The Merkel Cell Polyomavirus T Antigens Function as Tumor Promoters in Murine Skin

 ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
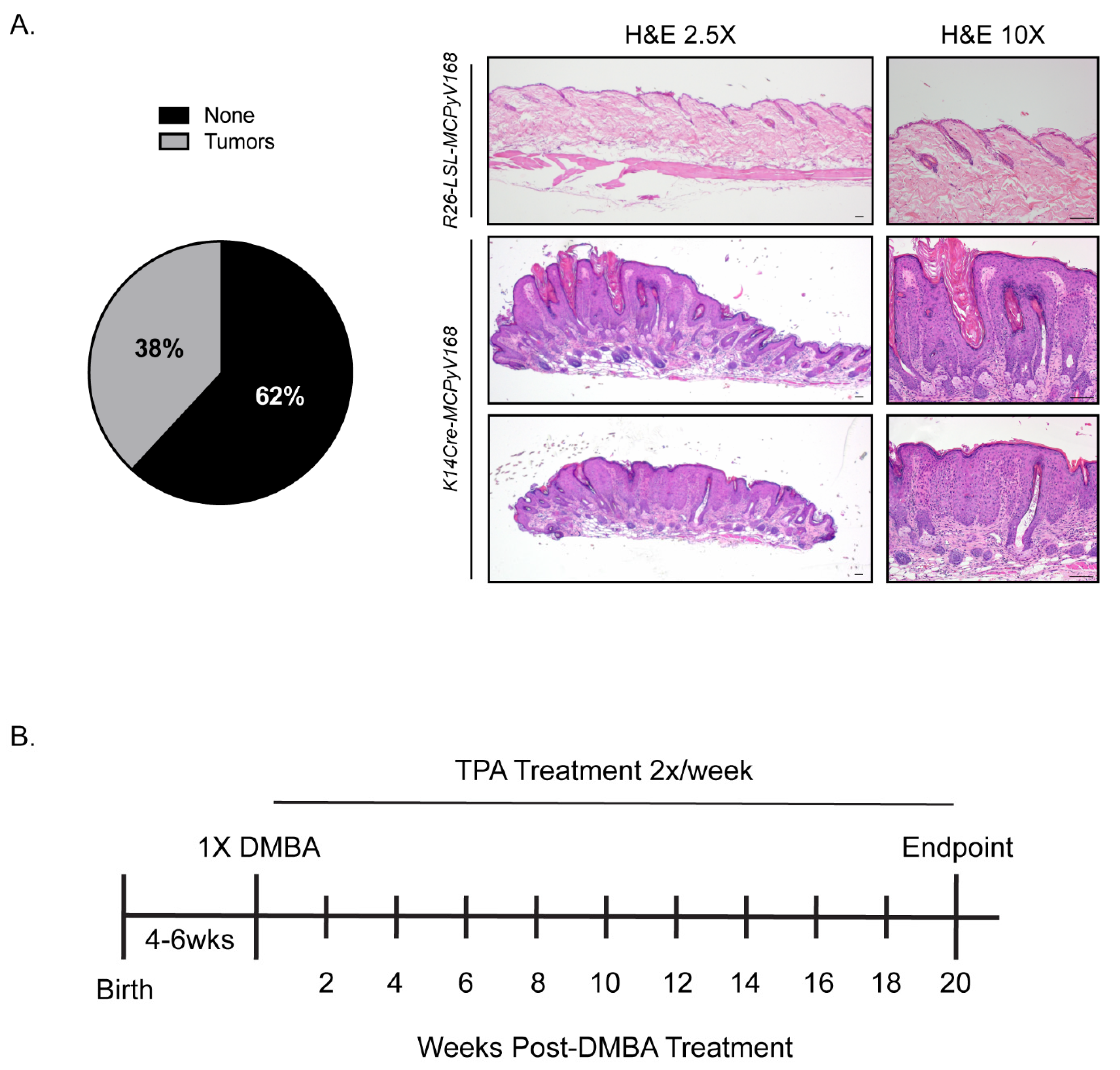
2.1. Model Validation and Experimental Overview of Studies to Determine the Role of the MCPyV T Antigens in Skin Carcinogenesis
2.2. The MCPyV T Antigens Function as Tumor Promoters, Not Initiators, in Murine Skin
2.3. The MCPyV T Antigens Synergize with Chemical Carcinogens to Exacerbate Skin Tumorigenesis
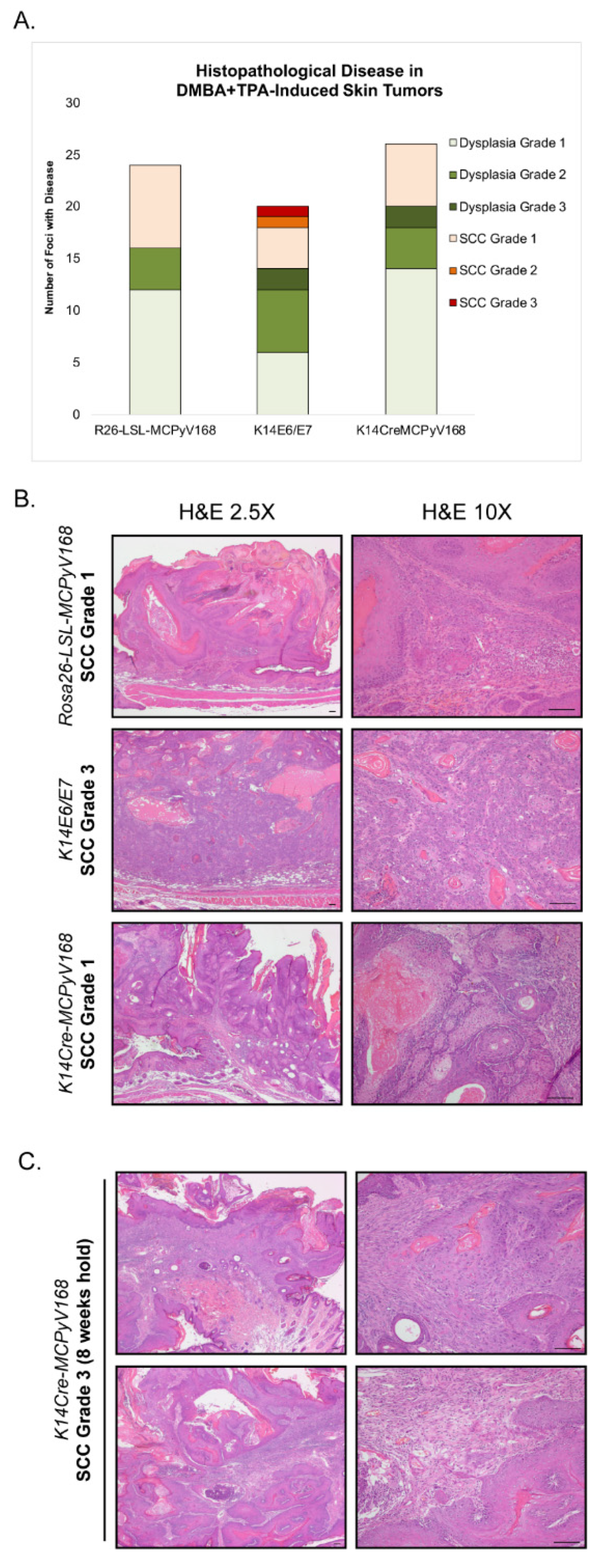
2.4. Assessment of Malignant Progression
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Genotyping
4.3. Skin Carcinogenesis Studies
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Lambert, P.F. The interwoven story of the small DNA tumor viruses. Virology 2009, 384, 255. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pipas, J.M. DNA tumor viruses and their contributions to molecular biology. J. Virol. 2019, 93, e01524-18. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; Banerjee, S.; Robertson, E.S. The role of gammaherpesviruses in cancer pathogenesis. Pathogens 2016, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Hausen, H.Z. Papillomaviruses in the causation of human cancers-A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Genet. 2013, 11, 264–276. [Google Scholar] [CrossRef]
- Toker, C. Trabecular carcinoma of the skin. Arch. Dermatol 1972, 105, 107–110. [Google Scholar] [CrossRef]
- Hodgson, N.C. Merkel cell carcinoma: Changing incidence trends. J. Surg. Oncol. 2004, 89, 1–4. [Google Scholar] [CrossRef]
- Paulson, K.G.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463.e2. [Google Scholar] [CrossRef]
- Harms, P.W.; on behalf of the International Workshop on Merkel Cell Carcinoma Research (IWMCC) Working Group; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef]
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Peñas, P.F.; Nghiem, P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: The AEIOU features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.J.; Paulson, K.G.; Wipf, G.C.; Miranda, D.; Madeleine, M.M.; Johnson, L.G.; Lemos, B.D.; Lee, S.; Warcola, A.H.; Iyer, J.G.; et al. Association of Merkel cell polyomavirus-specific antibodies with Merkel cell carcinoma. J. Natl. Cancer Inst. 2009, 101, 1510–1522. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Hedman, L.; Mattila, P.S.; Jartti, T.; Ruuskanen, O.; Söderlund-Venermo, M.; Hedman, K. Serological evidence of Merkel cell polyomavirus primary infections in childhood. J. Clin. Virol. 2011, 50, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of Human Seroresponsiveness to Merkel Cell Polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef]
- Tolstov, Y.L.; Knauer, A.; Chen, J.-G.; Kensler, T.W.; Kingsley, L.A.; Moore, P.S.; Chang, Y. Asymptomatic primary Merkel cell polyomavirus infection among adults. Emerg. Infect. Dis. 2011, 17, 1371–1380. [Google Scholar] [CrossRef]
- Tolstov, Y.L.; Pastrana, D.V.; Feng, H.; Becker, J.C.; Jenkins, F.J.; Moschos, S.; Chang, Y.; Buck, C.B.; Moore, P.S. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer 2009, 125, 1250–1256. [Google Scholar] [CrossRef]
- Touzé, A.; Gaitan, J.; Arnold, F.; Cazal, R.; Fleury, M.J.; Combelas, N.; Sizaret, P.-Y.; Guyetant, S.; Maruani, A.; Baay, M.; et al. Generation of Merkel cell polyomavirus (MCV)-like particles and their application to detection of MCV antibodies. J. Clin. Microbiol. 2010, 48, 1767–1770. [Google Scholar] [CrossRef]
- Viscidi, R.P.; Rollison, D.E.; Sondak, V.K.; Silver, B.; Messina, J.L.; Giuliano, A.R.; Fulp, W.; Ajidahun, A.; Rivanera, D. Age-specific seroprevalence of Merkel cell polyomavirus, BK virus, and JC virus. Clin. Vaccine Immunol. 2011, 18, 1737–1743. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Dominguez, M.; Greune, L.; Soria-Martinez, L.; Pfleiderer, M.M.; Schowalter, R.; Buck, C.B.; Blaum, B.S.; Schmidt, M.A.; Schelhaas, M. Infectious entry of Merkel cell polyomavirus. J. Virol. 2019, 93, 93. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Chang, Y.; Moore, P.S. Protein-mediated viral latency is a novel mechanism for Merkel cell polyomavirus persistence. Proc. Natl. Acad. Sci. USA 2017, 114, E4040–E4047. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Krump, N.A.; Buck, C.B.; You, J. Merkel cell polyomavirus infection and detection. J. Vis. Exp. 2019, e58950. [Google Scholar] [CrossRef]
- Neumann, F.; Borchert, S.; Schmidt, C.; Reimer, R.; Hohenberg, H.; Fischer, N.; Grundhoff, A. Replication, gene expression and particle production by a consensus Merkel cell polyomavirus (MCPyV) genome. PLoS ONE 2011, 6, e29112. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Reinhold, W.C.; Buck, C.B. Entry tropism of BK and Merkel cell polyomaviruses in cell culture. PLoS ONE 2012, 7, e42181. [Google Scholar] [CrossRef]
- Angermeyer, S.; Hesbacher, S.; Becker, J.C.; Schrama, D.; Houben, R. Merkel Cell polyomavirus-Positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J. Investig. Dermatol. 2013, 133, 2059–2064. [Google Scholar] [CrossRef]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef]
- Nwogu, N.; Ortiz, L.E.; Kwun, H.J. Surface charge of Merkel cell polyomavirus small T antigen determines cell transformation through allosteric FBW7 WD40 domain targeting. Oncogenesis 2020, 9, 1–12. [Google Scholar] [CrossRef]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.S.; et al. Merkel cell polyomavirus small T antigen induces cancer and embryonic Merkel cell proliferation in a transgenic mouse model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, M.E.; Cheng, J.; Bronson, R.T.; Lambert, P.F.; DeCaprio, J.A. Tumorigenic activity of Merkel cell polyomavirus T antigens expressed in the stratified epithelium of mice. Cancer Res. 2015, 75, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Eberl, M.; Wilbert, D.M.; Meireles, J.; Bichakjian, C.K.; Saunders, T.L.; Wong, S.Y.; Dlugosz, A.A. Merkel cell polyomavirus small T antigen initiates Merkel cell carcinoma-like tumor development in mice. Cancer Res. 2017, 77, 3151–3157. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Vozheiko, T.D.; Weick, J.W.; Wilbert, D.M.; Saunders, T.L.; Ermilov, A.N.; Bichakjian, C.K.; Johnson, T.M.; et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J. Investig. Dermatol. 2015, 135, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Wieland, U.; Kreuter, A.; Pawlita, M. C-terminal deletions of Merkel cell polyomavirus large T-antigen, a highly specific surrogate marker for virally induced malignancy. Int. J. Cancer 2012, 131, 2863–2868. [Google Scholar] [CrossRef]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.-J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef]
- Rodig, S.J.; Cheng, J.; Wardzala, J.; Dorosario, A.; Scanlon, J.J.; Laga, A.C.; Martinez-Fernandez, A.; Barletta, J.A.; Bellizzi, A.M.; Sadasivam, S.; et al. Improved detection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J. Clin. Investig. 2012, 122, 4645–4653. [Google Scholar] [CrossRef]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef]
- Laude, H.C.; Jonchère, B.; Maubec, E.; Carlotti, A.; Marinho, E.; Couturaud, B.; Peter, M.; Sastre-Garau, X.; Avril, M.-F.; Dupin, N.; et al. Distinct Merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with Merkel cell carcinoma. PLoS Pathog. 2010, 6, e1001076. [Google Scholar] [CrossRef]
- Bhatia, K.; Goedert, J.J.; Modali, R.; Preiss, L.; Ayers, L.W. Merkel cell carcinoma subgroups by Merkel cell polyomavirus DNA relative abundance and oncogene expression. Int. J. Cancer 2009, 126, 2240–2246. [Google Scholar] [CrossRef]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [PubMed]
- Katano, H.; Ito, H.; Suzuki, Y.; Nakamura, T.; Sato, Y.; Tsuji, T.; Matsuo, K.; Nakagawa, H.; Sata, T. Detection of Merkel cell polyomavirus in Merkel cell carcinoma and Kaposi’s sarcoma. J. Med. Virol. 2009, 81, 1951–1958. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Arora, R.; Kwun, H.J.; Feng, H.; Sarid, R.; Fernández-Figueras, M.-T.; Tolstov, Y.; Gjoerup, O.; Mansukhani, M.M.; Swerdlow, S.H.; et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int. J. Cancer 2009, 125, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Busam, K.J.; Jungbluth, A.A.; Rekthman, N.; Coit, D.; Pulitzer, M.; Bini, J.; Arora, R.; Hanson, N.C.; Tassello, J.A.; Frosina, D.; et al. Merkel cell polyomavirus expression in Merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am. J. Surg. Pathol. 2009, 33, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Peitsch, W.K.; Zapatka, M.; Kneitz, H.; Houben, R.; Eib, S.; Haferkamp, S.; Moore, P.S.; Shuda, M.; Thompson, J.F.; et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J. Investig. Dermatol. 2011, 131, 1631–1638. [Google Scholar] [CrossRef]
- Shuda, M.; Chang, Y.; Moore, P.S. Merkel cell polyomavirus-Positive Merkel cell carcinoma requires viral small T-antigen for cell proliferation. J. Investig. Dermatol. 2014, 134, 1479–1481. [Google Scholar] [CrossRef]
- Houben, R.; Adam, C.; Baeurle, A.; Hesbacher, S.; Grimm, J.; Angermeyer, S.; Henzel, K.; Hauser, S.; Elling, R.; Bröcker, E.-B.; et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int. J. Cancer 2011, 130, 847–856. [Google Scholar] [CrossRef]
- Schrama, D.; Hesbacher, S.; Angermeyer, S.; Schlosser, A.; Haferkamp, S.; Aue, A.; Adam, C.; Weber, A.; Schmidt, M.; Houben, R. Serine 220 phosphorylation of the Merkel cell polyomavirus large T antigen crucially supports growth of Merkel cell carcinoma cells. Int. J. Cancer 2015, 138, 1153–1162. [Google Scholar] [CrossRef]
- Fan, K.; Gravemeyer, J.; Ritter, C.; Rasheed, K.; Gambichler, T.; Moens, U.; Shuda, M.; Schrama, D.; Becker, J.C. MCPyV Large T antigen-induced atonal homolog 1 is a lineage-dependency oncogene in Merkel cell carcinoma. J. Investig. Dermatol. 2020, 140, 56–65.e3. [Google Scholar] [CrossRef]
- Harold, A.; Amako, Y.; Hachisuka, J.; Bai, Y.; Li, M.Y.; Kubat, L.; Gravemeyer, J.; Franks, J.; Gibbs, J.R.; Park, H.J.; et al. Conversion of Sox2-dependent Merkel cell carcinoma to a differentiated neuron-like phenotype by T antigen inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 20104–20114. [Google Scholar]
- Arora, R.; Shuda, M.; Guastafierro, A.; Feng, H.; Toptan, T.; Tolstov, Y.; Normolle, D.; Vollmer, L.L.; Vogt, A.; Dömling, A.; et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci. Transl. Med. 2012, 4, 133ra56. [Google Scholar] [CrossRef]
- Kervarrec, T.; Samimi, M.; Hesbacher, S.; Berthon, P.; Wobser, M.; Sallot, A.; Sarma, B.; Schweinitzer, S.; Gandon, T.; Destrieux, C.; et al. Merkel cell polyomavirus T antigens induce Merkel cell-like differentiation in GLI1-expressing epithelial cells. Cancers 2020, 12, 1989. [Google Scholar] [CrossRef]
- Pietropaolo, V.; Prezioso, C.; Moens, U. Merkel cell polyomavirus and Merkel cell carcinoma. Cancers 2020, 12, 1774. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.L.; Angel, J.M.; Kiguchi, K.; DiGiovanni, J. Multi-stage chemical carcinogenesis in mouse skin: Fundamentals and applications. Nat. Protoc. 2009, 4, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Boutwell, R.K. Model systems for defining initiation, promotion, and progression of skin neoplasms. Prog. Clin. Boil. Res. 1989, 298, 3–15. [Google Scholar]
- Maufort, J.P.; Williams, S.M.G.; Pitot, H.C.; Lambert, P. Human Papillomavirus 16 E5 oncogene contributes to two stages of skin carcinogenesis. Cancer Res. 2007, 67, 6106–6112. [Google Scholar] [CrossRef]
- Song, S.; Liem, A.; Miller, J.A.; Lambert, P. Human Papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology 2000, 267, 141–150. [Google Scholar] [CrossRef]
- Starrett, G.J.; Marcelus, C.; Cantalupo, P.G.; Katz, J.P.; Cheng, J.; Akagi, K.; Thakuria, M.; Rabinowits, G.; Wang, L.C.; Symer, D.E.; et al. Merkel cell polyomavirus exhibits dominant control of the tumor genome and transcriptome in virus-associated Merkel cell carcinoma. mBio 2017, 8, e02079-16. [Google Scholar] [CrossRef]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 1–22. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef]
- Schrama, D.; Sarosi, E.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; König, E.; Utikal, J.; Becker, J.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef]
- Kwun, H.J.; Wendzicki, J.A.; Shuda, Y.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen induces genome instability by E3 ubiquitin ligase targeting. Oncogene 2017, 36, 6784–6792. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A.J.; et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef] [PubMed]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortés-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; DeCaprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2018, 116, 1027–1032. [Google Scholar]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2015, 7, 3403–3415. [Google Scholar] [CrossRef] [PubMed]
- González-Vela, M.D.C.; Curiel-Olmo, S.; Derdak, S.; Beltran, S.; Santibañez, M.; Martínez, N.; Castillo-Trujillo, A.; Gut, M.; Sánchez-Pacheco, R.; Almaraz, C.; et al. Shared oncogenic pathways implicated in both virus-positive and UV-induced Merkel cell carcinomas. J. Investig. Dermatol. 2017, 137, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.-M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The distinctive mutational spectra of polyomavirus-negative Merkel cell carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef]
- Cohen, P.R.; Tomson, B.N.; Elkin, S.K.; Marchlik, E.; Carter, J.L.; Kurzrock, R. Genomic portfolio of Merkel cell carcinoma as determined by comprehensive genomic profiling: Implications for targeted therapeutics. Oncotarget 2016, 7, 23454–23467. [Google Scholar] [CrossRef]
- Hafner, C.; Houben, R.; Baeurle, A.; Ritter, C.; Schrama, D.; Landthaler, M.; Becker, J.C. Activation of the PI3K/AKT pathway in Merkel cell carcinoma. PLoS ONE 2012, 7, e31255. [Google Scholar] [CrossRef]
- Nardi, V.; Song, Y.C.; Santamaria-Barria, J.A.; Cosper, A.K.; Lam, Q.; Faber, A.C.; Boland, G.M.; Yeap, B.Y.; Bergethon, K.; Scialabba, V.L.; et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin. Cancer Res. 2012, 18, 1227–1236. [Google Scholar] [CrossRef]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. 2015, 75, 5228–5234. [Google Scholar] [CrossRef]
- Quintanilla, M.; Brown, K.; Ramsden, M.; Balmain, A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nat. Cell Biol. 1986, 322, 78–80. [Google Scholar] [CrossRef]
- Shin, M.-K.; Payne, S.N.; Bilger, A.; Matkowskyj, K.A.; Carchman, E.; Meyer, D.S.; Bentires-Alj, M.; Deming, D.A.; Lambert, P. Activating mutations in Pik3ca contribute to anal carcinogenesis in the presence or absence of HPV-16 oncogenes. Clin. Cancer Res. 2019, 25, 1889–1900. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, M.K.; Pitot, H.C.; Liem, A.; Schweizer, J.; Mahoney, C.; Lambert, P. A Mouse model for human anal cancer. Cancer Prev. Res. 2010, 3, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Kemp, C.J. Multistep skin cancer in mice as a model to study the evolution of cancer cells. Semin. Cancer Biol. 2005, 15, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Kervarrec, T.; Samimi, M.; Guyétant, S.; Sarma, B.; Chéret, J.; Blanchard, E.; Berthon, P.; Schrama, D.; Houben, R.; Touzé, A. Histogenesis of Merkel cell carcinoma: A comprehensive review. Front. Oncol. 2019, 9, 451. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Mascre, G.; Youseff, K.K.; Harel, I.; Michaux, C.; De Geest, N.; Szpalski, C.; Achouri, Y.; Bloch, W.; Hassan, B.A.; et al. Epidermal progenitors give rise to Merkel cells during embryonic development and adult homeostasis. J. Cell Biol. 2009, 187, 91–100. [Google Scholar] [CrossRef]
- Hausen, A.Z.; Rennspiess, D.; Winnepenninckx, V.; Speel, E.-J.M.; Kurz, A.K. Early B-cell differentiation in Merkel cell carcinomas: Clues to cellular ancestry. Cancer Res. 2013, 73, 4982–4987. [Google Scholar] [CrossRef]
- Morrison, K.M.; Miesegaes, G.R.; Lumpkin, E.A.; Maricich, S.M. Mammalian Merkel cells are descended from the epidermal lineage. Dev. Biol. 2009, 336, 76–83. [Google Scholar] [CrossRef]
- Ostrowski, S.M.; Wright, M.C.; Bolock, A.M.; Geng, X.; Maricich, S.M. Ectopic Atoh1 expression drives Merkel cell production in embryonic, postnatal and adult mouse epidermis. Development 2015, 142, 2533–2544. [Google Scholar] [CrossRef]
- Kervarrec, T.; Aljundi, M.; Appenzeller, S.; Samimi, M.; Maubec, E.; Cribier, B.; Deschamps, L.; Sarma, B.; Sarosi, E.-M.; Berthon, P.; et al. Polyomavirus-positive Merkel cell carcinoma derived from a Trichoblastoma suggests an epithelial origin of this Merkel cell carcinoma. J. Investig. Dermatol. 2020, 140, 976–985. [Google Scholar] [CrossRef]
- Dowlatshahi, M.; Huang, V.; Gehad, A.E.; Jiang, Y.; Calarese, A.; Teague, J.E.; Dorosario, A.A.; Cheng, J.; Nghiem, P.; Schanbacher, C.F.; et al. Tumor-specific T cells in human Merkel cell carcinomas: A possible role for Tregs and T-cell exhaustion in reducing T-cell responses. J. Investig. Dermatol. 2013, 133, 1879–1889. [Google Scholar] [CrossRef]
- Herber, R.; Liem, A.; Pitot, H.; Lambert, P. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J. Virol. 1996, 70, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Pitot, H.C.; Lambert, P. The human Papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J. Virol. 1999, 73, 5887–5893. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Experimental Group | Treatment | |||
|---|---|---|---|---|
| TPA Only | DMBA Only | DMBA+TPA | No Treatment | |
| ROSA26-LSL-MCPyV168 | 17 | 22 (18) | 23 (19) | 0 |
| K14Cre-MCPyV168 | 12 | 14 | 17 (16) | 28 (22) |
| K14E6/E7 | 13 | 10 | 22 (16) | 0 |
| Disease Grade | Experimental Groups | ||
|---|---|---|---|
| R26-LSL-MCPyV168 (n = 8 mice, n = 24 foci) | K14E6/E7 (n = 6 mice, n = 20 foci) | K14Cre-MCPyV168 (n = 7 mice, n = 26 foci) | |
| Dysplasia Grade 1 | 12 | 6 | 14 |
| Dysplasia Grade 2 | 4 | 6 | 4 |
| Dysplasia Grade 3 | 0 | 2 | 2 |
| SCC Grade 1 | 8 | 4 | 6 |
| SCC Grade 2 | 0 | 1 | 0 |
| SCC Grade 3 | 0 | 1 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spurgeon, M.E.; Liem, A.; Buehler, D.; Cheng, J.; DeCaprio, J.A.; Lambert, P.F. The Merkel Cell Polyomavirus T Antigens Function as Tumor Promoters in Murine Skin. Cancers 2021, 13, 222. https://doi.org/10.3390/cancers13020222
Spurgeon ME, Liem A, Buehler D, Cheng J, DeCaprio JA, Lambert PF. The Merkel Cell Polyomavirus T Antigens Function as Tumor Promoters in Murine Skin. Cancers. 2021; 13(2):222. https://doi.org/10.3390/cancers13020222
Chicago/Turabian StyleSpurgeon, Megan E., Amy Liem, Darya Buehler, Jingwei Cheng, James A. DeCaprio, and Paul F. Lambert. 2021. "The Merkel Cell Polyomavirus T Antigens Function as Tumor Promoters in Murine Skin" Cancers 13, no. 2: 222. https://doi.org/10.3390/cancers13020222
APA StyleSpurgeon, M. E., Liem, A., Buehler, D., Cheng, J., DeCaprio, J. A., & Lambert, P. F. (2021). The Merkel Cell Polyomavirus T Antigens Function as Tumor Promoters in Murine Skin. Cancers, 13(2), 222. https://doi.org/10.3390/cancers13020222