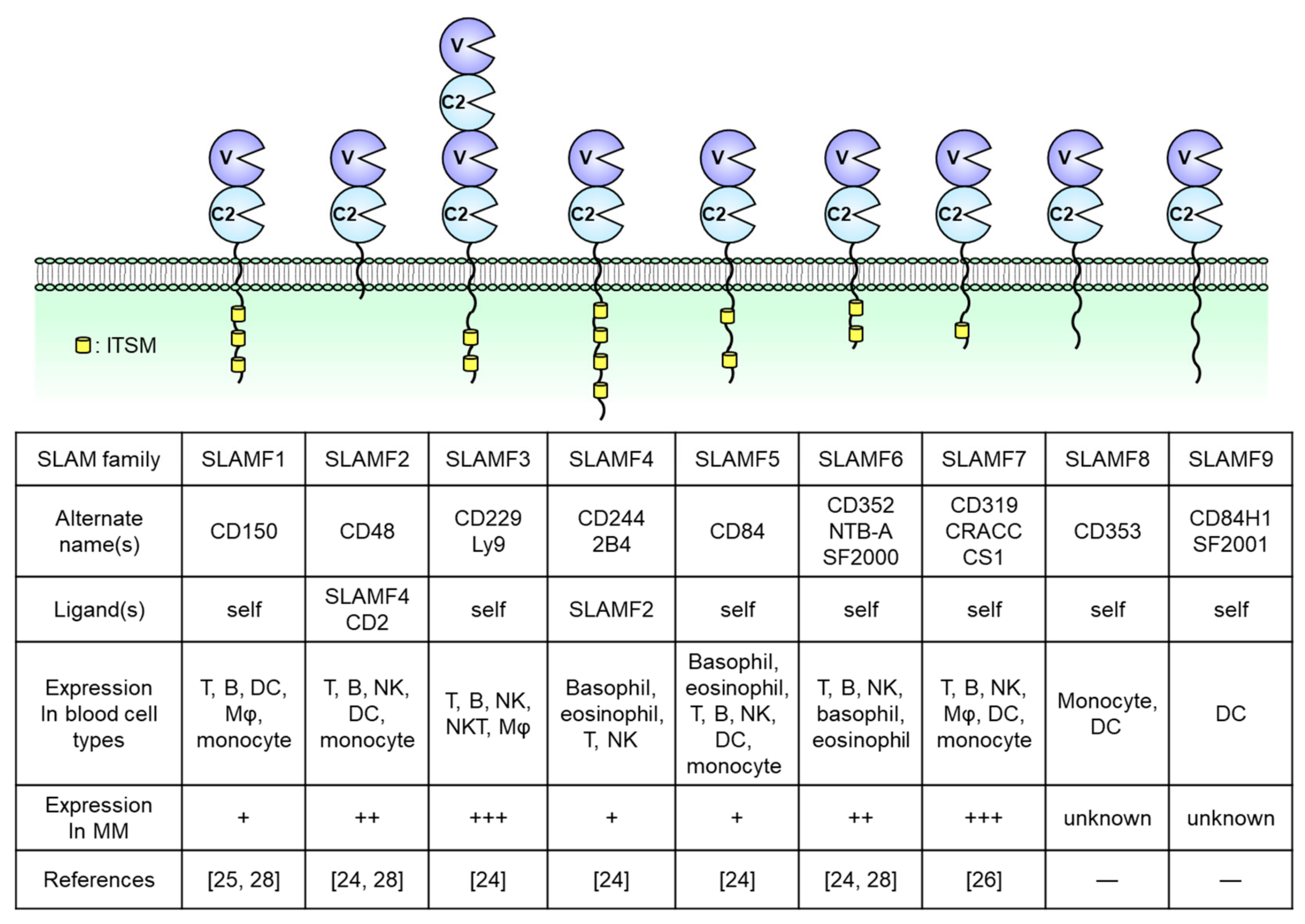
2. Overview of SLAM Family Receptors
3. SLAMF2 (CD48)
4. SLAMF3 (CD229, Ly9)
5. SLAMF6 (CD352, NTB-A)
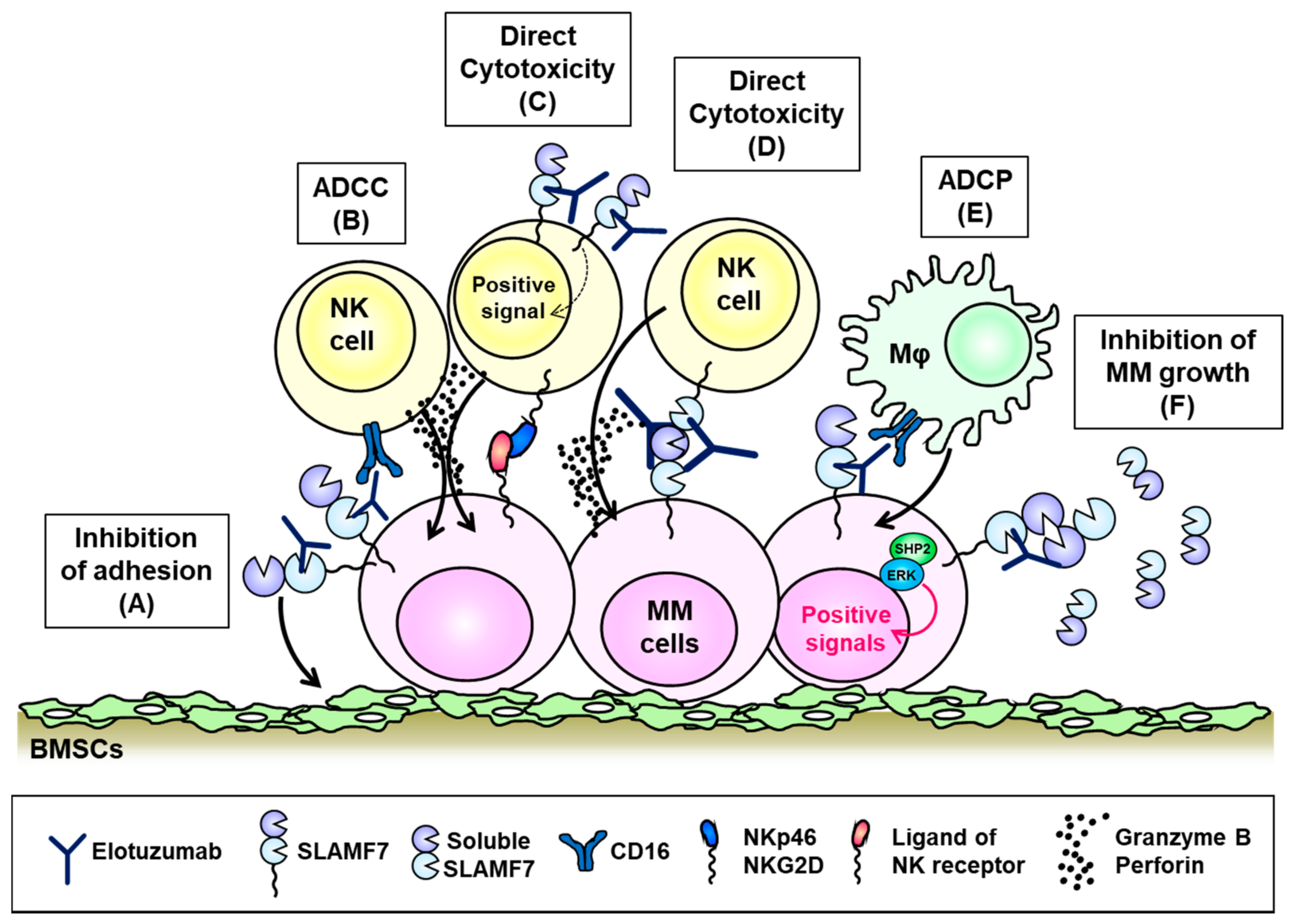
6. SLAMF7 (CD319, CS1)
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rajkumar, S.V. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2018, 93. [Google Scholar] [CrossRef]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Prideaux, S.M.; Conway-O’Brien, E.; Chevassut, T.J. The genetic architecture of multiple myeloma. Adv. Hematol. 2014, 2014, 864058. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef]
- Gooding, S.; Edwards, C.M. New approaches to targeting the bone marrow microenvironment in multiple myeloma. Curr. Opin. Pharmacol. 2016, 28, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Ishibashi, M.; Sunakawa, M.; Inokuchi, K. Immunotherapy for Multiple Myeloma. Cancers 2019, 11, 9. [Google Scholar] [CrossRef]
- Swan, D.; Lynch, K.; Gurney, M.; O’Dwyer, M. Current and emerging immunotherapeutic approaches to the treatment of multiple myeloma. Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef]
- Moreau, P.; Zamagni, E.; Mateos, M.V. Treatment of patients with multiple myeloma progressing on frontline-therapy with lenalidomide. Blood Cancer J. 2019, 9, 38. [Google Scholar] [CrossRef]
- Neri, P.; Bahlis, N.J.; Lonial, S. New Strategies in Multiple Myeloma: Immunotherapy as a Novel Approach to Treat Patients with Multiple Myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5959–5965. [Google Scholar] [CrossRef]
- Anderson, K.C. Progress and Paradigms in Multiple Myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5419–5427. [Google Scholar] [CrossRef]
- Ma, J.; Li, Q.; Yu, Z.; Cao, Z.; Liu, S.; Chen, L.; Li, H.; Gao, S.; Yan, T.; Wang, Y.; et al. Immunotherapy Strategies Against Multiple Myeloma. Technol. Cancer Res. Treat. 2017, 16, 717–726. [Google Scholar] [CrossRef]
- Hosen, N.; Matsunaga, Y.; Hasegawa, K.; Matsuno, H.; Nakamura, Y.; Makita, M.; Watanabe, K.; Yoshida, M.; Satoh, K.; Morimoto, S.; et al. The activated conformation of integrin β(7) is a novel multiple myeloma-specific target for CAR T cell therapy. Nat. Med. 2017, 23, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [PubMed]
- Veillette, A.; Latour, S. The SLAM family of immune-cell receptors. Curr. Opin. Immunol. 2003, 15, 277–285. [Google Scholar] [CrossRef]
- Veillette, A. SLAM-family receptors: Immune regulators with or without SAP-family adaptors. Cold Spring Harb. Perspect. Biol. 2010, 2, a002469. [Google Scholar] [CrossRef]
- Schwartzberg, P.L.; Mueller, K.L.; Qi, H.; Cannons, J.L. SLAM receptors and SAP influence lymphocyte interactions, development and function. Nat. Rev. Immunol. 2009, 9, 39–46. [Google Scholar] [CrossRef]
- Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L. SLAM family receptors and SAP adaptors in immunity. Annu. Rev. Immunol. 2011, 29, 665–705. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Deenick, E.K. The role of SAP and SLAM family molecules in the humoral immune response. Ann. N. Y. Acad. Sci. 2011, 1217, 32–44. [Google Scholar] [CrossRef]
- Fouquet, G.; Marcq, I.; Debuysscher, V.; Bayry, J.; Rabbind-Singh, A.; Bengrine, A.; Nguyen-Khac, E.; Naassila, M.; Bouhlal, H. Signaling lymphocytic activation molecules Slam and cancers: Friends or foes? Oncotarget 2018, 9, 16248–16262. [Google Scholar] [CrossRef] [PubMed]
- Detre, C.; Keszei, M.; Romero, X.; Tsokos, G.C.; Terhorst, C. SLAM family receptors and the SLAM-associated protein (SAP) modulate T cell functions. Semin. Immunopathol. 2010, 32, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Veillette, A. SLAM family receptors in normal immunity and immune pathologies. Curr. Opin. Immunol. 2016, 38, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Morra, M.; Lu, J.; Poy, F.; Martin, M.; Sayos, J.; Calpe, S.; Gullo, C.; Howie, D.; Rietdijk, S.; Thompson, A.; et al. Structural basis for the interaction of the free SH2 domain EAT-2 with SLAM receptors in hematopoietic cells. EMBO J. 2001, 20, 5840–5852. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Quintero, L.A.; Roncagalli, R.; Guo, H.; Latour, S.; Davidson, D.; Veillette, A. EAT-2, a SAP-like adaptor, controls NK cell activation through phospholipase Cγ, Ca++, and Erk, leading to granule polarization. J. Exp. Med. 2014, 211, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Veillette, A. How do SAP family deficiencies compromise immunity? Trends Immunol. 2010, 31, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, M.; Shaw, A.S. SAP signaling: A dual mechanism of action. Immunity 2012, 36, 899–901. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, S.; Dong, Z. NK cell recognition of hematopoietic cells by SLAM-SAP families. Cell. Mol. Immunol. 2019, 16, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Atanackovic, D.; Panse, J.; Hildebrandt, Y.; Jadczak, A.; Kobold, S.; Cao, Y.; Templin, J.; Meyer, S.; Reinhard, H.; Bartels, K.; et al. Surface molecule CD229 as a novel target for the diagnosis and treatment of multiple myeloma. Haematologica 2011, 96, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Gordiienko, I.M.; Shlapatska, L.M.; Kovalevska, L.M.; Sidorenko, S.P. Differential expression of CD150/SLAMF1 in normal and malignant B cells on the different stages of maturation. Exp. Oncol. 2016, 38, 101–107. [Google Scholar] [CrossRef]
- Hsi, E.D.; Steinle, R.; Balasa, B.; Szmania, S.; Draksharapu, A.; Shum, B.P.; Huseni, M.; Powers, D.; Nanisetti, A.; Zhang, Y.; et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 2775–2784. [Google Scholar] [CrossRef]
- Radhakrishnan, S.V.; Bhardwaj, N.; Luetkens, T.; Atanackovic, D. Novel anti-myeloma immunotherapies targeting the SLAM family of receptors. Oncoimmunology 2017, 6, e1308618. [Google Scholar] [CrossRef]
- Muccio, V.E.; Saraci, E.; Gilestro, M.; Gattei, V.; Zucchetto, A.; Astolfi, M.; Ruggeri, M.; Marzanati, E.; Passera, R.; Palumbo, A.; et al. Multiple myeloma: New surface antigens for the characterization of plasma cells in the era of novel agents. Cytom. Part B Clin. Cytom. 2016, 90, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.; Eck, M.J.; Terhorst, C. The SAP and SLAM families in immune responses and X-linked lymphoproliferative disease. Nat. Rev. Immunol. 2003, 3, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Sayos, J.; Wu, C.; Morra, M.; Wang, N.; Zhang, X.; Allen, D.; van Schaik, S.; Notarangelo, L.; Geha, R.; Roncarolo, M.G.; et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature 1998, 395, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, e56–e65. [Google Scholar] [CrossRef] [PubMed]
- Smetana, J.; Oppelt, J.; Štork, M.; Pour, L.; Kuglík, P. Chromothripsis 18 in multiple myeloma patient with rapid extramedullary relapse. Mol. Cytogenet. 2018, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Sklavenitis-Pistofidis, R.; Reidy, M.; Huynh, D.; Salem, K.Z.; Park, J.; Glavey, S.; Leleu, X.; Roo, D.; Ghobrial, I.M.; Manier, S. Founding Precision Therapy in 1q-Amplified Multiple Myeloma. Blood 2018, 132 (Suppl. 1), 1007. [Google Scholar] [CrossRef]
- McArdel, S.L.; Terhorst, C.; Sharpe, A.H. Roles of CD48 in regulating immunity and tolerance. Clin. Immunol. 2016, 164, 10–20. [Google Scholar] [CrossRef]
- Brown, M.H.; Boles, K.; van der Merwe, P.A.; Kumar, V.; Mathew, P.A.; Barclay, A.N. 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. J. Exp. Med. 1998, 188, 2083–2090. [Google Scholar] [CrossRef]
- Arulanandam, A.R.; Moingeon, P.; Concino, M.F.; Recny, M.A.; Kato, K.; Yagita, H.; Koyasu, S.; Reinherz, E.L. A soluble multimeric recombinant CD2 protein identifies CD48 as a low affinity ligand for human CD2: Divergence of CD2 ligands during the evolution of humans and mice. J. Exp. Med. 1993, 177, 1439–1450. [Google Scholar] [CrossRef]
- Moran, M.; Miceli, M.C. Engagement of GPI-linked CD48 contributes to TCR signals and cytoskeletal reorganization: A role for lipid rafts in T cell activation. Immunity 1998, 9, 787–796. [Google Scholar] [CrossRef]
- Kis-Toth, K.; Tsokos, G.C. Engagement of SLAMF2/CD48 prolongs the time frame of effective T cell activation by supporting mature dendritic cell survival. J. Immunol. 2014, 192, 4436–4442. [Google Scholar] [CrossRef] [PubMed]
- Meinke, S.; Watzl, C. NK cell cytotoxicity mediated by 2B4 and NTB-A is dependent on SAP acting downstream of receptor phosphorylation. Front. Immunol. 2013, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Morandi, B.; Costa, R.; Falco, M.; Parolini, S.; de Maria, A.; Ratto, G.; Mingari, M.C.; Melioli, G.; Moretta, A.; Ferlazzo, G. Distinctive lack of CD48 expression in subsets of human dendritic cells tunes NK cell activation. J. Immunol. 2005, 175, 3690–3697. [Google Scholar] [CrossRef] [PubMed]
- Agresta, L.; Hoebe, K.H.N.; Janssen, E.M. The Emerging Role of CD244 Signaling in Immune Cells of the Tumor Microenvironment. Front. Immunol. 2018, 9, 2809. [Google Scholar] [CrossRef] [PubMed]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8⁺ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef]
- Wu, Y.; Kuang, D.M.; Pan, W.D.; Wan, Y.L.; Lao, X.M.; Wang, D.; Li, X.F.; Zheng, L. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 2013, 57, 1107–1116. [Google Scholar] [CrossRef]
- Tan, J.; Chen, S.; Lu, Y.; Yao, D.; Xu, L.; Zhang, Y.; Yang, L.; Chen, J.; Lai, J.; Yu, Z.; et al. Higher PD-1 expression concurrent with exhausted CD8+ T cells in patients with de novo acute myeloid leukemia. Chin. J. Cancer Res. = Chung-Kuo Yen Cheng Yen Chiu 2017, 29, 463–470. [Google Scholar] [CrossRef]
- Hosen, N.; Ichihara, H.; Mugitani, A.; Aoyama, Y.; Fukuda, Y.; Kishida, S.; Matsuoka, Y.; Nakajima, H.; Kawakami, M.; Yamagami, T.; et al. CD48 as a novel molecular target for antibody therapy in multiple myeloma. Br. J. Haematol. 2012, 156, 213–224. [Google Scholar] [CrossRef]
- Ashour, R.; Ri, M.; Aly, S.S.; Yoshida, T.; Tachita, T.; Kanamori, T.; Aoki, S.; Kinoshita, S.; Narita, T.; Totani, H.; et al. Expression analysis of two SLAM family receptors, SLAMF2 and SLAMF7, in patients with multiple myeloma. Int. J. Hematol. 2019, 110, 69–76. [Google Scholar] [CrossRef]
- Lewis, T.S.; Olson, D.; Gordon, K.; Sandall, S.; Quick, M.; Finn, M.; Westendorf, L.; Linares, G.; Leiske, C.; Nesterova, A.; et al. SGN-CD48A: A Novel Humanized Anti-CD48 Antibody-Drug Conjugate for the Treatment of Multiple Myeloma. Blood 2016, 76, 4470. [Google Scholar] [CrossRef]
- Olson, D.J.; Liu, B.A.; Zaval, M.; Cao, A.; Gurgel, J.; Cochran, J.; Stevens, N.; Anderson, M.; Lewis, T.S. Additional mechanisms of action of SGN-CD48A in multiple myeloma and improved antitumor activity in combination with daratumumab. Cancer Res. 2018, 78, 5619. [Google Scholar] [CrossRef]
- Sintes, J.; Vidal-Laliena, M.; Romero, X.; Tovar, V.; Engel, P. Characterization of mouse CD229 (Ly9), a leukocyte cell surface molecule of the CD150 (SLAM) family. Tissue Antigens 2007, 70, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Sandrin, M.S.; Henning, M.M.; Lo, M.F.; Baker, E.; Sutherland, G.R.; McKenzie, I.F. Isolation and characterization of cDNA clones for Humly9: The human homologue of mouse Ly9. Immunogenetics 1996, 43, 13–19. [Google Scholar] [CrossRef]
- de la Fuente, M.A.; Tovar, V.; Villamor, N.; Zapater, N.; Pizcueta, P.; Campo, E.; Bosch, J.; Engel, P. Molecular characterization and expression of a novel human leukocyte cell-surface marker homologous to mouse Ly-9. Blood 2001, 97, 3513–3520. [Google Scholar] [CrossRef]
- Romero, X.; Zapater, N.; Calvo, M.; Kalko, S.G.; de la Fuente, M.A.; Tovar, V.; Ockeloen, C.; Pizcueta, P.; Engel, P. CD229 (Ly9) lymphocyte cell surface receptor interacts homophilically through its N-terminal domain and relocalizes to the immunological synapse. J. Immunol. 2005, 174, 7033–7042. [Google Scholar] [CrossRef]
- Cannons, J.L.; Yu, L.J.; Hill, B.; Mijares, L.A.; Dombroski, D.; Nichols, K.E.; Antonellis, A.; Koretzky, G.A.; Gardner, K.; Schwartzberg, P.L. SAP regulates T(H)2 differentiation and PKC-theta-mediated activation of NF-kappaB1. Immunity 2004, 21, 693–706. [Google Scholar] [CrossRef]
- Sayós, J.; Martín, M.; Chen, A.; Simarro, M.; Howie, D.; Morra, M.; Engel, P.; Terhorst, C. Cell surface receptors Ly-9 and CD84 recruit the X-linked lymphoproliferative disease gene product SAP. Blood 2001, 97, 3867–3874. [Google Scholar] [CrossRef]
- Simarro, M.; Lanyi, A.; Howie, D.; Poy, F.; Bruggeman, J.; Choi, M.; Sumegi, J.; Eck, M.J.; Terhorst, C. SAP increases FynT kinase activity and is required for phosphorylation of SLAM and Ly9. Int. Immunol. 2004, 16, 727–736. [Google Scholar] [CrossRef]
- Graham, D.B.; Bell, M.P.; McCausland, M.M.; Huntoon, C.J.; van Deursen, J.; Faubion, W.A.; Crotty, S.; McKean, D.J. Ly9 (CD229)-deficient mice exhibit T cell defects yet do not share several phenotypic characteristics associated with SLAM- and SAP-deficient mice. J. Immunol. 2006, 176, 291–300. [Google Scholar] [CrossRef]
- Tembhare, P.R.; Ghogale, S.; Tauro, W.; Badrinath, Y.; Deshpande, N.; Kedia, S.; Cherian, K.; Patkar, N.V.; Chatterjee, G.; Gujral, S.; et al. Evaluation of CD229 as a new alternative plasma cell gating marker in the flow cytometric immunophenotyping of monoclonal gammopathies. Cytom. Part B Clin. Cytom. 2018, 94, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Yousef, S.; Kovacsovics-Bankowski, M.; Salama, M.E.; Bhardwaj, N.; Steinbach, M.; Langemo, A.; Kovacsovics, T.; Marvin, J.; Binder, M.; Panse, J.; et al. CD229 is expressed on the surface of plasma cells carrying an aberrant phenotype and chemotherapy-resistant precursor cells in multiple myeloma. Hum. Vaccines Immunother. 2015, 11, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Takahashi, R.; Tsubota, A.; Sasaki, M.; Handa, H.; Imai, Y.; Tanaka, N.; Tsukune, Y.; Tanosaki, S.; Ito, S.; et al. SLAMF3-Mediated Signaling via ERK Pathway Activation Promotes Aggressive Phenotypic Behaviors in Multiple Myeloma. Mol. Cancer Res. MCR 2020, 18, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Pojero, F.; Flores-Montero, J.; Sanoja, L.; Pérez, J.J.; Puig, N.; Paiva, B.; Bottcher, S.; van Dongen, J.J.; Orfao, A. Utility of CD54, CD229, and CD319 for the identification of plasma cells in patients with clonal plasma cell diseases. Cytom. Part B Clin. Cytom. 2016, 90, 91–100. [Google Scholar] [CrossRef]
- Ijaz, M.; Wang, F.; Shahbaz, M.; Jiang, W.; Fathy, A.H.; Nesa, E.U. The Role of Grb2 in Cancer and Peptides as Grb2 Antagonists. Protein Pept. Lett. 2018, 24, 1084–1095. [Google Scholar] [CrossRef]
- Lin, Q.; Zhao, J.; Song, Y.; Liu, D. Recent updates on CAR T clinical trials for multiple myeloma. Mol. Cancer 2019, 18, 154. [Google Scholar] [CrossRef]
- Cunninghame-Graham, D.S.; Vyse, T.J.; Fortin, P.R.; Montpetit, A.; Cai, Y.C.; Lim, S.; McKenzie, T.; Farwell, L.; Rhodes, B.; Chad, L.; et al. Association of LY9 in UK and Canadian SLE families. Genes Immun. 2008, 9, 93–102. [Google Scholar] [CrossRef]
- Margraf, S.; Garner, L.I.; Wilson, T.J.; Brown, M.H. A polymorphism in a phosphotyrosine signalling motif of CD229 (Ly9, SLAMF3) alters SH2 domain binding and T-cell activation. Immunology 2015, 146, 392–400. [Google Scholar] [CrossRef]
- Ishibashi, M.; Sunakawa-Kii, M.; Kaito, Y.; Kinoshita, R.; Asayama, T.; Kuribayashi, Y.; Inokuchi, K.; Morita, R.; Tamura, H. The SLAMF3 rs509749 polymorphism correlates with malignant potential in multiple myeloma. Exp. Hematol. 2020. [Google Scholar] [CrossRef]
- Radhakrishnan, S.V.; Luetkens, T.; Scherer, S.D.; Davis, P.; Vander-Mause, E.R.; Olson, M.L.; Yousef, S.; Panse, J.; Abdiche, Y.; Li, K.D.; et al. CD229 CAR T cells eliminate multiple myeloma and tumor propagating cells without fratricide. Nat. Commun. 2020, 11, 798. [Google Scholar] [CrossRef]
- Cao, E.; Ramagopal, U.A.; Fedorov, A.; Fedorov, E.; Yan, Q.; Lary, J.W.; Cole, J.L.; Nathenson, S.G.; Almo, S.C. NTB-A receptor crystal structure: Insights into homophilic interactions in the signaling lymphocytic activation molecule receptor family. Immunity 2006, 25, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, M.A.; Adam, K.; Strazza, M.; Tocheva, A.S.; Peled, M.; Mor, A. SLAMF6 clustering is required to augment T cell activation. PLoS ONE 2019, 14, e0218109. [Google Scholar] [CrossRef] [PubMed]
- Valdez, P.A.; Wang, H.; Seshasayee, D.; van Lookeren-Campagne, M.; Gurney, A.; Lee, W.P.; Grewal, I.S. NTB-A, a new activating receptor in T cells that regulates autoimmune disease. J. Biol. Chem. 2004, 279, 18662–18669. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Kis-Toth, K.; Thai, T.H.; Terhorst, C.; Tsokos, G.C. SLAMF6-driven co-stimulation of human peripheral T cells is defective in SLE T cells. Autoimmunity 2011, 44, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Radomir, L.; Cohen, S.; Kramer, M.P.; Bakos, E.; Lewinsky, H.; Barak, A.; Porat, Z.; Bucala, R.; Stepensky, P.; Becker-Herman, S.; et al. T Cells Regulate Peripheral Naive Mature B Cell Survival by Cell-Cell Contact Mediated through SLAMF6 and SAP. J. Immunol. 2017, 199, 2745–2757. [Google Scholar] [CrossRef]
- Lewis, T.; Olson, D.J.; Gordon, K.A.; Sandall, S.L.; Miyamoto, J.; Westendorf, L.; Linares, G.; Leiske, C.; Kostner, H.; Stone, I.; et al. SGN-CD352A: A novel humanized anti-CD352 antibody-drug conjugate for the treatment of multiple myeloma. Cancer Res. 2016, 76, 1195. [Google Scholar] [CrossRef]
- Veillette, A.; Guo, H. CS1, a SLAM family receptor involved in immune regulation, is a therapeutic target in multiple myeloma. Crit. Rev. Oncol. Hematol. 2013, 88, 168–177. [Google Scholar] [CrossRef]
- Cruz-Munoz, M.E.; Dong, Z.; Shi, X.; Zhang, S.; Veillette, A. Influence of CRACC, a SLAM family receptor coupled to the adaptor EAT-2, on natural killer cell function. Nat. Immunol. 2009, 10, 297–305. [Google Scholar] [CrossRef]
- Tassi, I.; Colonna, M. The cytotoxicity receptor CRACC (CS-1) recruits EAT-2 and activates the PI3K and phospholipase Cgamma signaling pathways in human NK cells. J. Immunol. 2005, 175, 7996–8002. [Google Scholar] [CrossRef]
- Tai, Y.T.; Dillon, M.; Song, W.; Leiba, M.; Li, X.F.; Burger, P.; Lee, A.I.; Podar, K.; Hideshima, T.; Rice, A.G.; et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 2008, 112, 1329–1337. [Google Scholar] [CrossRef]
- Ritchie, D.; Colonna, M. Mechanisms of Action and Clinical Development of Elotuzumab. Clin. Transl. Sci. 2018, 11, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Szmania, S.; van Rhee, F. Profile of elotuzumab and its potential in the treatment of multiple myeloma. Blood Lymphat. Cancer Targets Ther. 2014, 2014, 15–27. [Google Scholar] [CrossRef]
- Weisel, K. Spotlight on elotuzumab in the treatment of multiple myeloma: The evidence to date. OncoTargets Ther. 2016, 9, 6037–6048. [Google Scholar] [CrossRef]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. CII 2013, 62, 1841–1849. [Google Scholar] [CrossRef] [PubMed]
- Pazina, T.; James, A.M.; MacFarlane, A.W.T.; Bezman, N.A.; Henning, K.A.; Bee, C.; Graziano, R.F.; Robbins, M.D.; Cohen, A.D.; Campbell, K.S. The anti-SLAMF7 antibody elotuzumab mediates NK cell activation through both CD16-dependent and -independent mechanisms. Oncoimmunology 2017, 6, e1339853. [Google Scholar] [CrossRef] [PubMed]
- Pazina, T.; James, A.M.; Colby, K.B.; Yang, Y.; Gale, A.; Jhatakia, A.; Kearney, A.Y.; Graziano, R.F.; Bezman, N.A.; Robbins, M.D.; et al. Enhanced SLAMF7 Homotypic Interactions by Elotuzumab Improves NK Cell Killing of Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 1633–1646. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, A.T.; Glavey, S.V.; Bezman, N.A.; Jhatakia, A.; Guerriero, J.L.; Manier, S.; Moschetta, M.; Mishima, Y.; Roccaro, A.; Detappe, A.; et al. Antibody-Dependent Cellular Phagocytosis by Macrophages is a Novel Mechanism of Action of Elotuzumab. Mol. Cancer Ther. 2018, 17, 1454–1463. [Google Scholar] [CrossRef]
- Zonder, J.A.; Mohrbacher, A.F.; Singhal, S.; van Rhee, F.; Bensinger, W.I.; Ding, H.; Fry, J.; Afar, D.E.; Singhal, A.K. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 2012, 120, 552–559. [Google Scholar] [CrossRef]
- van Rhee, F.; Szmania, S.M.; Dillon, M.; van Abbema, A.M.; Li, X.; Stone, M.K.; Garg, T.K.; Shi, J.; Moreno-Bost, A.M.; Yun, R.; et al. Combinatorial efficacy of anti-CS1 monoclonal antibody elotuzumab (HuLuc63) and bortezomib against multiple myeloma. Mol. Cancer Ther. 2009, 8, 2616–2624. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Lonial, S.; Betts, K.A.; Chen, C.; Zichlin, M.L.; Brun, A.; Signorovitch, J.E.; Makenbaeva, D.; Mekan, S.; Sy, O.; et al. Elotuzumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: Extended 4-year follow-up and analysis of relative progression-free survival from the randomized ELOQUENT-2 trial. Cancer 2018, 124, 4032–4043. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Cruz-Munoz, M.E.; Wu, N.; Robbins, M.; Veillette, A. Immune cell inhibition by SLAMF7 is mediated by a mechanism requiring src kinases, CD45, and SHIP-1 that is defective in multiple myeloma cells. Mol. Cell. Biol. 2015, 35, 41–51. [Google Scholar] [CrossRef]
- Kikuchi, J.; Hori, M.; Iha, H.; Toyama-Sorimachi, N.; Hagiwara, S.; Kuroda, Y.; Koyama, D.; Izumi, T.; Yasui, H.; Suzuki, A.; et al. Soluble SLAMF7 promotes the growth of myeloma cells via homophilic interaction with surface SLAMF7. Leukemia 2020, 34, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Soeda, S.; Sasaki, M.; Handa, H.; Imai, Y.; Tanaka, N.; Tanosaki, S.; Ito, S.; Odajima, T.; Sugimori, H.; et al. Clinical impact of serum soluble SLAMF7 in multiple myeloma. Oncotarget 2018, 9, 34784–34793. [Google Scholar] [CrossRef]
- Xie, Z.; Gunaratne, J.; Cheong, L.L.; Liu, S.C.; Koh, T.L.; Huang, G.; Blackstock, W.P.; Chng, W.J. Plasma membrane proteomics identifies biomarkers associated with MMSET overexpression in T(4;14) multiple myeloma. Oncotarget 2013, 4, 1008–1018. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Lonial, S.; White, D.; Moreau, P.; Palumbo, A.; San-Miguel, J.; Shpilberg, O.; Anderson, K.; Grosicki, S.; Spicka, I.; et al. Elotuzumab plus lenalidomide/dexamethasone for relapsed or refractory multiple myeloma: ELOQUENT-2 follow-up and post-hoc analyses on progression-free survival and tumour growth. Br. J. Haematol. 2017, 178, 896–905. [Google Scholar] [CrossRef]
- Chu, J.; He, S.; Deng, Y.; Zhang, J.; Peng, Y.; Hughes, T.; Yi, L.; Kwon, C.H.; Wang, Q.E.; Devine, S.M.; et al. Genetic modification of T cells redirected toward CS1 enhances eradication of myeloma cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 3989–4000. [Google Scholar] [CrossRef]
- Gogishvili, T.; Danhof, S.; Prommersberger, S.; Rydzek, J.; Schreder, M.; Brede, C.; Einsele, H.; Hudecek, M. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7(+) normal lymphocytes. Blood 2017, 130, 2838–2847. [Google Scholar] [CrossRef]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132. [Google Scholar] [CrossRef]
- Mathur, R.; Zhang, Z.; He, J.; Galetto, R.; Gouble, A.; Chion-Sotinel, I.; Filipe, S.; Gariboldi, A.; Veeramachaneni, T.; Manasanch, E.E.; et al. Universal SLAMF7-Specific CAR T-Cells As Treatment for Multiple Myeloma. Blood 2017, 130 (Suppl. 1), 502. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishibashi, M.; Morita, R.; Tamura, H. Immune Functions of Signaling Lymphocytic Activation Molecule Family Molecules in Multiple Myeloma. Cancers 2021, 13, 279. https://doi.org/10.3390/cancers13020279
Ishibashi M, Morita R, Tamura H. Immune Functions of Signaling Lymphocytic Activation Molecule Family Molecules in Multiple Myeloma. Cancers. 2021; 13(2):279. https://doi.org/10.3390/cancers13020279
Chicago/Turabian StyleIshibashi, Mariko, Rimpei Morita, and Hideto Tamura. 2021. "Immune Functions of Signaling Lymphocytic Activation Molecule Family Molecules in Multiple Myeloma" Cancers 13, no. 2: 279. https://doi.org/10.3390/cancers13020279
APA StyleIshibashi, M., Morita, R., & Tamura, H. (2021). Immune Functions of Signaling Lymphocytic Activation Molecule Family Molecules in Multiple Myeloma. Cancers, 13(2), 279. https://doi.org/10.3390/cancers13020279