Wilms’ Tumor 1 (WT1): A Novel Immunomarker of Dermatofibrosarcoma Protuberans—An Immunohistochemical Study on a Series of 114 Cases of Bland-Looking Mesenchymal Spindle Cell Lesions of the Dermis/Subcutaneous Tissues


 ,
, 
 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Clinical Data of the Cohort of Cases Included in the Study
2.2. Evaluation of Immunohistochemical Expression of WT1
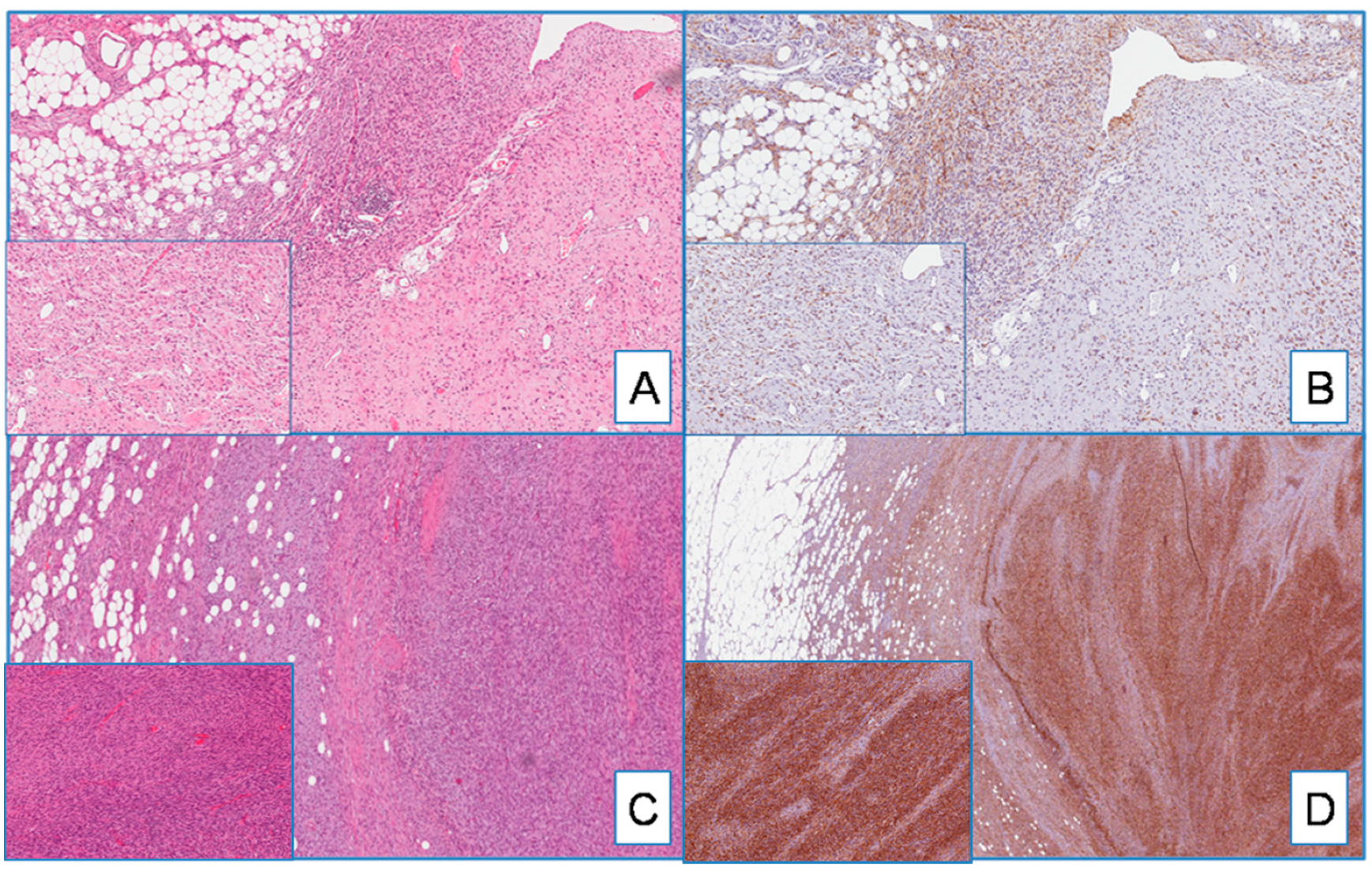
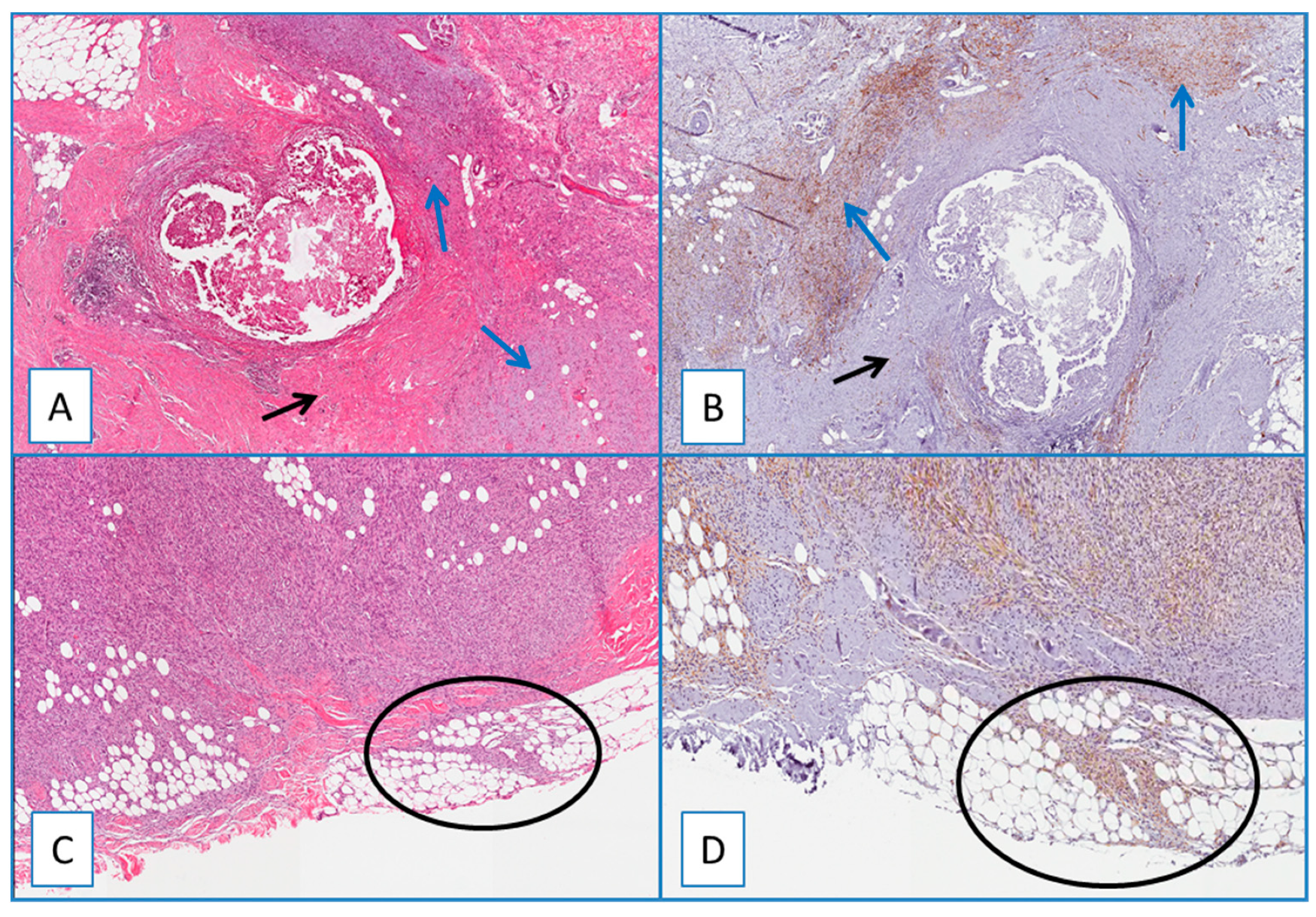
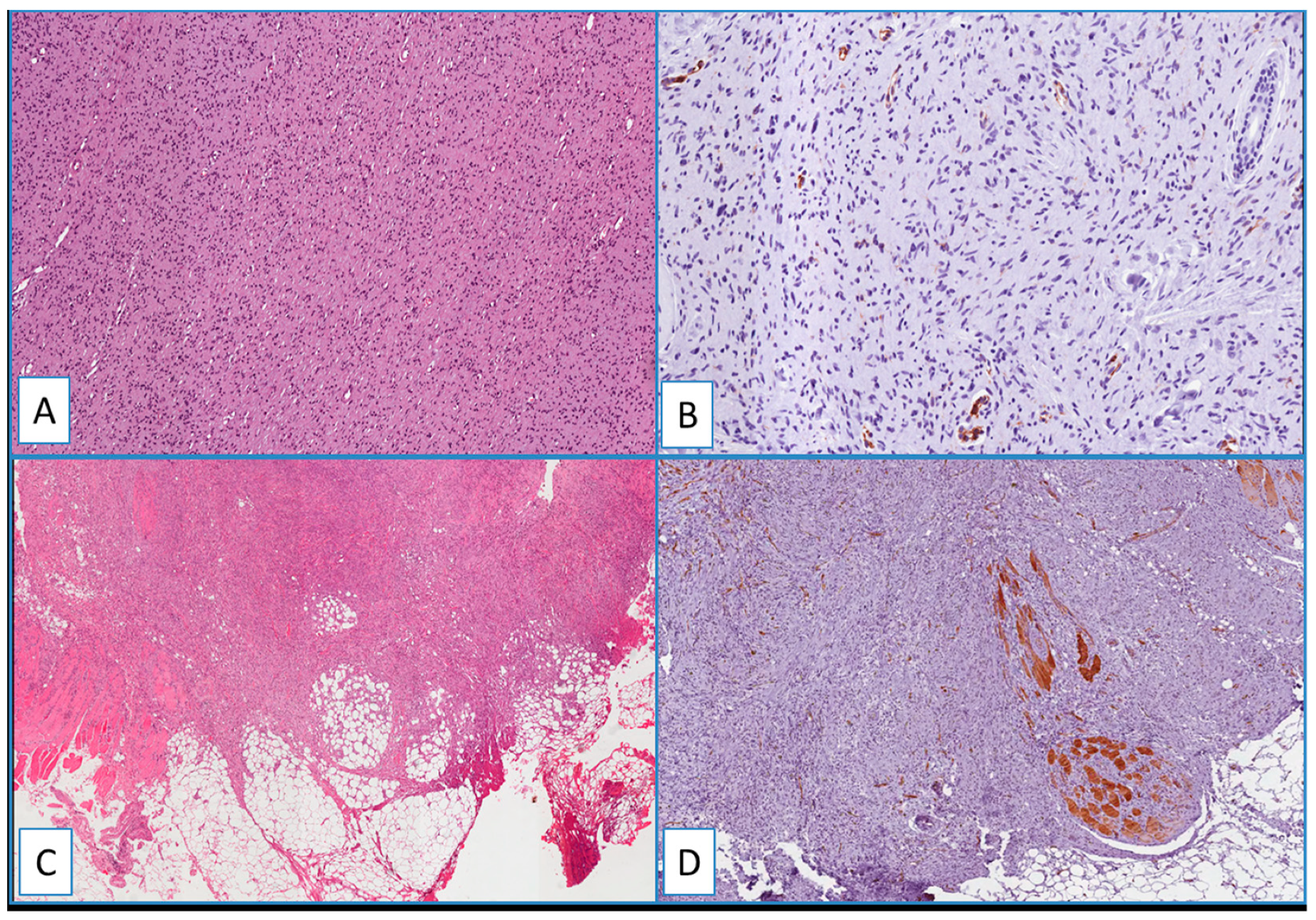
2.3. DFSP
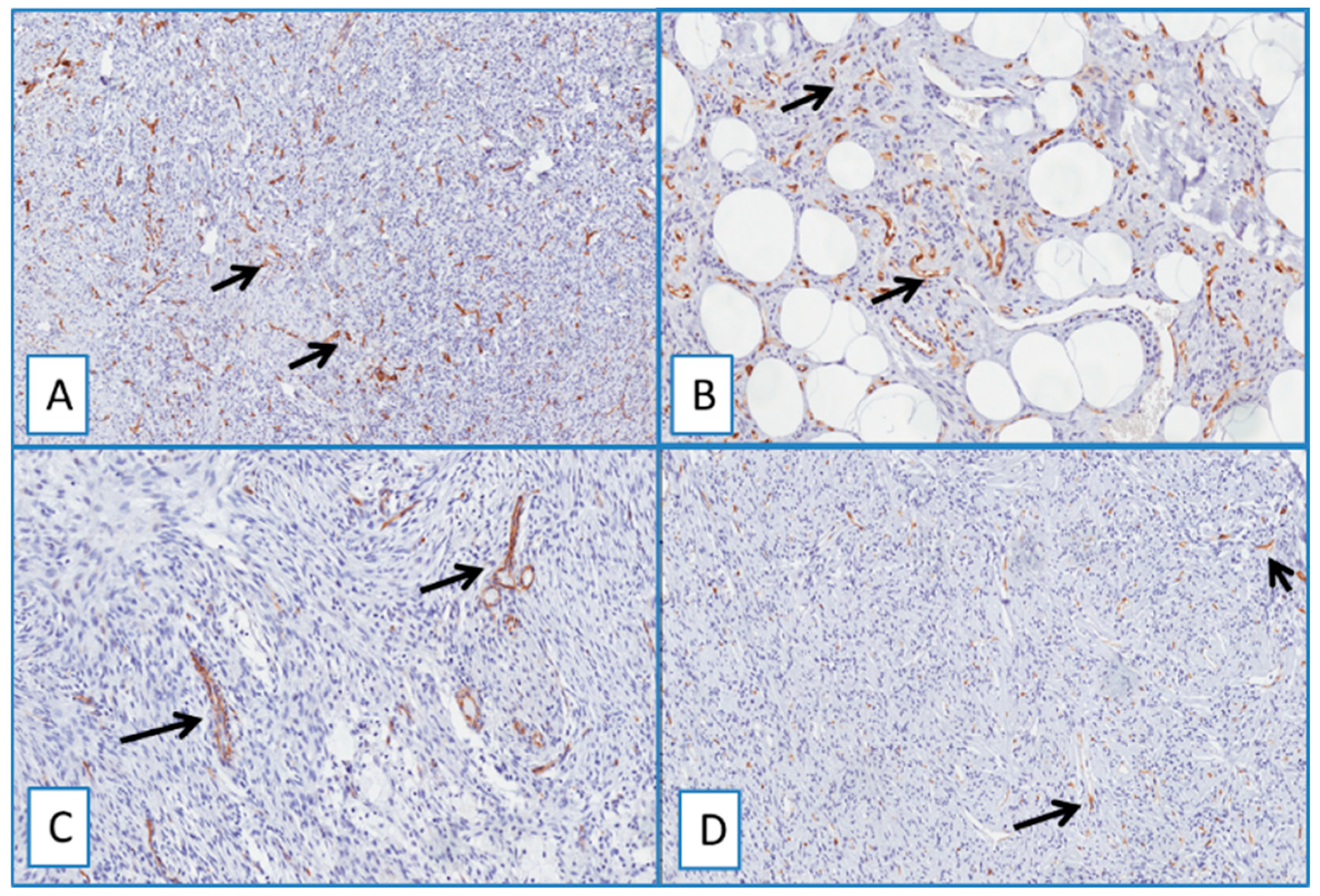
2.4. Non-DFSP Lesions
3. Discussion
3.1. Diagnostic Utility of WT1 in Distinguishing DFSP from Its Morphological Mimickers
3.2. Diagnostic Utility of WT1 in Recurrent/Residual DFSP
4. Materials and Methods
- Fifty-seven cases of DFSP; 11 of these cases were recurrent lesions; 2 primary cases exhibited an additional fibrosarcomatous overgrowth, while 2 primary and one recurrent tumor contained a minority of giant cell fibroblastoma component);
- Fifteen cases of dermatofibroma (classic type and cellular variants);
- Five cases of deep fibrous histiocytoma
- Eight cases of dermal scars;
- Five cases of spindle cell lipoma;
- Six cases of nodular fasciitis;
- Five cases of cutaneous leiomyomas;
- Eight cases of neurofibroma;
- Five cases of solitary fibrous tumor.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Degos, R.; Mouly, R.; Civatte, J.; Chérif-Cheikh, J.L.; Lautmann, F. Dermato-fibro-sarcome de Darier-Ferrand, datant de 70 ans, opéré au stade ultime de tumeur monstrueuse [Darier-Ferrand dermato-fibrosarcoma of 70 years’ duration operated on in the last stage of massive tumor]. Bull. Soc. Fr. Dermatol. Syphiligr. 1967, 74, 190–191. [Google Scholar] [PubMed]
- McKee, P.H.; Fletcher, C.D. Dermatofibrosarcoma protuberans presenting in infancy and childhood. J. Cutan. Pathol. 1991, 18, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Petoin, D.S.; Verola, O.; Banzet, P.; Dufourmentel, C.; Servant, J.M. Dermatofibrosarcome de Darier et Ferrand. Etude de 96 cas sur 15 ans [Darier-Ferrand dermatofibrosarcoma. Study of 96 cases over 15 years]. Chirurgie 1985, 111, 132–138. [Google Scholar] [PubMed]
- Taylor, H.B.; Helwig, E.B. Dermatofibrosarcoma protuberans. A study of 115 cases. Cancer 1962, 15, 717–725. [Google Scholar] [CrossRef]
- Thway, K.; Noujaim, J.; Jones, R.L.; Fisher, C. Dermatofibrosarcoma protuberans: Pathology, genetics, and potential therapeutic strategies. Ann. Diagn. Pathol. 2016, 25, 64–71. [Google Scholar] [CrossRef]
- Allen, A.; Ahn, C.; Sangüeza, O.P. Dermatofibrosarcoma Protuberans. Dermatol. Clin. 2019, 37, 483–488. [Google Scholar] [CrossRef]
- Vecchio, G.M.; Broggi, G.; Mulè, A.; Piombino, E.; Magro, G. Dermatofibrosarcoma protuberans: A tumor in the wide spectrum of the bland-looking spindle cell lesions of the breast. Pathologica 2019, 111, 87–91. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Puzzo, L.; Piombino, E.; Bartoloni, G.; Broggi, G.; Vecchio, G.M. Practical approach to diagnosis of bland-looking spindle cell lesions of the breast. Pathologica 2019, 111, 344–360. [Google Scholar] [CrossRef]
- Sadullahoğlu, C.; Dere, Y.; Atasever, T.R.; Öztop, M.T.; Karaaslan, Ö. The Role of CD34 and D2-40 in the Differentiation of Dermatofibroma and Dermatofibrosarcoma Protuberans. Turk. Patoloji. Derg. 2017, 1, 223–227. [Google Scholar] [CrossRef]
- Morimitsu, Y.; Hisaoka, M.; Okamoto, S.; Hashimoto, H.; Ushijima, M. Dermatofibrosarcoma protuberans and its fibrosarcomatous variant with areas of myoid differentiation: A report of three cases. Histopathology 1998, 32, 547–551. [Google Scholar] [CrossRef]
- Sanz-Trelles, A.; Ayala-Carbonero, A.; Rodrigo-Fernández, I.; Weil-Lara, B. Leiomyomatous nodules and bundles of vascular origin in the fibrosarcomatous variant of dermatofibrosarcoma protuberans. J. Cutan. Pathol. 1998, 25, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Goldblum, J.R.; Reith, J.D.; Weiss, S.W. Sarcomas arising in dermatofibrosarcoma protuberans: A reappraisal of biologic behavior in eighteen cases treated by wide local excision with extended clinical follow up. Am. J. Surg. Pathol. 2000, 24, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, A.; O’Brien, K.P.; Sjöblom, T.; Pietras, K.; Buchdunger, E.; Collins, V.P.; Heldin, C.H.; Dumanski, J.P.; Ostman, A. The dermatofibrosarcoma protuberans-associated collagen type Ialpha1/platelet-derived growth factor (PDGF) B-chain fusion gene generates a transforming protein that is processed to functional PDGF-BB. Cancer Res. 1999, 59, 3719–3723. [Google Scholar] [PubMed]
- Mentzel, T.; Beham, A.; Katenkamp, D.; Dei Tos, A.P.; Fletcher, C.D. Fibrosarcomatous (“high-grade”) dermatofibrosarcoma protuberans: Clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am. J. Surg. Pathol. 1998, 22, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Wrotnowski, U.; Cooper, P.H.; Shmookler, B.M. Fibrosarcomatous change in dermatofibrosarcoma protuberans. Am. J. Surg. Pathol. 1988, 12, 287–293. [Google Scholar] [CrossRef]
- Connelly, J.H.; Evans, H.L. Dermatofibrosarcoma protuberans. A clinicopathologic review with emphasis on fibrosarcomatous areas. Am. J. Surg. Pathol. 1992, 16, 921–925. [Google Scholar] [CrossRef]
- Lopez, L.V.; Yatsenko, S.A.; Burgess, M.; Schoedel, K.; Rao, U.N.M. Dermatofibrosarcoma protuberans with fibrosarcomatous transformation: Our experience, molecular evaluation of selected cases, and short literature review. Int. J. Dermatol. 2019, 58, 1246–1252. [Google Scholar] [CrossRef]
- Abbott, J.J.; Oliveira, A.M.; Nascimento, A.G. The prognostic significance of fibrosarcomatous transformation in dermatofibrosarcoma protuberans. Am. J. Surg. Pathol. 2006, 30, 436–443. [Google Scholar] [CrossRef]
- Broggi, G.; Salvatorelli, L.; Reibaldi, M.; Bonfiglio, V.; Longo, A.; Russo, A.; Caltabiano, R.; Magro, G. Solitary fibrous tumor of the orbital region: Report of a case with emphasis on the diagnostic utility of STAT-6. Pathologica 2020, 112, 195–199. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Piombino, E.; Broggi, G.; Castorina, S. Solitary fibrous tumor with atypical features of the paravesical space: Benign clinical course at the 10-years follow-up. Report of a case and review of the literature. Pathologica 2020, 112, 200–209. [Google Scholar] [CrossRef]
- Call, K.M.; Glaser, T.; Ito, C.Y.; Buckler, A.J.; Pelletier, J.; Haber, D.A.; Rose, E.A.; Kral, A.; Yeger, H.; Lewis, W.H.; et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990, 60, 509–520. [Google Scholar] [CrossRef]
- Tsuta, K.; Kato, Y.; Tochigi, N.; Hoshino, T.; Takeda, Y.; Hosako, M.; Maeshima, A.M.; Asamura, H.; Kondo, T.; Matsuno, Y. Comparison of different clones (WT49 versus 6F-H2) of WT-1 antibodies for immunohistochemical diagnosis of malignant pleural mesothelioma. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Gessler, M.; Poustka, A.; Cavenee, W.; Neve, R.L.; Orkin, S.H.; Bruns, G.A. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature 1990, 343, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.L.; van der Eb, A.J.; Jochemsen, A.G. The Wilms’ tumor 1 gene: Oncogene or tumor suppressor gene? Int. Rev. Cytol. 1998, 181, 151–212. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Haber, D.A. Wilms tumor and the WT1 gene. Exp. Cell Res. 2001, 264, 74–99. [Google Scholar] [CrossRef] [PubMed]
- Huff, V. Wilms’ tumours: About tumour suppressor genes, an oncogene and a chameleon gene. Nat. Rev. Cancer 2011, 11, 111–121. [Google Scholar] [CrossRef]
- Loke, S.L.; Neckers, L.M.; Schwab, G.; Jaffe, E.S. c-myc protein in normal tissue. Effects of fixation on its apparent subcellular distribution. Am. J. Pathol. 1988, 131, 29–37. [Google Scholar]
- Royds, J.A.; Sharrard, R.M.; Wagner, B.; Polacarz, S.V. Cellular localisation of c-myc product in human colorectal epithelial neoplasia. J. Pathol. 1992, 166, 225–233. [Google Scholar] [CrossRef]
- Ramani, P.; Cowell, J.K. The expression pattern of Wilms’ tumour gene (WT1) product in normal tissues and paediatric renal tumours. J. Pathol. 1996, 179, 162–168. [Google Scholar] [CrossRef]
- Lawley, L.P.; Cerimele, F.; Weiss, S.W.; North, P.; Cohen, C.; Kozakewich, H.P.; Mulliken, J.B.; Arbiser, J.L. Expression of Wilms tumor 1 gene distinguishes vascular malformations from proliferative endothelial lesions. Arch. Dermatol. 2005, 141, 1297–1300. [Google Scholar] [CrossRef]
- Timár, J.; Mészáros, L.; Orosz, Z.; Albini, A.; Rásó, E. WT1 expression in angiogenic tumours of the skin. Histopathology 2005, 47, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, S.; Oji, Y.; Horiuchi, T.; Kanda, T.; Kitagawa, M.; Takeuchi, T.; Kawano, K.; Kuwae, Y.; Yamauchi, A.; Okumura, M.; et al. Immunohistochemical detection of WT1 protein in a variety of cancer cells. Mod. Pathol. 2006, 19, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Salvatorelli, L.; Calabrese, G.; Parenti, R.; Vecchio, G.M.; Puzzo, L.; Caltabiano, R.; Musumeci, G.; Magro, G. Immunohistochemical Expression of Wilms’ Tumor 1 Protein in Human Tissues: From Ontogenesis to Neoplastic Tissues. Appl. Sci. 2020, 10, 40. [Google Scholar] [CrossRef]
- Parenti, R.; Puzzo, L.; Vecchio, G.M.; Gravina, L.; Salvatorelli, L.; Musumeci, G.; Vasquez, E.; Magro, G. Immunolocalization of Wilms’ Tumor protein (WT1) in developing human peripheral sympathetic and gastroenteric nervous system. Acta Histochem. 2014, 116, 48–54. [Google Scholar] [CrossRef]
- Al Dhaybi, R.; Powell, J.; McCuaig, C.; Kokta, V. Differentiation of vascular tumors from vascular malformations by expression of Wilms tumor 1 gene: Evaluation of 126 cases. J. Am. Acad. Dermatol. 2010, 63, 1052–1057. [Google Scholar] [CrossRef]
- Trindade, F.; Tellechea, O.; Torrelo, A.; Requena, L.; Colmenero, I. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am. J. Dermatopathol. 2011, 33, 569–572. [Google Scholar] [CrossRef]
- Carpentieri, D.F.; Nichols, K.; Chou, P.M.; Matthews, M.; Pawel, B.; Huff, D. The expression of WT1 in the differentiation of rhabdomyosarcoma from other pediatric small round blue cell tumors. Mod. Pathol. 2002, 15, 1080–1086. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Puzzo, L.; Musumeci, G.; Bisceglia, M.; Parenti, R. Oncofetal expression of Wilms’ tumor 1 (WT1) protein in human fetal, adult and neoplastic skeletal muscle tissues. Acta Histochem. 2015, 117, 492–504. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Vecchio, G.M.; Musumeci, G.; Rita, A.; Parenti, R. Cytoplasmic expression of Wilms tumor transcription factor-1 (WT1): A useful immunomarker for young-type fibromatoses and infantile fibrosarcoma. Acta Histochem. 2014, 116, 1134–1140. [Google Scholar] [CrossRef]
- Magro, G.; Spadola, S.; Motta, F.; Palazzo, J.; Catalano, F.; Vecchio, G.M.; Salvatorelli, L. STAT6 expression in spindle cell lesions of the breast: An immunohistochemical study of 48 cases. Pathol. Res. Pract. 2018, 214, 1544–1549. [Google Scholar] [CrossRef]
- Cammarata, F.P.; Forte, G.I.; Broggi, G.; Bravatà, V.; Minafra, L.; Pisciotta, P.; Calvaruso, M.; Tringali, R.; Tomasello, B.; Torrisi, F.; et al. Molecular Investigation on a Triple Negative Breast Cancer Xenograft Model Exposed to Proton Beams. Int. J. Mol. Sci. 2020, 21, 6337. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis/Number of Cases | Positive Cases (%) | Staining Extension Cases (%) | Staining Intensity Cases (%) |
|---|---|---|---|
| Dermatofibrosarcoma protuberans (n = 57) | 54/57 (95%) | Diffuse: 42/57 (75%) Heterogeneous: 9/57 (15%) Focal: 6/57 (6%) | Strong: 53/57 (93%) Weak: 4/57 (7%) |
| Dermatofibroma (n = 15) | 0/15 (0%) | No staining | No staining |
| Deep fibrous histiocytoma (n = 5) | 0/5 (0%) | No staining | No staining |
| Dermal scars (n = 8) | 0/8 (0%) | No staining | No staining |
| Spindle cell lipoma (n = 5) | 0/5 (0%) | No staining | No staining |
| Nodular fasciitis (n = 6) | 0/6 (0%) | No staining | No staining |
| Cutaneous leiomyomas (n = 5) | 0/5 (0%) | No staining | No staining |
| Neurofibroma (n = 8) | 8/8 (100%) | Heterogeneous 8/8 (100%) | Weak/moderate 8/8 (100%) |
| Solitary fibrous tumor (n =5) | 0/5 (0%) | No staining | No staining |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piombino, E.; Broggi, G.; Barbareschi, M.; Castorina, S.; Parenti, R.; Bartoloni, G.; Salvatorelli, L.; Magro, G. Wilms’ Tumor 1 (WT1): A Novel Immunomarker of Dermatofibrosarcoma Protuberans—An Immunohistochemical Study on a Series of 114 Cases of Bland-Looking Mesenchymal Spindle Cell Lesions of the Dermis/Subcutaneous Tissues. Cancers 2021, 13, 252. https://doi.org/10.3390/cancers13020252
Piombino E, Broggi G, Barbareschi M, Castorina S, Parenti R, Bartoloni G, Salvatorelli L, Magro G. Wilms’ Tumor 1 (WT1): A Novel Immunomarker of Dermatofibrosarcoma Protuberans—An Immunohistochemical Study on a Series of 114 Cases of Bland-Looking Mesenchymal Spindle Cell Lesions of the Dermis/Subcutaneous Tissues. Cancers. 2021; 13(2):252. https://doi.org/10.3390/cancers13020252
Chicago/Turabian StylePiombino, Eliana, Giuseppe Broggi, Mattia Barbareschi, Sergio Castorina, Rosalba Parenti, Giovanni Bartoloni, Lucia Salvatorelli, and Gaetano Magro. 2021. "Wilms’ Tumor 1 (WT1): A Novel Immunomarker of Dermatofibrosarcoma Protuberans—An Immunohistochemical Study on a Series of 114 Cases of Bland-Looking Mesenchymal Spindle Cell Lesions of the Dermis/Subcutaneous Tissues" Cancers 13, no. 2: 252. https://doi.org/10.3390/cancers13020252
APA StylePiombino, E., Broggi, G., Barbareschi, M., Castorina, S., Parenti, R., Bartoloni, G., Salvatorelli, L., & Magro, G. (2021). Wilms’ Tumor 1 (WT1): A Novel Immunomarker of Dermatofibrosarcoma Protuberans—An Immunohistochemical Study on a Series of 114 Cases of Bland-Looking Mesenchymal Spindle Cell Lesions of the Dermis/Subcutaneous Tissues. Cancers, 13(2), 252. https://doi.org/10.3390/cancers13020252