Simple Summary
Cisplatin is a widely used chemotherapy drug, but its use and efficacy are limited by its nephrotoxicity. HIF has protective effects against kidney injury during cisplatin chemotherapy, but it may attenuate the anti-cancer effect of cisplatin. In this review, we describe the role and regulation of HIF in cisplatin-induced nephrotoxicity and highlight the therapeutic potential of targeting HIF in chemotherapy.
Abstract
Cisplatin is a highly effective, broad-spectrum chemotherapeutic drug, yet its clinical use and efficacy are limited by its side effects. Particularly, cancer patients receiving cisplatin chemotherapy have high incidence of kidney problems. Hypoxia-inducible factor (HIF) is the “master” transcription factor that is induced under hypoxia to trans-activate various genes for adaptation to the low oxygen condition. Numerous studies have reported that HIF activation protects against AKI and promotes kidney recovery in experimental models of cisplatin-induced acute kidney injury (AKI). In contrast, little is known about the effects of HIF on chronic kidney problems following cisplatin chemotherapy. Prolyl hydroxylase (PHD) inhibitors are potent HIF inducers that recently entered clinical use. By inducing HIF, PHD inhibitors may protect kidneys during cisplatin chemotherapy. However, HIF activation by PHD inhibitors may reduce the anti-cancer effect of cisplatin in tumors. Future studies should test PHD inhibitors in tumor-bearing animal models to verify their effects in kidneys and tumors.
1. Introduction
Cisplatin is an effective and broad-spectrum chemotherapeutic agent for various kind of tumors, such as testicular, ovarian, head and neck, bladder, lung, breast, and cervical cancer [1,2]. Though being used worldwide, the therapeutic efficacy of cisplatin is limited, to some extent, by its side effects in normal tissues including ototoxicity, neurotoxicity, and nephrotoxicity [3]. Among them, cisplatin-induced kidney injury or nephrotoxicity is life-threatening and has attracted great attention with numerous studies focusing on its underlying mechanisms and potential therapeutic strategies [4,5]. Cisplatin can induce both acute kidney injury (AKI) [5,6] and the chronic kidney diseases (CKD) following AKI [7,8,9].
Hypoxia is a common factor involved in the development of renal pathology in both AKI and CKD [10]. Hypoxia-inducible factor(HIF), firstly discovered in the studies of erythropoietin(EPO), is generally considered as the core regulator for maintaining oxygen homeostasis due to its crucial role in cellular sensing and adaption to hypoxia [11,12,13]. As a transcription factor, HIF binds to DNA in a sequence-specific manner to promote or repress the transcription of multiple genes. HIF is widely involved in various biological processes, such as oxygen sensing, angiogenesis, vasodilation, erythropoiesis, metabolism, inflammation, and cell-cycle regulation [14,15]. Several studies have reported that HIF may participate in cisplatin-induced nephrotoxicity. However, most of these studies focus on cisplatin-induced AKI and find that HIF activation may protect against tubular cell injury and promote kidney recovery [16,17,18,19]. The lack of information of HIF in AKI to CKD progression and chronic kidney problems following cisplatin exposure make it an urgent need to study the regulation of HIF in these conditions.
Hypoxia is a known condition in solid tumors due to abnormal cancer cellular proliferation, expansion of tumor size and disruption of angiogenesis. As such, HIF is activated in tumors for cellular adaptation, and studies have found that inhibition of HIF may be an anti-cancer strategy under some circumstances [20]. In cisplatin chemotherapy, the activation of HIF for kidney protection may therefore antagonize the anti-tumor effects. Thus, when considering HIF activation as a kidney-protective strategy in cisplatin chemotherapy, the effect on cisplatin’s anti-tumor efficiency has to be taken into consideration.
In this review, we briefly introduce the major characteristics of HIF and cisplatin-induced nephrotoxicity, summarize the studies investigating how HIF participates in the renal pathological process after cisplatin exposure, and propose future research directions pertaining to HIF in cisplatin-induced AKI and CKD. We especially discuss the possible therapeutic strategies by activating or targeting HIF for cisplatin-induced nephrotoxicity as well as their influences on the anti-cancer effect of cisplatin.
2. Characteristics of HIF
2.1. Structure of HIF
HIF is a protein heterodimer complex of two subunits, HIF-α and HIF-β [12,21]. While HIF-α expression is oxygen sensitive and induced by hypoxia, HIF-β is constitutively expressed. HIF-α has three isoforms, ie. HIF-1α, HIF-2α and HIF-3α. HIF-1α is widely expressed in various cell types, while HIF-2α is mainly found in some specific cell types, such as vascular endothelial cells, renal interstitial cells, type II pneumocytes, liver parenchymal cells, and cells of the myeloid lineage [22,23,24]. HIF-3α is mainly found in the thymus, cerebellar Purkinje cells, and corneal epithelium of the eye [25,26,27]. HIF-β is stably expressed in an oxygen-independent manner in most cell types [13,28]. The HIF-α subunit binds to HIF-β to form the corresponding members of the HIF family. Although both known as essential regulators of hypoxia-induced transcriptional activities, HIF-1 and HIF-2 may have some differences in their roles. While HIF-1 is mostly reported to maintain the initial adaptation to hypoxia, HIF-2 mainly works under the long-term hypoxic conditions [29]. HIF-3α has various splice forms, which may either negatively or positively regulate the transcription activities of HIF-1 and HIF-2, and the function of these splices remains largely unclear [26,27,30].
Both HIF-α and HIF-β share the basic helix-loop-helix-PER-ARNT-SIM (bHLH/PAS) domains at their N-termini, by which the two subunits heterodimerize with each other and bind to specific DNA sequences [31]. All isoforms of HIF-α and HIF-β contain the transactivation domain (TAD) at their C-termini for transcription regulation, whereas HIF-1α and HIF-2α also have N-terminal TAD (N-TAD) [32]. Notably, HIF-1α and HIF-2α contain an oxygen-dependent degradation domain (ODDD) that mediates oxygen-induced inactivation and degradation.
2.2. Regulation of HIF
As HIF-β is stably expressed, the regulation of HIF expression and function mainly depends upon HIF-α. HIF-α can be modulated at multiple levels including transcriptional, translational, and post-translational regulations.
At the transcriptional level, HIF-α mRNA expression may be induced by hypoxia or ischemia [13]. Nuclear factor kappa B (NF-κB) pathway is reported to regulate HIF-α mRNA transcription directly [33]. Yamaguchi et al. [34] further reported that NF-κB pathway regulates CCAAT/enhancer-binding protein δ(CEBPD), which may directly promote HIF-1α transcription by binding to its promoter in both acute and chronic hypoxic kidneys.
At the translational level, although the overall protein synthesis is suppressed during cellular hypoxia, the synthesis of HIF-1α and HIF-2α is not reduced [13]. Stimuli such as oncogene activation, tumor suppressor inactivation, growth factors, and varieties of cytokines may also promote HIF-α expression [35]. Growth factor heregulin has been reported to induce HIF-α translation to maintain the balance between oxygen consumption and delivery [36]. Recent studies have shown that microRNAs also contribute to the regulation of HIF-α expression [37,38,39].
Nonetheless, the most critical mechanism regulating HIF-α expression is the post-translational degradation (Figure 1). In the presence of oxygen, HIF-1α or HIF-2α are hydroxylated at the sites of two proline residues (P402/P564 and P405/P531 for HIF-1α and HIF-2α in human, respectively) in ODDD domain by prolyl hydroxylase (PHD). The hydroxylated HIF-α is preferentially recognized by the Von Hippel-Lindau tumor suppressor protein (pVHL) [40]. pVHL binds to Elongin C and then forms the VCB-Cul2 E3 ligase complex that is composed of pVHL and elongin C, elongin B, cullin-2, and Ring-Box 1 (Rbx1). Thereby, HIF-α is ubiquitinated by the VCB-Cul2 E3 ligase complex and is targeted to proteasome for degradation [41,42]. Besides, the existence of oxygen also enables the enzymatic activity of factor inhibiting HIF-1 (FIH). FIH hydroxylates an asparagine residue (N803 in huamn HIF-1α) in the C-terminal of HIF-α and prevents the recruitment and binding of HIF-α coactivator p300 and CREB-binding protein (CBP) [43]. In hypoxia, the lack of oxygen prevents the hydroxylation response of PHD and, as a result, HIF-α is not ubiquitinated for degradation, leading to the accumulation of HIF-α protein. In this regard, PHD inhibitors have received intensive interests for their potent ability of inducing HIF [44,45]. Several oxygen-independent mechanisms for HIF stabilization are also involved in the regulation of HIF-α. Receptor for activated C kinase 1 (RACK1) was reported to competitively bind HIF-1α against heat shock protein 90 (HSP90) to induce oxygen-independent degradation of HIF-1α [46]. In addition, Garth Powis et al. [47] reported that hypoxia-associated factor(HAF) mediates HIF-1α degradation independent of pVHL or oxygen. Pharmacological depletion of Fe2+ also induces HIF because Fe2+ is a cofactor of PHD and FIH [35].
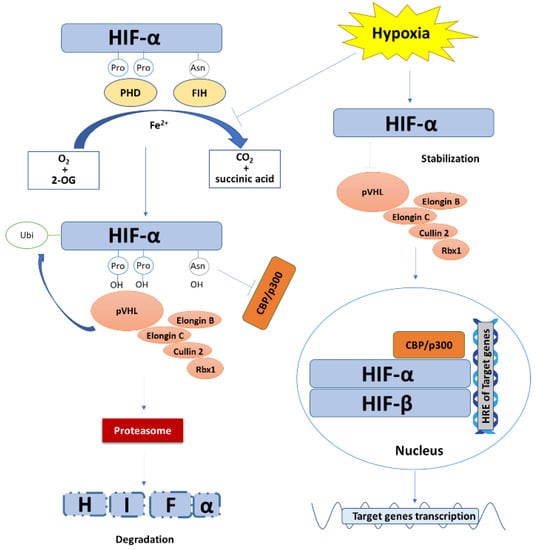
Figure 1.
The degradation, stabilization, and transcriptional function of HIF. Under normoxic condition, with Fe2+ as a cofactor and oxygen and 2-oxoglutaric acid (2-OG) as substrates, PHD and FIH hydroxylate HIF-α at the site of two proline residues and an asparagine residue in ODDD, respectively, accompanied by the generation of carbon dioxide and succinic acid as by-products. The prolyl hydroxylated HIF-α is preferentially recognized and ubiquitinated by VCB-Cul2 E3 ligase complex and is then targeted to proteasome for degradation. Meanwhile, hydroxylation of the asparagine residue of HIF-α prevents its recruitment and binding to coactivator p300 and CBP. Under hypoxic stress, the hydroxylation reaction of PHD and FIH is inhibited. The VCB-Cul2 E3 ligase complexes are not able to recognize and ubiquitinate HIF-α, leading to the stabilization and accumulation of HIF-α. HIF-α then translocates into the nucleus, heterodimerizes with HIF-β, and recruits coactivator p300 and CBP to specifically bind to the hypoxia response elements (HRE) of target genes, resulting in gene transcription.
2.3. Functions of HIF
The major function of HIF depends on its transcriptional activity (Figure 1). HIF-α translocates into the nuclei and binds to HIF-β via the bHLH/PAS domains to form the HIF heterodimer. The heterodimerized HIF recruits coactivator p300 and CBP and specifically binds to the HRE [5′-RCGTG-3′ (R = A or G)] in the target gene promotor to regulate the transcription of a wide range of genes [13,35,40,48].
As a transcription factor, HIF regulates the expression of approximately 400 target genes in various biological processes [13,49]. Especially, HIF is a critical regulator in maintaining oxygen homeostasis and has a close relationship with the reactive oxygen species (ROS) signaling pathway, protecting organs and tissues against oxidative damages [50,51]. Under hypoxia, ROS can either promote HIF-1α transcription [52] or stabilize HIF-1α protein [53]. Conversely, HIF-1 activation may alleviate ROS production. Overexpression of HIF-1α was shown to protect against 1,4- benzoquinone-induced apoptosis by reducing ROS level in K562 cells [54]. Jiang et al. showed that HIF-1α stabilization by dihydrotanshinone I (DI) pretreatment has a cardio-protective effect after ischemic injury by inducing nuclear factor E2-related factor 2 (Nrf2) expression and consequent activation of antioxidant enzymes to reduce ROS [55].
In addition, HIF may affect autophagy through its regulation of BCL2 Interacting Protein 3 (BNIP3)-related pathway [56,57]. BNIP3 or BCL2 Interacting Protein 3 Like (BNIP3L) is transcriptionally upregulated by HIF during hypoxia. The induced BNIP3/BNIP3L then competes with B-cell lymphoma 2 (Bcl2) and B-cell lymphoma-extra large (BCL-xL) to bind with Beclin1. The dissociation of Beclin1 from Bcl2 or Bcl-xl may promote the activation of autophagy.
Under hypoxic conditions, HIF also promotes angiogenesis by up-regulating the genes encoding angiogenic growth factors and cytokines, which recruit and regulate bone marrow-derived angiogenic cells (BMDACs) [58]. Transgenic mice overexpressing HIF-1α have an increased level of vascular endothelial growth factor (VEGF), a key mediator of angiogenesis in normal and pathological conditions, and exhibit significant induction of hypervascularity [59]. In this regard, Rankin et al. [60] demonstrated that Hif-1α deficiency has little effect on VEGF expression, while HIF-2α activation is demonstrated to induce VEGF. Warnecke et al. [61] reported that both HIF-1α and HIF-2α may contribute to VEGF activation, although HIF-1α is significantly more effective.
EPO, a potent growth factor for erythropoiesis, is reported to be preferentially regulated by HIF-2α [61,62]. Under hypoxia conditions, HIF enhances EPO synthesis by promoting its transcription via direct binding to the HRE of EPO gene, which is coordinated with iron metabolism. HIF also regulates the bone marrow microenvironment to promote the maturation and proliferation of erythroid progenitor. Thus, HIF promotes erythropoiesis at multiple levels, and the therapeutic strategies of activating HIF are effective in the treatment of anemia [63].
HIF is also closely related to immune responses and inflammation induction. Under hypoxic stress, activated HIF plays an important role in regulating the survival and differentiation of immune cells and in controlling the expression of pro-inflammatory factors [28]. HIF-1α-dependent NF-κB activity is directly involved in the regulation of neutrophil survival under hypoxic stress [64]. Cramer et al. [65] reported that HIF-1α is crucial for energy metabolism and function of myeloid cell. Inactivation of HIF-1α markedly suppresses the motility, invasiveness, aggregation, and anti-bacterial efficiency of macrophages, while stabilization of HIF-1α by VHL inhibition causes significantly enhanced inflammation. Also, HIF-1α induction by hypoxia or lipopolysaccharide (LPS) stimuli promotes glycolysis in dendritic cell (DC), which results in DC maturation for innate immune response activation [66]. Meanwhile, LPS-induced HIF-1α can contribute to the development of sepsis by the induction of multiple inflammatory cytokines [67]. In addition, Kojima et al. [68] reported an essential role of HIF-1α in B cell development where HIF-1α deficiency leads to abnormal peritoneal B-1 cells, aberrant maturation of B-2 cells, and autoimmunity. HIF-1α was also reported to participate in myeloid-derived suppressor cells (MDSC)-mediated T cell activation via the transcriptional up-regulation of programmed death ligand 1 (PD-L1) [69].
In tumorigenesis, HIF-1 participates in glucose metabolism, cancer cell survival and proliferation, angiogenesis, as well as metastasis [70]. HIF-1 activation in tumor cells up-regulates the expression of some glycolytic enzymes and transporters, including 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), M2 isoform of pyruvate kinase (PKM2), and glucose transporter 1 and 3 (GLUT1, GLUT3) [71]. PFKFB3 accumulation has been found to protect against cisplatin-induced apoptosis via enhanced glycolysis when accumulated in cells [72]. Emerging evidence has identified GLUT1 and GLUT3 as two major contributors to the accelerated glucose metabolism in tumor cells. Up-regulated GLUT1 and GLUT3 expression is associated with tumor resistance during anti-cancer therapy [73]. In addition, Azoitei et al. [74] reported that PKM2 accumulates in hypoxic pancreatic tumors to promote HIF-1α-induced VEGF secretion via the NF-κB pathway, leading to enhanced angiogenesis. Overall, HIF plays a crucial role in tumor maintenance, progression, and treatment resistance. Thus, targeting HIF-1 has become a therapeutic strategy for tumor treatment [75].
In addition to the transcriptional regulation of gene expression, emerging evidence indicates that HIF can also function independently of its transcriptional activity. In various tumor cells, even without DNA binding domain, HIF-1α can promote the ubiquitination of Dicer to facilitate its degradation by autophagy-lysosomal pathway. The downregulation of Dicer by HIF leads to the decrease of multiple tumor suppressive microRNAs and enhanced tumor metastasis [76]. In acute myeloid leukemia, HIF-1α can stimulate cell differentiation without HIF-1β, implicating its non-transactivation function [77]. Hubbi et al. [78] have shown that HIF-1α can function as a inhibitor of DNA replication, which is independent of its transcriptional activity. Under hypoxic condition, HIF-1α directly binds to cell division cycle 6 (Cdc6) and promotes its interaction with the minichromosome maintenance complex, leading to suppressed firing of replication origins and cell cycle arrest. HIF-1α was also reported to induce cell cycle arrest via the activation of p21 by displacing cellular Myc (c-Myc) and preventing its inhibitory effect on p21 expression [79]. In addition, HIF-1α may translocate to mitochondria under oxidative stresses to suppress ROS generation, mitochondrial membrane potential loss, mitochondrial DNA-encoded gene expression, and cell death [80]. The mitochondrially translocated HIF-1α is associated with the outer membrane, indicating its non-transactivation function.
3. Cisplatin-Induced Nephrotoxicity
Cisplatin-induced nephrotoxicity, firstly reported in the clinical trials of cisplatin chemotherapy, is one of the major side effects that limit cisplatin’s use and therapeutic efficacy [5]. Patients with solid tumors and malignant lymphomas who received cisplatin treatment have shown significant kidney injury [81,82]. A retrospective cohort study has shown that in intravenous cisplatin-treated patients with head and neck cancer, esophageal cancer, or gastric cancer, 41 out of 182 patients develop cisplatin nephrotoxicity during the initial administration. 71 cases have been observed in the total 442 cycles, with 8 patients developing multiple episodes of nephrotoxicity [83]. Other studies have shown nephrotoxicity development during cisplatin treatment of different cancers including ovarian cancer [84,85], hepatocellular carcinoma [86], lung cancer [87], colon cancer [88], breast cancer [89], and others [90]. Though various clinical manifestations of cisplatin induced-nephrotoxicity have been observed, in this review we mainly focus on cisplatin-induced AKI and CKD.
3.1. Cisplatin-Induced AKI
Among the multiple renal manifestations induced by cisplatin, AKI occurs in 20–40% of cancer patients following cisplatin treatment [4]. In days after cisplatin exposure, these patients show decreased glomerular filtration rate (GFR), increased level of blood urea nitrogen (BUN) and serum creatine, as well as disruption of fluid and electrolyte homeostasis [91]. The mechanism of cisplatin-induced AKI is a very complex process, which is the focus of our discussion in the following sections.
3.1.1. Cisplatin Uptake and Metabolism
The uptake and accumulation of cisplatin in proximal tubules, especially in the S3 segment, causes massive renal tubular damage [92]. Cisplatin enters the cells not only by passive diffusion through cellular membrane, but also by active transportation [91,93]. Two membrane transporters, the copper transport protein (Ctr1) and the organic cation transporter 2(OCT2), contribute to the uptake of cisplatin in tubular cells [94,95]. Once cisplatin enters kidney tubular cells, it goes through a series of bio-activation processes to generate various toxic metabolites. For example, γ-glutamyl transpeptidase (GGT) and cysteine-S-conjugate β-lyase may catalyze the reactions to generate glutathione-cisplatin conjugate, cysteinyl-glycine-conjugate, cysteine-conjugate, and reactive thiol. Blocking either GGT or cysteine-S-conjugate β-lyase results in reduced cisplatin-induced nephrotoxicity [96,97].
3.1.2. Cisplatin Induced Cell Death
Cisplatin triggers the activation of multiple signaling pathways, which eventually results in cellular injury and death. The most well-known toxic effect of cisplatin is DNA damage [5,6]. After binding to DNA with high affinity, cisplatin causes cross-links between and inside the strands of DNA, and impedes DNA synthesis and replication, thereby leading to cell-cycle arrest and DNA damage response [98,99]. Cisplatin induces several types of renal tubular cell death, such as necroptosis, ferroptosis, and apoptosis.
Necroptosis, a programmed form of necrosis, is one of the major contributors of proximal tubular cell death in cisplatin-induced AKI. Cisplatin stimulates receptor-interacting protein 1 (RIP1)/receptor-interacting protein 1 (RIP3)/mixed lineage kinase domain-like protein (MLKL)-dependent necroptosis of tubular cells and induces pro-inflammatory cytokines release, and the latter is capable to further promote necroptosis [100]. Pharmacological [100,101,102] or genetic [100,103] inhibition of the RIP1/RIP3/MLKL pathway can ameliorate tubular cell damage, inflammatory responses, and cisplatin-induced AKI. Other strategies that alleviate necroptosis have also shown a protective effect on cisplatin induced AKI [104,105].
Ferroptosis is an iron-dependent form of nonapoptotic cell death characterized by the iron-catalyzed accumulation of lethal lipid ROS and differs from apoptosis, necrosis, and autophagy in morphological, biochemical, and genetic features [106]. Ferroptosis also plays a role in the pathogenesis of cisplatin-induced AKI and the inhibition of ferroptosis reduces cisplatin-induced AKI [107]. Lu et al. [108] have shown that Ras homolog enriched in brain (Rheb1) deletion in renal tubules promotes cisplatin-induced AKI partially through enhanced ferroptosis. Ferroptosis, featured by lipid peroxidation, can be attenuated by vitamin D receptor (VDR) activation, which also ameliorates cisplatin-induced AKI [109]. Similarly, other anti-ferroptosis agents that prevent lipid peroxidation and alleviate ferroptosis improve renal function, diminish renal tubular damage, and attenuate cisplatin-induced AKI [110].
Additionally, cisplatin has been well-known to activate various pathways of apoptosis to cause renal damage. Cisplatin evokes the extrinsic apoptotic pathway of death receptor by mediators including Fas and tumor necrosis factor α (TNF-α) [91,111,112]. Cisplatin also induces mitochondrial dysfunction to cause intrinsic apoptosis through caspase-9 activation [5,113,114]. In addition, cisplatin also promotes apoptosis by elevated caspase-12 activation via the endoplasmic reticulum (ER) stress pathway [115]. Apoptosis is a critical target for treatment of cisplatin-induced AKI due to its essential role in tubular cell death. Studies have reported that attenuated apoptosis is beneficial to cisplatin-induced AKI [90]. Kong et al. [116] has reported that EPO protects against cisplatin-induced AKI in rats by attenuating tubular cell apoptosis. Yang et al. [117] have reported that inhibition of apoptosis partially contributes to the reno-protective effect of Asiatic acid. However, although the pan caspase inhibitor zVAD-fmk has shown a reno-protective role against ischemic AKI mice by inhibiting apoptosis [118], it increases cisplatin-induced necrotic cell death and impairs autophagic flux, leading to aggravated kidney dysfunction in cisplatin-induced AKI [119].
3.1.3. Other Mechanisms
The tumor suppressor p53 plays an essential role in cisplatin-induced cell death, due to its central role in DNA damage response, apoptosis induction, cell cycle arrest, and oncogene inactivation [91,99,120,121]. Autophagy is a cytoprotective mechanism in cisplatin-induced AKI, and the inhibition of autophagy worsens AKI [122,123]. Inflammation also contributes significantly to cisplatin-induced nephrotoxicity. By activating Toll like receptors (TLRs) via the secretion of damage-associated molecular pattern molecules (DAMPs), cisplatin induces the release of a broad range of chemokines and cytokines, which leads to the infiltration of inflammatory cells and increased inflammatory responses in kidney tissues [5,90,91,124]. Oxidative stress has been considered to be another important injurious factor in cisplatin-induced nephrotoxicity. Through the depletion of antioxidants (e.g., glutathione), defective mitochondrial respiratory chain, and cytochrome P450 effects in microsomes, cisplatin induces cellular ROS generation, which may further trigger apoptosis and promote inflammatory responses [5,91,125]. In addition, endothelial dysfunction and vascular injury may be part of the side effects of cisplatin, leading to insufficient renal blood flow and hypoxic condition in kidney [126,127]. Accumulating evidence has demonstrated that the kidney tubules are vulnerable to hypoxic injury, due to their low oxygen tensions resulted from the unique renal vascular architecture, low capacity of anaerobic energy generation, as well as high consumption of oxygen required for cellular activities [128]. Hypoxia in kidney after cisplatin injury may further augment tubular injury. Moreover, epigenetic alterations including DNA methylation, histone modification, and noncoding RNAs are involved in acute kidney dysfunction after cisplatin injury [90,129].
3.2. Cisplatin Induced CKD
AKI patients are commonly observed to suffer the loss of kidney function, which may be transient [92]. However, even after initial recovery, AKI is an important risk factor for the development of CKD [130]. Although a single high dose of cisplatin mainly induces AKI, it may also have long term effects on kidney and act as a high-risk factor for CKD development [7]. Emerging evidence indicates that over 50% of patients receiving cisplatin chemotherapy may suffer a CKD progression [131]. In addition, in clinical practice, cancer patients often receive relatively low doses of cisplatin for several cycles. Recent studies have begun to investigate the chronic effect of repeated low-dose cisplatin treatment on kidney [9,132,133,134,135,136,137,138]. Sharp et al. [133] have reported that when compared to the AKI model, repeated low dose of cisplatin significantly enhances inflammatory cytokines and chemokines but has a lower level of cell death. Meanwhile, elevated expression of fibrotic markers and interstitial fibrosis occur. Long-term observation for 6 months reveals that the mice in the repeated dosing model have glomerular pathologies and persistent endothelial dysfunction [137]. The development of CKD may have some variations between different animal strains [138]. Similarly, Torres et al. [134] have reported that mice receiving two doses of cisplatin with a 2-week interval between injections followed by long-term observation show significant body weight decline and loss of renal function, as well as other CKD features including endothelial rarefaction, myofibroblast proliferation, and macrophage infiltration. Notably, “atubular” glomeruli have been observed and considered to be associated with degenerating tubules. Similar results have been reported in a few other in vivo and in vitro studies [9,135]. Although the severity of impairment in these studies may vary due to the different doses of cisplatin, these evidences indicate that repeated low-dose cisplatin treatment indeed leads to CKD progression, which is characterized by declined renal function, reduced kidney weight, chronic interstitial inflammation and fibrosis, endothelial dysfunction, as well as morphological changes such as atubular glomeruli.
According to the recent studies, CKD development is mainly due to the maladaptive repair after kidney injury. Kidney has the capacity of repairing damaged tubules. Depending on the severity of initial injury, there are two categories of kidney repair, normal complete repair and maladaptive incomplete repair [139]. When the injury is mild to moderate, kidney may undergo complete or adaptive repair and the injured tubular epithelia can proliferate and regenerate to restore the normal structure and function of kidney [139,140]. However, when the injury is severe or prolonged, the kidney may go through incomplete or maladaptive repair. Under this circumstance, CKD progression is associated with impaired tubular regeneration, endothelial dysfunction and prolonged hypoxia, persistent inflammation, interstitial fibrosis, epigenetic alteration, activated renin-angiotensin-aldosterone-system (RAAS), mitochondrial dysfunction, and cellular senescence [141,142,143,144]. Some of these changes have been reported in cisplatin-induced CKD [9,135,145,146,147,148].
4. HIF in Cisplatin-Induced AKI
Over the past few decades, several studies demonstrated the evidence for a reno-protective role of HIF in cisplatin-induced AKI [17]. These studies are summarized in Table 1.

Table 1.
Summary of studies about the role of HIF in cisplatin-induced AKI.
HIF induction in cisplatin-induced AKI has been reported by several studies though whether cisplatin can directly activate HIF is still controversial. Xu et al. [152] have reported the increases in renal HIF-1α mRNA in rats after cisplatin injection in a dose- and time-dependent manner. Meanwhile, multiple well-known HIF-targeted genes such as VEGF and Heme oxygenase-1 (HO-1) are also induced, indicating activation of the HIF pathway. Using “HIF-sensing” transgenic mice to reveal HIF activation, Tanaka et al. [16] have found increased activation of HIF-1 in renal tubules (mainly the proximal tubular cells in the outer medulla) and HIF-2 activation in peritubular area in rats after cisplatin injection. However, no HIF activation is observed in cultured proximal cell line of rat after cisplatin treatment [16]. Weidemann et al. failed to detect HIF activation either in vivo or in vitro [17]. Instead, cisplatin treatment may impair HIF activation as the downstream GLUT1 mRNA transcription is reduced in HKC-8 cells treated with cisplatin [17]. Thus, it is possible that in in vitro conditions, the existence of normal oxygen (21% O2 level usually used for cell cultivation) may prevent HIF activation with cisplatin injury. However, in in vivo conditions, cisplatin-induced vascular damage and renal blood flow reduction may break the balance between low delivery and high consumption of oxygen in renal tubules, leading to severe local hypoxia [127]. Though with some variations in different in vivo models, HIF is likely activated due to this local hypoxia after cisplatin injury.
Consistently, the protective effect of HIF activation has been reported in several studies. Tanaka et al. have shown that cobalt (HIF-1α inducer) attenuates cisplatin-induced AKI in rats. The protective role of HIF-1 has been further confirmed in vitro by HIF-1α siRNA or dominant negative HIF-1α expression [16]. Although HIF activation was not detected after cisplatin treatment by Weidemann et al., hypoxic preconditioning to induce HIF activation protected both cultured renal cells and kidneys in their study [17]. Their in vitro experiments indicated that the protection by hypoxic preconditioning is HIF-dependent as HIF-1α knockout abolishes the protective effect [17]. The reno-protection by HIF activation in cisplatin AKI models has been further confirmed in other studies using PHD inhibitor FG-4592, iron chelator deferiprone, HIf-1α transfected human adipose-derived stem cells, or PHD1 knockout [18,19,149,150,151]. Interestingly, cisplatin induced less apoptosis in hypoxic kidney cells than in normoxic cells, but this protective effect of hypoxia may not be directly related to HIF [17,153]. Our study indicates that this effect of hypoxia is related to P53 activation [153]. Overall, the in vivo and in vitro evidence implicates that HIF up-regulation can be an effective therapeutic strategy for preventing cisplatin-induced AKI.
Currently, the underlying mechanism for the reno-protection of HIF in cisplatin induced nephrotoxicity has been explored but not completely understood (Figure 2). In most of the studies, the reduction of renal tubular apoptosis has been reported. The anti-apoptotic effect of HIF-1 activation is accompanied with increased expression of BCL-2 family proteins (MCl1, survivin) [19]. In addition, HIF activation is also associated with anti-oxidative effects, showing less lipid peroxidation [149], which may be related to ferroptosis inhibition. Furthermore, HIF activation may protect endothelial cells because angiogenesis-related genes such as VEGF may be induced [150,151]. Finally, the anti-inflammation effect of HIF activation may also contribute to its reno-protection. HIF upregulation by PHD inhibitor or HIF-1α overexpression can significantly reduce multiple cytokine release after cisplatin injury, which can lead to less inflammatory cell infiltration and reduced renal injury [18,151].
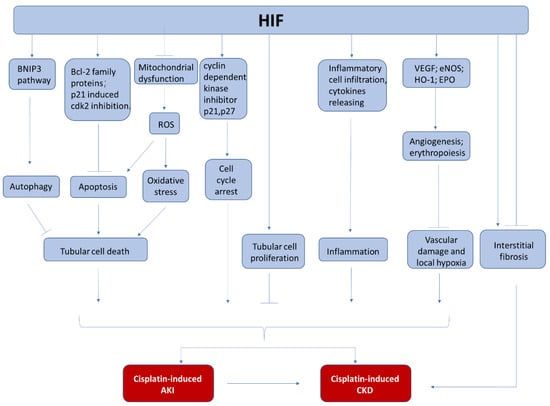
Figure 2.
The involvement of HIF in cisplatin-induced nephrotoxicity. During cisplatin chemotherapy, HIF may protect kidneys from AKI and CKD by the inhibition of tubular cell death, the regulation of cell proliferation, the suppression of kidney inflammation, and the attenuation of vascular damage.
Finally, HIF also interacts with other pathways that are involved in the development and progress of cisplatin-induced AKI. Goda et al. [154] have found that hypoxia upregulates p27 and p21 in a HIF-dependent manner and the enhanced p27 expression ultimately prevents retinoblastoma protein (Rb) hyperphosphorylation, making HIF a crucial determinant of hypoxia-induced cell cycle arrest. Thus, HIF may participate in regulating cell cycle arrest in cisplatin-induced nephrotoxicity. Besides, p21 is proved to prevent cell apoptosis in cisplatin-induced AKI by directly inhibiting cyclin-dependent kinase 2 (cdk2), which may be another pathway for HIF to exert its protection [155,156]. In addition, HIF has been reported to activate autophagy through BNIP3 pathway in other systems [56,157], and autophagy has been shown to protect kidney from cisplatin injury [158,159,160,161,162], although it is unclear about the HIF/BNIP3 pathway regulation in cisplatin-induced AKI.
In conclusion, current evidence indicates that HIF activation is beneficial to the kidney during cisplatin chemotherapy. Further investigation on HIF-1 and its interaction with other cellular process may reveal deeper understanding and novel therapeutic targets of cisplatin-induced AKI.
5. HIF in Cisplatin-Induced AKI to CKD Progression
Numerous evidences have demonstrated that tubulointerstitial hypoxia is not only an essential contributor to AKI but also a key player in CKD. Chronic hypoxia is considered as a common pathological condition in CKD [10,163]. Though studies have revealed that multiple pathophysiological mechanisms are related to cisplatin-induced AKI to CKD progression [9,135,145,146,147,148], the investigation focusing on the role of HIF in this field is lacking. Nevertheless, HIF is a potential player in this process because of its essential role in hypoxia and its interactions with multiple pathophysiological procedures involved in the progression of CKD (Figure 2).
It is well-known that HIF-1α plays a crucial role in kidney repair in various types of CKD [10]. After kidney injury, the surviving epithelial tubular cells, which are commonly considered to be responsible for tubular regeneration, go through dedifferentiation, proliferation, and re-differentiation to replace the lost neighboring cells [164]. The disruption of balance between tubular cell death and proliferation may result in maladaptive repair and lead to CKD progression [10]. In cisplatin-induced AKI, HIF-1α affects tubular cell apoptosis via the regulation of Bcl-2 family members [19] and mitochondrial function [16]. Meanwhile, HIF also regulates tubular cell proliferation through different downstream pathways, either positively or negatively [10]. Although HIF activation is mainly to alleviate epithelial tubular cell death and promote cell proliferation and renal recovery after cisplatin-induced AKI, its effect on tubular cell repair in cisplatin-induced AKI to CKD progression remains unclear and warrants further investigation.
Oxidative stress has been shown to be a major injurious factor during CKD development. DNA damage by oxidative stress and the associated DNA damage response may prevent the cell cycle progression during repair and induce G2/M arrest [147] and senescence [148]. Chemical treatment by semicarbazide sensitive amine oxidase inhibitor (PXS-4728A) [135], superoxide dismutase (SOD) mimetic (GC4419) [146], or general antioxidants like N-acetylcysteine (NAC) [148] to suppress oxidative stress significantly reduces cisplatin-induced CKD progression. Considering the antioxidative function of HIF in cisplatin-induced AKI [149], it is possible that HIF may benefit the tubular repair and regeneration to alleviate CKD progression.
Yamaguchi et al. [34] discovered CEBPD as a novel regulator of HIF-1 in acute model of cisplatin-induced nephrotoxicity and in models of other acute and chronic hypoxic kidney injury. CEBPD is not only a transcription factor that transactivates HIF, but is also induced by inflammatory signals like interleukin 1β (IL–1β) via the NF-κB pathway. Thus, CEBPD can act as a mediator by which inflammation affects HIF-1 in both acute and chronic kidney diseases [34]. Conversely, HIF has a role in inflammatory infiltration and responses [28,65,165]. Inflammation is a well-known feature of maladaptive repair in the process of CKD [10]. These evidences suggest a cross talk between inflammation and HIF pathways. Emerging evidence indicates that repeated low-dose cisplatin treatment induces significant interstitial inflammation, which contributes to the progression of CKD [133,134,135,137]. Apparently, more work on HIF is needed to explore its exact role in chronic inflammation in cisplatin-induced AKI to CKD progression.
Renal interstitial fibrosis is a common final pathological feature of CKD. Studies have reported that repeated low-dose cisplatin administration leads to significant interstitial fibrosis, a sign of AKI to CKD progression [9,133,134,135]. Numerous studies have demonstrated that HIF, particularly HIF-1α, is a key regulator of renal interstitial fibrosis in the progression of CKD, though controversial opinions may exist on whether it promotes or suppresses renal fibrosis [166]. On one hand, HIF activation may attenuate fibrosis in various types of CKD models. Kapitsinou et al. [167] have showed pharmacological activation of HIF before ischemia/reperfusion injury (IRI) improves kidney fibrosis, while no beneficial effect is observed by post-injury activation. They have also reported that PHD inhibitors suppress interstitial inflammation and fibrosis by activating endothelial HIF-2 and promote renal recovery after kidney IRI [168]. Kobayashi H et al. [169] have shown that systemic HIF activation suppresses unilateral ureteral obstruction (UUO)-induced kidney inflammation and fibrosis in mice. Several other studies have also reported that pharmacological activation of HIF-1α alleviates interstitial fibrosis and improves kidney recovery in the rat remnant kidney (RK) model of CKD [170,171]. On the other hand, there are emerging evidences showing a profibrotic role of HIF activation. Kimura et al. [171] have found that stable tubular expression of HIF-1α promotes fibrosis in mouse RK model, while HIF1-α inhibition ameliorates UUO-associated renal fibrosis in mice. Liu et al. [172] have reported that HIF-1α transcriptionally upregulates p53 to promote G2/M cell cycle arrest and renal fibrosis in hypoxic tubular cells and in UUO mice. Consistently, genetic inhibition of HIF-1α shows attenuated fibrosis in angiotensin II-induced renal injury [173] and in chronic ischemic renal injury [174].The reason for the controversial effects of HIF activation on fibrosis remains unknown, but it may be related to the differential roles of HIF in different renal cell types or in different pathological stages (injury vs. repair). Further research focusing on HIF’s role in development of fibrosis in cisplatin-induced AKI to CKD progression may bring us deeper insight into the pathological mechanisms and potential intervening strategies.
Furthermore, HIF participates in the pathogenesis of CKD-related complications, such as anemia [175,176] and vascular calcification [177], which can affect the clinical outcome of CKD regardless of the initial causes of kidney damage. Recently, several clinical trials have confirmed the beneficial effects of PHD inhibitors in treating CKD-related anemia [175,176,177,178,179]. In CKD patients, the stabilization of HIF by PHD inhibitors can improve the EPO level and attenuate anemia, an observation leading to clinical use of PHD inhibitors. Recent studies also showed a new series of orally active PHD2 inhibitor, such as 15i [180] and 17 [179] can significantly improve cisplatin-induced anemia in mice without apparent toxicity. However, HIF induction has been reported to enhance vascular calcification, which may deteriorate CKD [177]
6. Therapeutic Potential of HIF in Cisplatin Chemotherapy
In view of the role of HIF in cisplatin-induced nephrotoxicity, targeting or activating HIF and its related pathways may be a therapeutic strategy. PHD-pVHL pathway plays a vital role in HIF regulation, and pharmacological or genetic inhibition of PHD activity is under intensive research in kidney diseases [18,45,167,168,181,182,183,184,185]. The role of regulating HIF in cisplatin-induced nephrotoxicity is summarized in Table 1; besides the evidence that HIF accumulation by PHD inhibitors treatment protects against cisplatin-induced nephrotoxicity [18], studies have reported beneficial effects of PHD inhibitors in other kidney diseases including ischemic AKI [167,168], diabetic nephropathy [182,183], obesity related kidney disease [181], chronic tubulointerstitial nephritis [184], and remnant kidneys models of CKD [186]. A great breakthrough in this field is that PHD inhibitors have been proved to have a therapeutic effect in anemia, a complication of CKD that contributes to poor clinical outcome [187]. These inhibitors block enzymatic activity of PHD to stabilize HIFs, whose accumulation leads to increased transcription and expression of EPO, followed by enhanced erythropoiesis to alleviate anemia in CKD patients [45,176]. Recently, Wu et al. [180] found that an orally active PHD2 inhibitor 15i can bring the hemoglobin in cisplatin-induced anemia mice to a normal level with no obvious toxicity observed. Zhang et al. [179] also discovered that another PHD2 inhibitor 17 can significantly improve cisplatin-induced anemia in mice with an excellent safety property. Compared with the conventional erythropoiesis-stimulating agents (ESAs), PHD inhibitors have less side effects and are more convenient to be administered orally [45]. To date, following successful clinical trials, several small-molecule PHD inhibitors, including roxadustat (FG-4592), daprodustat (GSK1278863), and vadadustat (AKB-6548), are being used clinically for the treatment of anemia in CKD patients [188]. Meanwhile, PHD inhibitors are reported to benefit cardiovascular disease in CKD patients through HIF, and HIF activation improves myocardial remodeling, atherosclerosis and vascular injury [189,190].
However, most of the current investigations focusing on HIF’s role in cisplatin induced nephrotoxicity were conducted in tumor-free models that only have cisplatin exposure to study HIF’s effect on the pathophysiological mechanisms, while in clinical settings, cisplatin chemotherapy is always given to patients with tumor burden. When considering therapeutic strategies of cisplatin-induced nephrotoxicity, attention should be paid to their effects on both reno-protection and tumor sensitivity to cisplatin. Amifosten, the FDA-approved agent for the prevention of cisplatin-induced AKI in patients with non-small cell lung cancer (NSCLC) and advanced ovarian cancer, reduces renal toxicities of repeated cisplatin administration, with no reduction in antitumor efficacy [191]. Consistent with the experimental evidence that magnesium supplement significantly improved cisplatin-induced AKI and reduced tumor growth in mice with tumor xenograft [84,88], clinical data showed it may have a role as a protectant in cisplatin-induced AKI [192]. Our previous work found that pharmacological or genetic inhibition of PKCδ enhanced the chemotherapeutic effects of cisplatin in several xenograft and syngeneic mouse tumor models, while protecting kidneys from nephrotoxicity [85]. Sánchez-González et al. [193] reported that in rat breast adenocarcinoma model, quercetin ameliorated cisplatin-induced nephrotoxicity without interfering with its tumor toxicity. In view of the above, HIF’s role in tumor sensitivity to cisplatin should also be considered seriously when HIF-regulating strategies are studied as combination treatment of cisplatin chemotherapy. Notably, although HIF activation may have protective effect on kidney during cisplatin chemotherapy, it may attenuate the anti-cancer effects of cisplatin in tumors [194]. Intratumoral hypoxia is a common feature of solid tumors where vascular oxygen delivery cannot meet the oxygen consumption of rapidly growing cancer cells [194]. Generally, intratumoral hypoxia, together with functional alterations of tumor suppressor genes (most importantly, VHL) and oncogenes, upregulates HIF activity, which regulate multiple pathological processes and eventually result in the resistance to chemotherapy, radiation therapy, and targeted therapy [195,196]. Studies have revealed HIF overexpression in multiple tumors [20,197] and found that HIF is closely related to tumor resistance to cisplatin [198,199,200,201,202]. Zhang et al. [200] reported that HIF-1α may act as a mediator of the interaction between p53 and RAS signaling to participate in the cisplatin resistance in ovarian cancer and that silencing HIF-1α significantly decreased the cellular sensitivity to cisplatin. Keremu et al. [201] found dramatically elevated HIF-1α expression in cisplatin-resistant osteosarcoma cells under hypoxia. The inhibition of HIF-1α by miR-199a overexpression can sensitize the response of cisplatin-resistant cells. Ai et al. [202] reported that overexpression of degradation-resistant HIF-1α suppresses cisplatin-induced apoptosis while genetic knockdown of HIF-1α or pharmacological promotion of HIF-1α degradation enhances the response to cisplatin in ovarian cancer cells. Kizaka-Kondoh et al. [203] have demonstrated that HIF-1 activity is related to the invasion and metastasis of pancreatic cancer and the selective killing of HIF-1-actived cells via apoptosis by POP33 leads to suppressed peritoneal dissemination of cancer cells and improved mouse survival. Liu et al. [204] found that Oroxylin A directly binds to the bHLH-PAS domain of HIF-1α to prevent hypoxia-induced xeroderma pigmentosum group C (XPC) transcription, attenuating the cisplatin resistance in NSCLC. Parmakhtiar et al. [205] revealed that topotecan-induced HIF inhibition restores cisplatin and paclitaxel sensitivity in ovarian cancer via enhanced p53-mediated apoptosis. More studies showing HIF’s involvment in cancers during cisplatin chemotherapy are summarized in Table 2. In general, inhibiting HIF has been shown as a powerful strategy to reinforce anti-cancer efficiency of chemotherapeutic strategies including cisplatin [194,195,196,206]. Thus, in cisplatin chemotherapy, activating HIF is a double-edged sword. The failure to balance its effects on cisplatin’s anti-tumor function and nephrotoxicity may limit its application in cisplatin-induced AKI or CKD. For example, Erez et al. [207] reported that the overexpression of mouse PHD1 in colon carcinoma cells suppresses tumor growth by destabilizing HIF-1α. Bordoli et al. [208] demonstrated that PHD2 inhibition promotes tumor growth and that human biopsies with low-PHD2 protein expression level correlates significantly with shorter surviving time of breast cancer patients. Therefore, the clinical application of PHD inhibitors to reduce the side-effect of cisplatin requires further investigation and evaluation. A possible strategy is to develop drug delivery methods that can specifically regulate HIF activity in the kidney without systemic influence. Potential strategies for restrictive renal delivery have been studied, such as protein-based and peptide-based carriers, water-soluble polymeric carriers, small-molecule prodrugs, and nanoparticles [209]. However, renal specific HIF-regulating drug delivery has not been reported in the treatment of cisplatin-induced nephrotoxicity yet.
Table 2.
Summary of studies about therapeutic potential of HIF in cisplatin chemotherapy.
Meanwhile, because PHD has 3 isoforms (PHD1, PHD2, PHD3) that have different expression patterns and HIF-α targeting specificity in different cell types [10], cell type restricted PHD isoform-specific inhibitory strategy should also be taken into consideration for HIF regulation in cisplatin chemotherapy. Several studies have reported that endothelial PHD2 inhibition induced HIF-2α activation. It not only inhibits the intravasation and metastasis of cancer cells by normalizing the endothelial lining of tumor, improving the tumor perfusion, and increasing the chemotherapeutic drug delivery without affecting drug level in normal organs, but also alleviates the oxidative response to protect against cisplatin-induced nephrotoxicity and doxorubicin-induced cardiotoxicity [149,219]. However, further research is still needed to verify and extend these observations, especially examination of the long-term effect on cisplatin-induced CKD progression.
In addition, HIF-targeted therapies may enhance the efficacy of immunotherapies. HIF inhibition by topotecan can enhance the anti-tumor activity of bevacizumab in U251-HRE glioblastoma xenografts [220]. Kheshtchin et al. [221] reported that the combination of DC-based vaccination and PX-478, an inhibitor of oxygen-sensitive HIF-1α, enhances T cell effector functions and leads to inhibited tumor growth and enhanced survival in breast cancer mouse model. However, a recent clinical trial for NSCLC using the combination of evofosfamide, a hypoxia-targeted pro-drug, and tarloxotinib, has been stopped early due to futility [222]. Overall, it remains possible that the combination of HIF targeting therapy with immunotherapies may be a more effective anti-tumor strategy to replace the toxic therapies such as cisplatin in the future.
In conclusion, when considering therapeutic strategies activating or targeting HIF in cisplatin-induced nephrotoxicity, more comprehensive and rigorous work is still needed to identify novel chemicals or drug delivery techniques to achieve the maximal reno-protective effect without diminishing the anti-tumor efficacy of cisplatin.
7. Conclusions
In this review, we summarized the recent findings about HIF and cisplatin-induced acute and chronic nephrotoxicity and discussed the function of HIF in the injury development. The critical role of HIF in cisplatin nephropathy and the availability of potential clinical treatment such as PHD inhibitors make it a promising candidate for future therapeutics. However, the beneficial effect of HIF especially for cisplatin induced CKD is still waiting to be clarified by future study.
Author Contributions
S.L. and Z.D. contributed to the conceptualization, design and outline of this review. S.L. prepared the original draft with figures. S.L., L.W., X.H., Q.W. and Z.D. contributed to revision and editing. All authors have read and agreed to the published version of the manuscript.
Funding
The authors were partly supported by the grants from China Scholarship Council, the National Institutes of Health of USA (DK058831, DK087843), and Department of Veterans Administration of USA (000319). Z.D. is a recipient of Senior Research Career Scientist award of Department of Veterans Administration of USA.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorgan. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Steyger, P.S. An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 2015, 237, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Hamroun, A.; Lenain, R.; Bigna, J.J.; Speyer, E.; Bui, L.; Chamley, P.; Pottier, N.; Cauffiez, C.; Dewaeles, E.; Dhalluin, X.; et al. Prevention of Cisplatin-Induced Acute Kidney Injury: A Systematic Review and Meta-Analysis. Drugs 2019, 79, 1567–1582. [Google Scholar] [CrossRef]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2018, 31, 15–25. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; McMillan, K.L.; Wu, J.; Gillings, N.; Flores, B.; Moe, O.W.; Hu, M.C. Cisplatin nephrotoxicity as a model of chronic kidney disease. Lab. Investig. 2018, 98, 1105–1121. [Google Scholar] [CrossRef]
- Landau, S.I.; Guo, X.; Velazquez, H.; Torres, R.; Olson, E.; Garcia-Milian, R.; Moeckel, G.W.; Desir, G.V.; Safirstein, R. Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int. 2019, 95, 797–814. [Google Scholar] [CrossRef]
- Fu, Y.; Cai, J.; Li, F.; Liu, Z.; Shu, S.; Wang, Y.; Liu, Y.; Tang, C.; Dong, Z. Chronic effects of repeated low-dose cisplatin treatment in mouse kidneys and renal tubular cells. Am. J. Physiol. Physiol. 2019, 317, F1582–F1592. [Google Scholar] [CrossRef]
- Shu, S.; Wang, Y.; Zheng, M.; Liu, Z.; Cai, J.; Tang, C.; Dong, Z. Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair. Cells 2019, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Purification and Characterization of Hypoxia-inducible Factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen homeostasis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 336–361. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2019, 15, 21–32. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kojima, I.; Ohse, T.; Inagi, R.; Miyata, T.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. Hypoxia-inducible factor modulates tubular cell survival in cisplatin nephrotoxicity. Am. J. Physiol. Renal Physiol. 2005, 289, F1123–F1133. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Bernhardt, W.M.; Klanke, B.; Daniel, C.; Buchholz, B.; Câmpean, V.; Amann, K.; Warnecke, C.; Wiesener, M.S.; Eckardt, K.-U.; et al. HIF Activation Protects From Acute Kidney Injury. J. Am. Soc. Nephrol. 2008, 19, 486–494. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, X.; Zhang, Y.; Ding, G.; Zhu, C.; Huang, S.; Jia, Z.; Zhang, A. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 825–838. [Google Scholar] [CrossRef]
- Makhdoumi, P.; Abnous, K.; Mehri, S.; Etemad, L.; Imenshahidi, M.; Karimi, G. Oral deferiprone admin-istration ameliorates cisplatin-induced nephrotoxicity in rats. J. Pharm. Pharmacol. 2018, 70, 1357–1368. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Maltepe, E.; Lu, M.M.; Simon, C.; Bradfield, C.A. Expression of ARNT, ARNT2, HIF1 alpha, HIF2 alpha and Ah receptor mRNAs in the developing mouse. Mech. Dev. 1998, 73, 117–123. [Google Scholar] [CrossRef]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef]
- Wiesener, M.S.; Jürgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Hörstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Wide-spread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [CrossRef]
- Gu, Y.-Z.; Moran, S.M.; HogenEsch, J.B.; Wartman, L.; Bradfield, C.A. Molecular Characterization and Chromosomal Localization of a Third α-Class Hypoxia Inducible Factor Subunit, HIF3α. Gene Expr. 2018, 7, 205–213. [Google Scholar]
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nat. Cell Biol. 2001, 414, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- McGettrick, A.F.; O’Neill, L.A. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef]
- Jiang, B.H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Had-dad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Tanaka, T.; Eto, N.; Nangaku, M. Inflammation and hypoxia linked to renal injury by CCAAT/enhancer-binding protein δ. Kidney Int. 2015, 88, 262–275. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Pouysségur, J. HIF at a glance. J. Cell Sci. 2009, 122, 1055–1057. [Google Scholar] [CrossRef]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) Signaling Increases the Rate of Hypoxia-Inducible Factor 1α (HIF-1α) Synthesis: Novel Mechanism for HIF-1-Mediated Vascular Endothelial Growth Factor Expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef]
- Byun, Y.; Choi, Y.-C.; Jeong, Y.; Lee, G.; Yoon, S.; Jeong, Y.; Yoon, J.; Baek, A.K. MiR-200c downregulates HIF-1α and inhibits migration of lung cancer cells. Cell. Mol. Biol. Lett. 2019, 24, 1–14. [Google Scholar] [CrossRef]
- Liu, Y.; Nie, H.; Zhang, K.; Ma, D.; Yang, G.; Zheng, Z.; Liu, K.; Yu, B.; Zhai, C.; Yang, S. A feedback regulatory loop between HIF-1α and miR-21 in response to hypoxia in cardiomyocytes. FEBS Lett. 2014, 588, 3137–3146. [Google Scholar] [CrossRef]
- Wu, K.; Hu, M.; Chen, Z.; Xiang, F.; Chen, G.; Yan, W.; Peng, Q.; Chen, X. Asiatic acid enhances survival of human AC16 cardiomyocytes under hypoxia by upregulating miR-1290. IUBMB Life 2017, 69, 660–667. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-Inducible Factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Cardote, T.A.F.; Gadd, M.S.; Ciulli, A. Crystal Structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Lig-ase Complex. Structure 2017, 25, 901–911.e3. [Google Scholar] [CrossRef] [PubMed]
- Lisy, K.; Peet, D.J. Turn me on: Regulating HIF transcriptional activity. Cell Death Differ. 2008, 15, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Van Welden, S.; Selfridge, A.C.; Hindryckx, P. Intestinal hypoxia and hypoxia-induced signalling as therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 596–611. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Eckardt, K.-U. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat. Rev. Nephrol. 2016, 12, 157–168. [Google Scholar] [CrossRef]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 Competes with HSP90 for Binding to HIF-1α and Is Required for O2-Independent and HSP90 Inhibitor-Induced Degradation of HIF-1α. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. HAF: The new player in oxygen-independent HIF-1α degradation. Cell Cycle 2009, 8, 1359–1366. [Google Scholar] [CrossRef]
- JLee, W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free. Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef]
- Jain, T.; Nikolopoulou, E.A.; Xu, Q.; Qu, A. Hypoxia inducible factor as a therapeutic target for atherosclerosis. Pharmacol. Ther. 2018, 183, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Meng, X.; Pu, Y.; Sun, F.; Man, Z.; Zhang, J.; Yin, L.; Pu, Y. Overexpression of HIF-1a could partially protect K562 cells from 1,4-benzoquinone induced toxicity by inhibiting ROS, apoptosis and enhancing glycolysis. Toxicol. In Vitro 2019, 55, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zeng, H.; Ni, L.; Qi, L.; Xu, Y.; Xia, L.; Yu, Y.; Liu, B.; Yang, H.; Hao, H.; et al. HIF-1α Preconditioning Potentiates Antioxidant Activity in Ischemic Injury: The Role of Sequential Administration of Di-hydrotanshinone I and Protocatechuic Aldehyde in Cardioprotection. Antioxid. Redox Signal. 2019, 31, 227–242. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factor 1 and Cardiovascular Disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef]
- Elson, D.A.; Thurston, G.; Huang, L.E.; Ginzinger, D.G.; McDonald, D.M.; Johnson, R.S.; Arbeit, J.M. In-duction of hypervascularity without leakage or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1alpha. Genes Dev. 2001, 15, 2520–2532. [Google Scholar] [CrossRef]
- Rankin, E.B.; Higgins, D.F.; Walisser, J.A.; Johnson, R.S.; Bradfield, C.A.; Haase, V.H. Inactivation of the Arylhydrocarbon Receptor Nuclear Translocator (Arnt) Suppresses von Hippel-Lindau Disease-Associated Vascular Tumors in Mice. Mol. Cell. Biol. 2005, 25, 3163–3172. [Google Scholar] [CrossRef]
- Warnecke, C.; Zaborowska, Z.; Kurreck, J.; Erdmann, V.A.; Frei, U.; Wiesener, M.; Eckardt, K.U. Differ-entiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: Erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J. 2004, 18, 1462–1464. [Google Scholar] [CrossRef]
- Scortegagna, M.; Ding, K.; Zhang, Q.; Oktay, Y.; Bennett, M.J.; Bennett, M.; Shelton, J.M.; Richardson, J.A.; Moe, O.; Garcia, J.A. HIF-2alpha regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood 2005, 105, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1α Is Essential for Myeloid Cell-Mediated Inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Gläsner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Cejudo-Martin, P.; Doedens, A.; Zinkernagel, A.S.; Johnson, R.S.; Nizet, V. Cutting Edge: Essential Role of Hypoxia Inducible Factor-1α in Development of Lipopolysaccharide-Induced Sepsis. J. Immunol. 2007, 178, 7516–7519. [Google Scholar] [CrossRef]
- Kojima, H.; Gu, H.; Nomura, S.; Caldwell, C.C.; Kobata, T.; Carmeliet, P.; Semenza, G.L.; Sitkovsky, M.V. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1 -deficient chimeric mice. Proc. Natl. Acad. Sci. USA 2002, 99, 2170–2174. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Marín-Hernández, A.; Gallardo-Pérez, J.C.; Ralph, S.J.; Rodríguez-Enríquez, S.; Moreno-Sánchez, R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-L.; Liu, J.-P.; Bao, R.-X.; Yan, G.; Feng, X.; Xu, Y.-P.; Sun, Y.-P.; Yan, W.; Ling, Z.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ancey, P.B.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef] [PubMed]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 promotes tumor angiogenesis by regulating HIF-1α through NF-κB activation. Mol. Cancer 2016, 15, 3. [Google Scholar] [CrossRef]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef]
- Lai, H.H.; Li, J.N.; Wang, M.Y.; Huang, H.Y.; Croce, C.M.; Sun, H.L.; Lyu, Y.J.; Kang, J.W.; Chiu, C.F.; Hung, M.C.; et al. HIF-1α promotes autophagic proteolysis of Dicer and enhances tumor me-tastasis. J. Clin. Investig. 2018, 128, 625–643. [Google Scholar] [CrossRef]
- Song, L.P.; Zhang, J.; Wu, S.F.; Huang, Y.; Zhao, Q.; Cao, J.P.; Wu, Y.L.; Wang, L.S.; Chen, G.Q. Hypoxia-inducible factor-1alpha-induced differentiation of myeloid leukemic cells is its transcriptional activity independent. Oncogene 2008, 27, 519–527. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Kshitiz; Gilkes, D.M.; Rey, S.; Wong, C.C.; Luo, W.; Kim, D.H.; Dang, C.V.; Levchenko, A.; Semenza, G.L. A nontranscriptional role for HIF-1α as a direct inhibitor of DNA replication. Sci. Signal. 2013, 6, ra10. [Google Scholar] [CrossRef]
- Koshiji, M.; Kageyama, Y.; Pete, E.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1α induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef]
- Li, H.-S.; Zhou, Y.-N.; Li, L.; Li, S.-F.; Long, D.; Chen, X.-L.; Zhang, J.-B.; Feng, L.; Li, Y. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef]
- Dentino, M.; Luft, F.C.; Yum, M.N.; Williams, S.D.; Einhorn, L.H. Long term effect of Cis-Diamminedichloride platinum (CDDP) on renal function and structure in man. Cancer 1978, 41, 1274–1281. [Google Scholar] [CrossRef]
- Gonzalez-Vitale, J.C.; Hayes, D.M.; Cvitkovic, E.; Sternberg, S.S. The renal pathology in clinical trials of Cis-platinum (II) diamminedichloride. Cancer 1977, 39, 1362–1371. [Google Scholar] [CrossRef]
- Komaki, K.; Kusaba, T.; Tanaka, M.; Kado, H.; Shiotsu, Y.; Matsui, M.; Shiozaki, A.; Nakano, H.; Ishikawa, T.; Fujiwara, H.; et al. Lower blood pressure and risk of cisplatin nephrotoxicity: A retrospective cohort study. BMC Cancer 2017, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Solanki, M.H.; Chatterjee, P.K.; Xue, X.; Gupta, M.; Rosales, I.; Yeboah, M.M.; Kohn, N.; Metz, C.N. Magnesium protects against cisplatin-induced acute kidney injury without compromising cisplatin-mediated killing of an ovarian tumor xenograft in mice. Am. J. Physiol. Renal Physiol. 2015, 309, F35–F47. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Dong, G.; Jiang, M.; Huang, S.; Kumar, M.V.; Messing, R.O.; Dong, Z. Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Investig. 2011, 121, 2709–2722. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Khan, M.N.; Li, Q.; Chen, X.; Wei, J.; Wang, B.; Cheng, J.-W.; Gordon, J.R.; Li, F. G31P, CXCR1/2 inhibitor, with cisplatin inhibits the growth of mice hepatocellular carcinoma and mitigates high-dose cis-platin-induced nephrotoxicity. Oncol. Rep. 2015, 33, 751–757. [Google Scholar] [CrossRef]
- Ravichandran, K.; Holditch, S.; Brown, C.N.; Wang, Q.; Ozkok, A.; Weiser-Evans, M.C.; Nemenoff, R.; Miyazaki, M.; Thiessen-Philbrook, H.; Parikh, C.R.; et al. IL-33 deficiency slows cancer growth but does not protect against cisplatin-induced AKI in mice with cancer, American journal of physiology. Renal Physiol. 2018, 314, F356–F366. [Google Scholar] [CrossRef]
- Kumar, G.; Solanki, M.H.; Xue, X.; Mintz, R.; Madankumar, S.; Chatterjee, P.K.; Metz, C.N. Magnesium improves cisplatin-mediated tumor killing while protecting against cisplatin-induced nephrotoxicity. Am. J. Physiol. Physiol. 2017, 313, F339–F350. [Google Scholar] [CrossRef]
- Mundhe, N.A.; Kumar, P.; Ahmed, S.; Jamdade, V.S.; Mundhe, S.; Lahkar, M. Nordihydroguaiaretic acid ameliorates cisplatin induced nephrotoxicity and potentiates its anti-tumor activity in DMBA induced breast cancer in female Sprague–Dawley rats. Int. Immunopharmacol. 2015, 28, 634–642. [Google Scholar] [CrossRef]
- Holditch, S.J.; Brown, C.N.; Lombardi, A.M.; Nguyen, K.N.; Edelstein, C.L. Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2019, 20, 3011. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, M.K.; Burkhardt, G.; Kohler, H. Insights into potential cellular mechanisms of cisplatin nephrotoxicity and their clinical application. Nephrol. Dial. Transplant. 1997, 12, 2478–2480. [Google Scholar] [CrossRef] [PubMed]
- Gately, D.P.; Howell, S.B. Cellular accumulation of the anticancer agent cisplatin: A review. Br. J. Cancer 1993, 67, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef] [PubMed]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef]
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of Cisplatin to a nephrotoxin in proximal tubule cells. J. Am. Soc. Nephrol. 2013, 14, 1–10. [Google Scholar] [CrossRef]
- Townsend, D.M.; Hanigan, M.H. Inhibition of gamma-glutamyl transpeptidase or cysteine S-conjugate beta-lyase activity blocks the nephrotoxicity of cisplatin in mice. J. Pharmacol. Exp. Ther. 2002, 300, 142–148. [Google Scholar] [CrossRef]
- Zamble, D.B.; Lippard, S.J. Cisplatin and DNA repair in cancer chemotherapy. Trends Biochem. Sci. 1995, 20, 435–439. [Google Scholar] [CrossRef]
- Yan, M.; Tang, C.; Ma, Z.; Huang, S.; Dong, Z. DNA damage response in nephrotoxic and ischemic kidney injury. Toxicol. Appl. Pharmacol. 2016, 313, 104–108. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, H.; Shao, J.; Wu, J.; Zhou, L.; Zhang, Z.; Wang, Y.; Huang, Z.; Ren, J.; Liu, S.; et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2015, 26, 2647–2658. [Google Scholar] [CrossRef]
- Wang, J.N.; Liu, M.M.; Wang, F.; Wei, B.; Yang, Q.; Cai, Y.T.; Chen, X.; Liu, X.Q.; Jiang, L.; Li, C.; et al. RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin. Sci. (Lond.) 2019, 133, 1609–1627. [Google Scholar] [CrossRef] [PubMed]
- Tristão, V.R.; Pessoa, E.A.; Nakamichi, R.; Reis, L.A.; Batista, M.C.; Junior, M.D.S.D.; Monte, J.C.M. Synergistic effect of apoptosis and necroptosis inhibitors in cisplatin-induced nephrotoxicity. Apoptosis 2015, 21, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, X.Q.; Ma, Q.; Yang, Q.; Gao, L.; Li, H.D.; Wang, J.N.; Wei, B.; Wen, J.; Li, J.; et al. hsa-miR-500a-3P alleviates kidney injury by targeting MLKL-mediated necroptosis in renal epithelial cells. FASEB J. 2019, 33, 3523–3535. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Ren, G.L.; Wei, B.; Jin, J.; Huang, X.R.; Shao, W.; Li, J.; Meng, X.M.; Lan, H.Y. Conditional knockout of TGF-βRII /Smad2 signals protects against acute renal injury by alleviating cell necroptosis, apoptosis and inflammation. Theranostics 2019, 9, 8277–8293. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.F.; Wang, J.N.; Li, Z.; Wei, B.; Jin, J.; Gao, L.; Li, H.D.; Li, J.; Chen, H.Y.; Meng, X.M. 7-Hydroxycoumarin protects against cisplatin-induced acute kidney injury by inhibiting necroptosis and promoting Sox9-mediated tubular epithelial cell proliferation. Phytomedicine 2020, 69, 153202. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Deng, F.; Sharma, I.; Dai, Y.; Yang, M.; Kanwar, Y.S. Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J. Clin. Investig. 2019, 129, 5033–5049. [Google Scholar] [CrossRef]
- Lu, Q.; Wang, M.; Gui, Y.; Hou, Q.; Gu, M.; Liang, Y.; Xiao, B.; Zhao, A.Z.; Dai, C. Rheb1 protects against cisplatin-induced tubular cell death and acute kidney injury via maintaining mitochondrial homeostasis. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, H.; Yi, B.; Yang, S.; Liu, J.; Hu, J.; Wang, J.; Cao, K.; Zhang, W. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Sato, E.; Ito, J.; Yamada, K.-I.; Suzuki, C.; Oikawa, Y.; Matsuhashi, T.; Kikuchi, K.; Toyohara, T.; Suzuki, T.; et al. Drugs Repurposed as Antiferroptosis Agents Suppress Organ Damage, Including AKI, by Functioning as Lipid Peroxyl Radical Scavengers. J. Am. Soc. Nephrol. 2019, 31, 280–296. [Google Scholar] [CrossRef]
- Tsuruya, K.; Ninomiya, T.; Tokumoto, M.; Hirakawa, M.; Masutani, K.; Taniguchi, M.; Fukuda, K.; Kanai, H.; Kishihara, K.; Hirakata, H.; et al. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int. 2003, 63, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S.; Koji, T.; Kumatori, A.; Taguchi, T. Cisplatin-induced apoptosis in human proximal tubular epithelial cells is associated with the activation of the Fas/Fas ligand system. Histochem. Cell Biol. 1999, 111, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin−DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; De Leon, M.; Devarajan, P. Cisplatin induces apoptosis in LLC-PK1 cells via activation of mitochondrial pathways. J. Am. Soc. Nephrol. 2002, 13, 858–865. [Google Scholar] [PubMed]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Zhuo, L.; Gao, C.; Shi, S.; Huang, Z.; Li, W.; Wang, N.; Hao, L. Erythropoietin protects against cisplatin-induced nephrotoxicity by attenuating endoplasmic reticulum stress-induced apoptosis. J. Nephrol. 2012, 26, 219–227. [Google Scholar] [CrossRef]
- Yang, C.; Guo, Y.; Huang, T.S.; Zhao, J.; Huang, X.J.; Tang, H.X.; An, N.; Pan, Q.; Xu, Y.Z.; Liu, H.F. Asiat-ic acid protects against cisplatin-induced acute kidney injury via anti-apoptosis and anti-inflammation. Biomed. Pharmacother. 2018, 107, 1354–1362. [Google Scholar] [CrossRef]
- Daemen, M.A.; van Veer, C.; Denecker, G.; Heemskerk, V.H.; Wolfs, T.G.; Clauss, M.; Vandenabeele, P.; Buurman, W.A. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J. Clin. Investig. 1999, 104, 541–549. [Google Scholar] [CrossRef]
- Herzog, C.; Yang, C.; Holmes, A.; Kaushal, G.P. zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am. J. Physiol. Renal Physiol. 2012, 303, F1239–F1250. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.; Wang, J.; Du, Q.; Yu, J.; Zhang, L.; Dong, Z. Regulation of PUMA-α by p53 in cisplatin-induced renal cell apoptosis. Oncogene 2006, 25, 4056–4066. [Google Scholar] [CrossRef]
- Wei, Q.; Dong, G.; Yang, T.; Megyesi, J.; Price, P.M.; Dong, Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am. J. Physiol. Physiol. 2007, 293, F1282–F1291. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Autophagy is associated with apoptosis in cisplatin in-jury to renal tubular epithelial cells. Am. J. Physiol. Renal Physiol. 2008, 294, F777–F787. [Google Scholar] [CrossRef] [PubMed]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. Biomed. Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef] [PubMed]
- Gluba, A.; Banach, M.; Hannam, S.; Mikhailidis, D.P.; Sakowicz, A.; Rysz, J. The role of Toll-like re-ceptors in renal diseases. Nat. Rev. Nephrol. 2010, 6, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Chirino, Y.I.; Pedraza-Chaverri, J. Role of oxidative and nitrosative stress in cisplatin-induced ne-phrotoxicity. Exp. Toxicol. Pathol. 2009, 61, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Troxell, M.L.; Higgins, J.P.; Kambham, N. Antineoplastic Treatment and Renal Injury: An Update on Renal Pathology Due to Cytotoxic and Targeted Therapies. Adv. Anat. Pathol. 2016, 23, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.; Safirstein, R. Reduced renal blood flow in early cisplatin-induced acute renal failure in the rat. Am. J. Physiol. Content 1985, 249, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.-U.; Bernhardt, W.W.; Weidemann, A.; Warnecke, C.; Rosenberger, C.; Wiesener, M.M.; Willam, C. Role of hypoxia in the pathogenesis of renal disease. Kidney Int. 2005, 68, S46–S51. [Google Scholar] [CrossRef]
- Xiang, X.; Guo, C.; Tang, C.; Cai, J.; Dong, Z. Epigenetic Regulation in Kidney Toxicity: Insights From Cisplatin Nephrotoxicity. Semin. Nephrol. 2019, 39, 152–158. [Google Scholar] [CrossRef]
- Kurzhagen, J.T.; Dellepiane, S.; Cantaluppi, V.; Rabb, H. AKI: An increasingly recognized risk factor for CKD development and progression. J. Nephrol. 2020, 33, 1171–1187. [Google Scholar] [CrossRef]
- Latcha, S.; Jaimes, E.A.; Patil, S.; Glezerman, I.G.; Mehta, S.; Flombaum, C.D. Long-Term Renal Out-comes after Cisplatin Treatment. Clin. J. Am. Soc. Nephrol. 2016, 11, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Dong, Z. Rodent models of AKI-CKD transition. Am. J. Physiol. Physiol. 2018, 315, F1098–F1106. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.N.; Doll, M.A.; Dupre, T.V.; Shah, P.P.; Subathra, M.; Siow, D.; Arteel, G.E.; Megyesi, J.; Beverly, L.J.; Siskind, L. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am. J. Physiol. Physiol. 2016, 310, F560–F568. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.; Velazquez, H.; Chang, J.J.; Levene, M.J.; Moeckel, G.; Desir, G.V.; Safirstein, R. Three-Dimensional Morphology by Multiphoton Microscopy with Clearing in a Model of Cispla-tin-Induced CKD. J. Am. Soc. Nephrol. 2016, 27, 1102–1112. [Google Scholar] [CrossRef]
- Katagiri, D.; Hamasaki, Y.; Doi, K.; Negishi, K.; Sugaya, T.; Nangaku, M.; Noiri, E. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int. 2016, 89, 374–385. [Google Scholar] [CrossRef]
- Sharp, C.N.; Doll, M.; Dupre, T.V.; Beverly, L.J.; Siskind, L.J. Moderate aging does not exacerbate cis-platin-induced kidney injury or fibrosis despite altered inflammatory cytokine expression and immune cell infiltration. Am. J. Physiol. Renal Physiol. 2019, 316, F162–F172. [Google Scholar] [CrossRef]
- Sharp, C.N.; Doll, M.A.; Megyesi, J.; Oropilla, G.B.; Beverly, L.J.; Siskind, L.J. Subclinical kidney in-jury induced by repeated cisplatin administration results in progressive chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2018, 315, F161–F172. [Google Scholar] [CrossRef]
- Sears, S.M.; Sharp, C.N.; Krueger, A.; Oropilla, G.B.; Saforo, D.; Doll, M.A.; Megyesi, J.; Beverly, L.J.; Siskind, L.J. C57BL/6 mice require a higher dose of cisplatin to induce renal fibrosis and CCL2 correlates with cisplatin-induced kidney injury. Am. J. Physiol. Physiol. 2020, 319, F674–F685. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef]
- Kumar, S. Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int. 2018, 93, 27–40. [Google Scholar] [CrossRef]
- Fiorentino, M.; Grandaliano, G.; Gesualdo, L.; Castellano, G. Acute Kidney Injury to Chronic Kidney Disease Transition. Contrib. Nephrol. 2018, 193, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.L.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2016, 27, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, F.; Cirillo, L.; Roperto, R.M.; Romagnani, P.; Lazzeri, E. Molecular Mechanisms of the Acute Kidney Injury to Chronic Kidney Disease Transition: An Updated View. Int. J. Mol. Sci. 2019, 20, 4941. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Takahashi, M.; Yanagita, M. Pathophysiology of AKI to CKD progression. Semin. Nephrol. 2020, 40, 206–215. [Google Scholar] [CrossRef]
- Menshikh, A.; Scarfe, L.; Delgado, R.; Finney, C.; Zhu, Y.; Yang, H.; de Caestecker, M.P. Capillary rare-faction is more closely associated with CKD progression after cisplatin, rhabdomyolysis, and ische-mia-reperfusion-induced AKI than renal fibrosis. Am. J. Physiol. Renal Physiol. 2019, 317, F1383–F1397. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef]
- Kishi, S.; Brooks, C.R.; Taguchi, K.; Ichimura, T.; Mori, Y.; Akinfolarin, A.; Gupta, N.; Galichon, P.; Elias, B.C.; Suzuki, T.; et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J. Clin. Investig. 2019, 129, 4797–4816. [Google Scholar] [CrossRef]
- Li, C.; Xie, N.; Li, Y.; Liu, C.; Hou, F.; Wang, J. N-acetylcysteine ameliorates cisplatin-induced renal senescence and renal interstitial fibrosis through sirtuin1 activation and p53 deacetylation. Free Radic. Biol. Med. 2019, 130, 512–527. [Google Scholar] [CrossRef]
- De Oliveira, R.L.; Deschoemaeker, S.; Henze, A.T.; Debackere, K.; Finisguerra, V.; Takeda, Y.; Roncal, C.; Dettori, D.; Tack, E.; Jönsson, Y.; et al. Gene-targeting of Phd2 improves tumor response to chemo-therapy and prevents side-toxicity. Cancer Cell 2012, 22, 263–277. [Google Scholar]
- Wang, W.; Wang, W.; Jiang, Y.; Li, Z.; Cheng, J.; Liu, N.; Han, G.; Lu, S.; Zhang, J. Human adi-pose-derived stem cells modified by HIF-1α accelerate the recovery of cisplatin-induced acute renal injury in vitro. Biotechnol. Lett. 2014, 36, 667–676. [Google Scholar] [CrossRef]
- Wang, W.-W.; Li, Z.-Z.; Jiang, Y.; Cheng, J.; Lu, S.; Zhang, J.; Wang, W. Enhanced renoprotective effect of HIF-1α modified human adipose-derived stem cells on cisplatin-induced acute kidney injury in vivo. Sci. Rep. 2015, 5, 10851. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.Y.; Perlina, A.; Vu, H.; Troth, S.P.; Brennan, R.J.; Aslamkhan, A.G.; Xu, Q. Integrated pathway analysis of rat urine metabolic profiles and kidney transcriptomic profiles to elucidate the systems toxi-cology of model nephrotoxicants. Chem. Res. Toxicol. 2008, 21, 1548–1561. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Biju, M.P.; Wang, M.H.; Haase, V.H.; Dong, Z. Cytoprotective effects of hypoxia against cis-platin-induced tubular cell apoptosis: Involvement of mitochondrial inhibition and p53 suppression. J. Am. Soc. Nephrol. 2006, 17, 1875–1885. [Google Scholar] [CrossRef]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-Inducible Factor 1α Is Essential for Cell Cycle Arrest during Hypoxia. Mol. Cell. Biol. 2003, 23, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Price, P.M.; Yu, F.; Kaldis, P.; Aleem, E.; Nowak, G.; Safirstein, R.L.; Megyesi, J. Dependence of cisplatin-induced cell death in vitro and in vivo on cyclin-dependent kinase 2. J. Am. Soc. Nephrol. 2006, 17, 2434–2442. [Google Scholar] [CrossRef]
- Yu, F.; Megyesi, J.; Safirstein, R.L.; Price, P.M. Identification of the functional domain of p21WAF1/CIP1 that protects cells from cisplatin cytotoxicity. Am. J. Physiol. Physiol. 2005, 289, F514–F520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, D.; Hu, H.; Zhang, P.; Xie, R.; Cui, W. HIF-1α/BNIP3 signaling path-way-induced-autophagy plays protective role during myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 2019, 120, 109464. [Google Scholar] [CrossRef]
- Periyasamy-Thandavan, S.; Jiang, M.; Wei, Q.; Smith, R.; Yin, X.M.; Dong, Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008, 74, 631–640. [Google Scholar] [CrossRef]
- Mei, S.; Livingston, M.; Hao, J.; Li, L.; Mei, C.; Dong, Z. Autophagy is activated to protect against endotoxic acute kidney injury. Sci. Rep. 2016, 6, 22171. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.-Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-mediated mitophagy is ac-tivated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Livingston, M.J.; Dong, G.; Tang, C.; Su, Y.; Wu, G.; Yin, X.-M.; Dong, Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M. Chronic Hypoxia and Tubulointerstitial Injury: A Final Common Pathway to End-Stage Renal Failure. J. Am. Soc. Nephrol. 2005, 17, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Kusaba, T.; Humphreys, B.D. Who regenerates the kidney tubule? Nephrol. Dial. Transplant. 2014, 30, 903–910. [Google Scholar] [CrossRef]
- Kong, T.; Eltzschig, H.K.; Karhausen, J.; Colgan, S.P.; Shelley, C.S. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 10440–10445. [Google Scholar] [CrossRef]
- Liu, J.; Wei, Q.; Guo, C.; Dong, G.; Liu, Y.; Tang, C.; Dong, Z. Hypoxia, HIF, and Associated Signaling Networks in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18, 950. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Jaffe, J.; Michael, M.; Swan, C.E.; Duffy, K.J.; Erickson-Miller, C.L.; Haase, V.H. Pre-ischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am. J. Physiol. Renal Physiol. 2012, 302, F1172–F1179. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Sano, H.; Michael, M.; Kobayashi, H.; Davidoff, O.; Bian, A.; Yao, B.; Zhang, M.-Z.; Harris, R.C.; Duffy, K.J.; et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Investig. 2014, 124, 2396–2409. [Google Scholar] [CrossRef]
- Kobayashi, H.; Gilbert, V.; Liu, Q.; Kapitsinou, P.P.; Unger, T.L.; Rha, J.; Rivella, S.; Schlöndorff, D.; Haase, V.H. Myeloid cell-derived hypoxia-inducible factor attenuates inflammation in unilateral ureteral ob-struction-induced kidney injury. J. Immunol. 2012, 188, 5106–5115. [Google Scholar] [CrossRef]
- Song, Y.R.; You, S.J.; Lee, Y.M.; Chin, H.J.; Chae, D.W.; Oh, Y.K.; Joo, K.W.; Han, J.S.; Na, K.Y. Activa-tion of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrol. Dial. Transplant. 2010, 25, 77–85. [Google Scholar] [CrossRef]
- Kimura, K.; Iwano, M.; Higgins, D.F.; Yamaguchi, Y.; Nakatani, K.; Harada, K.; Kubo, A.; Akai, Y.; Rankin, E.B.; Neilson, E.G.; et al. Stable expression of HIF-1α in tubular epithelial cells promotes interstitial fibrosis. Am. J. Physiol. Physiol. 2008, 295, F1023–F1029. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, P.; Bai, M.; He, L.; Zhang, L.; Liu, T.; Yang, Z.; Duan, M.; Liu, M.; Liu, B.; et al. p53 upregulated by HIF-1α promotes hypoxia-induced G2/M arrest and renal fibrosis in vitro and in vivo. J. Mol. Cell Biol. 2019, 11, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Wang, Z.; Xia, M.; Li, P.L.; van Tassell, B.W.; Abbate, A.; Dhaduk, R.; Li, N. Silencing of hy-poxia-inducible factor-1α gene attenuated angiotensin II-induced renal injury in Sprague-Dawley rats. Hypertension 2011, 58, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, Q.; Li, P.-L.; Dhaduk, R.; Zhang, F.; Gehr, T.W.; Li, N. Silencing of hypoxia-inducible factor-1α gene attenuates chronic ischemic renal injury in two-kidney, one-clip rats. Am. J. Physiol. Physiol. 2014, 306, F1236–F1242. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Wish, J.B. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Sharma, N.; Dikdan, S. Hypoxia-Inducible Factor and Its Role in the Management of Anemia in Chronic Kidney Disease. Int. J. Mol. Sci. 2018, 19, 389. [Google Scholar] [CrossRef]
- Mokas, S.; Larivière, R.; Lamalice, L.; Gobeil, S.; Cornfield, D.N.; Agharazii, M.; Richard, D.E. Hypox-ia-inducible factor-1 plays a role in phosphate-induced vascular smooth muscle cell calcification. Kidney Int. 2016, 90, 598–609. [Google Scholar] [CrossRef]
- Martin, E.R.; Smith, M.T.; Maroni, B.J.; Zuraw, Q.C.; de Goma, E.M. Clinical Trial of Vadadustat in Pa-tients with Anemia Secondary to Stage 3 or 4 Chronic Kidney Disease. Am. J. Nephrol. 2017, 45, 380–388. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, Y.; Hu, T.; Wu, Y.; Li, Z.; Jiang, Z.; Yang, C.; Zhang, L.; You, Q. Discovery of Clinical Candidate (5-(3-(4-Chlorophenoxy)prop-1-yn-1-yl)-3-hydroxypicolinoyl)glycine, an Orally Bioavailable Prolyl Hydroxylase Inhibitor for the Treatment of Anemia. J. Med. Chem. 2020, 63, 10045–10060. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, Z.; Li, Z.; Gu, J.; You, Q.; Zhang, X. Click Chemistry-Based Discovery of [3-Hydroxy-5-(1H-1,2,3-triazol-4-yl)picolinoyl]glycines as Orally Active Hypoxia-Inducing Factor Prolyl Hydroxylase Inhibitors with Favorable Safety Profiles for the Treatment of Anemia. J. Med. Chem. 2018, 61, 5332–5349. [Google Scholar] [CrossRef]
- Saito, H.; Tanaka, T.; Sugahara, M.; Tanaka, S.; Fukui, K.; Wakashima, T.; Nangaku, M. Inhibition of prolyl hydroxylase domain (PHD) by JTZ-951 reduces obesity-related diseases in the liver, white adipose tissue, and kidney in mice with a high-fat diet. Lab. Investig. 2019, 99, 1217–1232. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Tanaka, S.; Tanaka, T.; Saito, H.; Ishimoto, Y.; Wakashima, T.; Ueda, M.; Fukui, K.; Shimizu, A.; Inagi, R.; et al. Prolyl Hydroxylase Domain Inhibitor Protects against Metabolic Disorders and Associated Kidney Disease in Obese Type 2 Diabetic Mice. J. Am. Soc. Nephrol. 2020, 31, 560–577. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Tanaka, T.; Saito, T.; Fukui, K.; Wakashima, T.; Susaki, E.A.; Ueda, H.R.; Nangaku, M. The oral hypoxia-inducible factor prolyl hydroxylase inhibitor enarodustat counteracts alterations in renal en-ergy metabolism in the early stages of diabetic kidney disease. Kidney Int. 2020, 97, 934–950. [Google Scholar] [CrossRef] [PubMed]
- Schley, G.; Klanke, B.; Kalucka, J.; Schatz, V.; Daniel, C.; Mayer, M.; Goppelt-Struebe, M.; Herrmann, M.; Thorsteinsdottir, M.; Palsson, R.; et al. Mononuclear phagocytes orchestrate prolyl hydroxylase inhibition-mediated renopro-tection in chronic tubulointerstitial nephritis. Kidney Int. 2019, 96, 378–396. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, G.; Schonfeld, M.P.; Tiwari, R.; Huang, S.; Torosyan, R.; Fields, T.; Park, J.; Susztak, K.; Kapitsinou, P.P. Inhibition of Endothelial PHD2 Suppresses Post-Ischemic Kidney Inflammation through Hy-poxia-Inducible Factor-1. J. Am. Soc. Nephrol. 2020, 31, 501–516. [Google Scholar] [CrossRef]
- Yu, X.; Fang, Y.; Liu, H.; Zhu, J.; Zou, J.; Xu, X.; Jiang, S.; Ding, X. The balance of beneficial and delete-rious effects of hypoxia-inducible factor activation by prolyl hydroxylase inhibitor in rat remnant kidney depends on the timing of administration. Nephrol. Dial. Transplant. 2012, 27, 3110–3119. [Google Scholar] [CrossRef]
- Joharapurkar, A.; Pandya, V.B.; Patel, V.J.; Desai, R.C.; Jain, M.R. Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases. J. Med. Chem. 2018, 61, 6964–6982. [Google Scholar] [CrossRef]
- Sanghani, N.S.; Haase, V.H. Hypoxia-Inducible Factor Activators in Renal Anemia: Current Clinical Experience. Adv. Chronic Kidney Dis. 2019, 26, 253–266. [Google Scholar] [CrossRef]
- Tanaka, T.; Eckardt, K.-U. HIF Activation Against CVD in CKD: Novel Treatment Opportunities. Semin. Nephrol. 2018, 38, 267–276. [Google Scholar] [CrossRef]
- Uchida, L.; Tanaka, T.; Saito, H.; Sugahara, M.; Wakashima, T.; Fukui, K.; Nangaku, M. Effects of a prolyl hydroxylase inhibitor on kidney and cardiovascular complications in a rat model of chronic kidney disease. Am. J. Physiol. Physiol. 2020, 318, F388–F401. [Google Scholar] [CrossRef]
- Kemp, G.; Rose, P.; Lurain, J.; Berman, M.; Manetta, A.; Roullet, B.; Homesley, H.; Belpomme, D.; Glick, J. Amifostine pretreatment for protection against cyclophosphamide-induced and cisplatin-induced toxicities: Results of a randomized control trial in patients with advanced ovarian cancer. J. Clin. Oncol. 1996, 14, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Crona, D.J.; Faso, A.; Nishijima, T.F.; McGraw, K.A.; Galsky, M.D.; Milowsky, M.I. A Systematic Re-view of Strategies to Prevent Cisplatin-Induced Nephrotoxicity. Oncologist 2017, 22, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-González, P.D.; López-Hernández, F.J.; Dueñas, M.; Prieto, M.; Sánchez-López, E.; Thomale, J.; Ruiz-Ortega, M.; López-Novoa, J.M.; Morales, A.I. Differential effect of quercetin on cisplatin-induced toxicity in kidney and tumor tissues. Food Chem. Toxicol. 2017, 107, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Sun, Y.; Guan, Z.; Liang, L.; Cheng, Y.; Zhou, J.; Li, J.; Xu, Y. HIF-1α/MDR1 pathway confers chemo-resistance to cisplatin in bladder cancer. Oncol. Rep. 2016, 35, 1549–1556. [Google Scholar] [CrossRef]
- Deben, C.; Deschoolmeester, V.; De Waele, J.; Jacobs, J.; Bossche, J.V.D.; Wouters, A.; Peeters, M.; Rolfo, C.; Smits, E.; Lardon, F.; et al. Hypoxia-Induced Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Mediated by HIF-1α and Mutant p53 and Can Be Overcome by Induction of Oxidative Stress. Cancers 2018, 10, 126. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, Z.; Yin, H.; Yang, G. Interaction between p53 and Ras signaling controls cisplatin re-sistance via HDAC4- and HIF-1α-mediated regulation of apoptosis and autophagy. Theranostics 2019, 9, 1096–1114. [Google Scholar] [CrossRef]
- Keremu, A.; Aini, A.; Maimaitirexiati, Y.; Liang, Z.; Aila, P.; Xierela, P.; Tusun, A.; Moming, H.; Yusufu, A. Overcoming cisplatin resistance in osteosarcoma through the miR-199a-modulated inhibition of HIF-1α. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Ai, Z.; Lu, Y.; Qiu, S.; Fan, Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016, 373, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Kizaka-Kondoh, S.; Itasaka, S.; Zeng, L.; Tanaka, S.; Zhao, T.; Takahashi, Y.; Shibuya, K.; Hirota, K.; Semenza, G.L.; Hiraoka, M. Selective Killing of Hypoxia-Inducible Factor-1–Active Cells Improves Survival in a Mouse Model of Invasive and Metastatic Pancreatic Cancer. Clin. Cancer Res. 2009, 15, 3433–3441. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Li, W.; Xu, Y.; Zhuo, Y.; Li, M.; He, Y.; Wang, X.; Guo, Q.; Zhao, L.; et al. Oroxylin A reverses hypoxia-induced cisplatin resistance through inhibiting HIF-1α mediated XPC transcription. Oncogene 2020, 39, 6893–6905. [Google Scholar] [CrossRef]
- Parmakhtiar, B.; Burger, R.A.; Kim, J.-H.; Fruehauf, J.P. HIF Inactivation of p53 in Ovarian Cancer Can Be Reversed by Topotecan, Restoring Cisplatin and Paclitaxel Sensitivity. Mol. Cancer Res. 2019, 17, 1675–1686. [Google Scholar] [CrossRef]
- Hussain, I.; Waheed, S.; Ahmad, K.A.; Pirog, J.E.; Syed, V. Scutellaria baicalensis targets the hypox-ia-inducible factor-1α and enhances cisplatin efficacy in ovarian cancer. J. Cell Biochem. 2018, 119, 7515–7524. [Google Scholar] [CrossRef]
- Erez, N.; Milyavsky, M.; Eilam, R.; Shats, I.; Goldfinger, N.; Rotter, V. Expression of prolyl-hydroxylase-1 (PHD1/EGLN2) suppresses hypoxia inducible factor-1alpha activation and inhibits tumor growth. Cancer Res. 2003, 63, 8777–8783. [Google Scholar] [PubMed]
- Bordoli, M.R.; Stiehl, D.P.; Borsig, L.; Kristiansen, G.; Hausladen, S.; Schraml, P.; Wenger, R.H.; Camenisch, G. Prolyl-4-hydroxylase PHD2- and hypoxia-inducible factor 2-dependent regulation of amphiregulin contributes to breast tumorigenesis. Oncogene 2011, 30, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Hu, Y.; Lin, J.; Fu, H.; Lim, L.Y.; Yuan, Z.-X. Targeting strategies for drug delivery to the kidney: From renal glomeruli to tubules. Med. Res. Rev. 2019, 39, 561–578. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.J.; Yuan, W.D.; Yuan, J.Q.; Yuan, K.; Wang, Y. Downregulation of HIF-2α Reverse the Chemo-therapy Resistance of Lung Adenocarcinoma A549 Cells to Cisplatin. Med. Sci. Monit. 2018, 24, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- He, S.-Y.; Min, L.; Chen, Q.; Liu, S.; Ma, Y. Hypoxia-induced increases in A549/CDDP cell drug resistance are reversed by RNA interference of HIF-1? expression. Mol. Med. Rep. 2011, 5, 228–232. [Google Scholar] [CrossRef][Green Version]
- Song, X.; Liu, X.; Chi, W.; Liu, Y.; Wei, L.; Wang, X.; Yu, J. Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1α gene. Cancer Chemother. Pharmacol. 2006, 58, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.Y.; Wang, G.P.; Gu, L.J.; Huang, S.H.; Chen, X.L.; Li, Y.; Cai, S.W. HIF-1α siRNA and cisplatin in combination suppress tumor growth in a nude mice model of esophageal squamous cell carcinoma. Asian Pac. J. Cancer Prev. 2012, 13, 473–477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fischer, C.; Leithner, A.; Wohlkoenig, C.; Quehenberger, F.; Bertsch, A.; Olschewski, A.; Olschewski, H.; Hrzenjak, A. Panobinostat reduces hypoxia-induced cisplatin resistance of non-small cell lung carcinoma cells via HIF-1α destabilization. Mol. Cancer 2015, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Li, Y.; Zeng, J.; Wang, B.; Ji, K.; Tang, Y.; Sun, Q. Knockdown of HIF-1α by siRNA-expressing plasmid delivered by attenuated Salmonella enhances the antitumor effects of cisplatin on prostate cancer. Sci. Rep. 2017, 7, 7546. [Google Scholar] [CrossRef]
- Su, W.; Huang, L.; Ao, Q.; Zhang, Q.; Tian, X.; Fang, Y.; Lu, Y. Noscapine sensitizes chemoresistant ovarian cancer cells to cisplatin through inhibition of HIF-1α. Cancer Lett. 2011, 305, 94–99. [Google Scholar] [CrossRef]
- Huang, Y.; Tian, Y.; Zhao, Y.; Xue, C.; Zhan, J.; Liu, L.; He, X.; Zhang, L. Efficacy of the hypox-ia-activated prodrug evofosfamide (TH-302) in nasopharyngeal carcinoma in vitro and in vivo. Cancer Commun. (Lond.) 2018, 38, 15. [Google Scholar] [CrossRef]
- Kim, H.; Cho, Y.; Kim, H.S.; Kang, D.; Cheon, D.; Kim, Y.J.; Chang, M.J.; Lee, K.M.; Chang, C.B.; Kang, S.B.; et al. A system-level approach identifies HIF-2α as a critical regulator of chondro-sarcoma progression. Nat. Commun. 2020, 11, 5023. [Google Scholar] [CrossRef]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef]
- Rapisarda, A.; Hollingshead, M.G.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Gehrs, B.; Raffeld, M.; Kinders, R.J.; Parchment, R.; et al. Increased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibition. Mol. Cancer Ther. 2009, 8, 1867–1877. [Google Scholar] [CrossRef]
- Kheshtchin, N.; Arab, S.; Ajami, M.; Mirzaei, R.; Ashourpour, M.; Mousavi, N.; Khosravianfar, N.; Jadidi-Niaragh, F.; Namdar, A.; Noorbakhsh, F.; et al. Inhibition of HIF-1α enhances anti-tumor effects of dendritic cell-based vaccination in a mouse model of breast cancer. Cancer Immunol. Immunother. 2016, 65, 1159–1167. [Google Scholar] [CrossRef]
- Salem, A.; Asselin, M.-C.; Reymen, B.; Jackson, A.; Lambin, P.; West, C.M.L.; O’Connor, J.P.; Faivre-Finn, C. Targeting Hypoxia to Improve Non–Small Cell Lung Cancer Outcome. J. Natl. Cancer Inst. 2017, 110, 14–30. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).