Molecular Targeting of HuR Oncoprotein Suppresses MITF and Induces Apoptosis in Melanoma Cells



 ,
,  ,
, 

Abstract
Simple Summary
Abstract
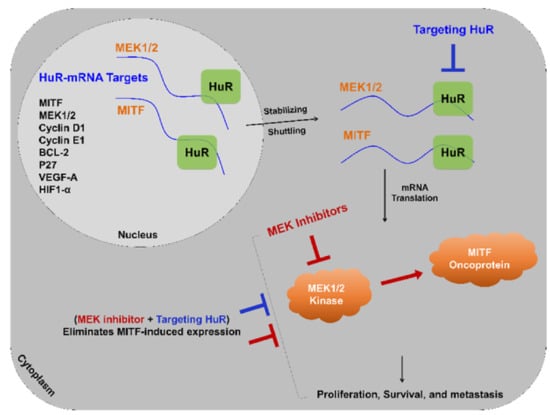
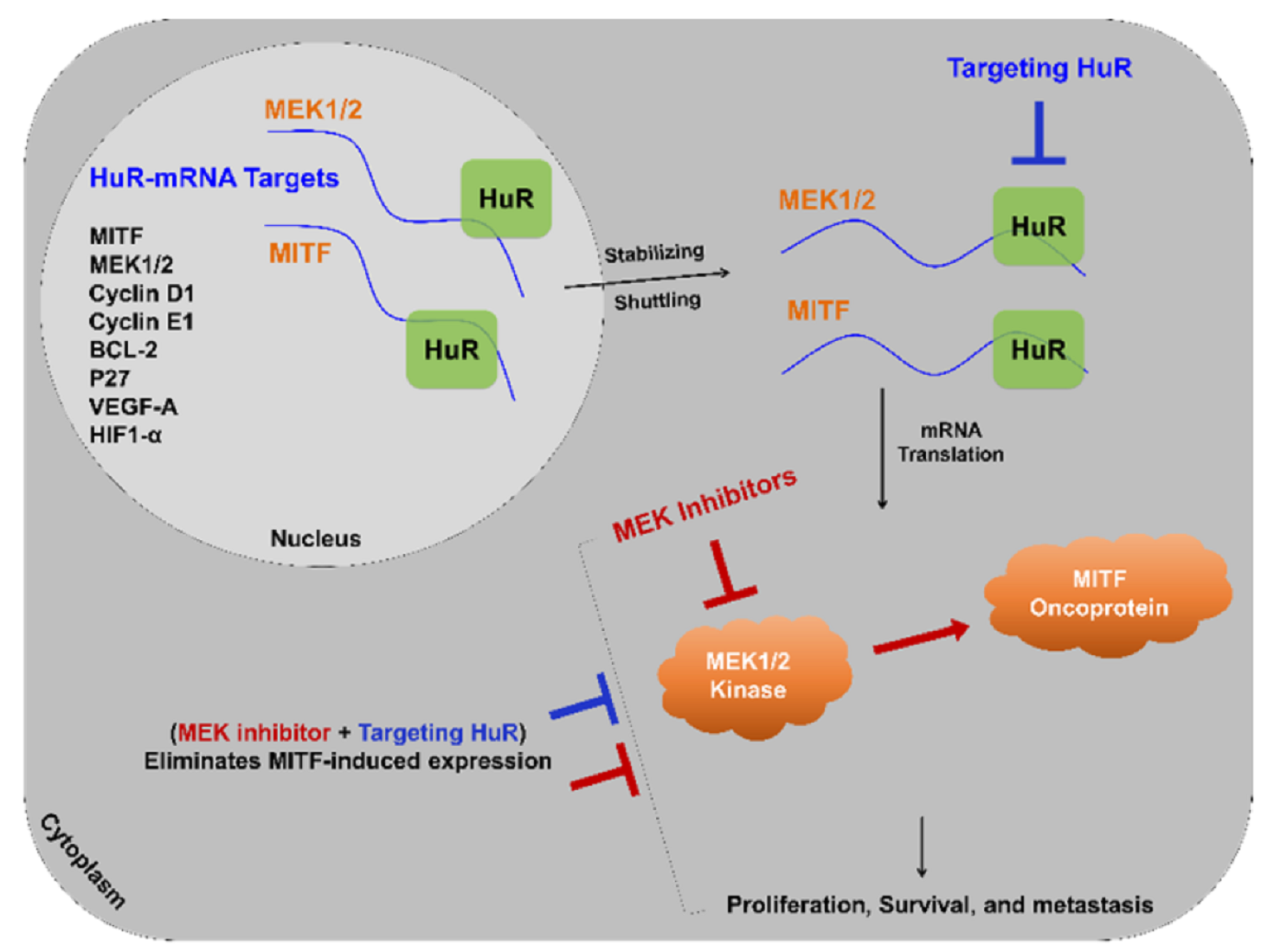{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Synthesis and Preparation of siRNA Containing Nanoparticles
2.3. Cell Viability Assay
2.4. Quantitative (q) RT-PCR Assay
- Human HuR
- Forward—5′ATGAAGACCACATGGCCGAAGACT 3′
- Reverse—5′ TGTGGTCATGAGTCCTTCCACGAT 3′
- Human BCL-2
- Forward—5′ ATG TGT GTG GAG AGC GTC AA 3′
- Reverse—5′ ACA GTT CCA CAA AGG CAT CC 3′
- Human 18S
- Forward—5′ tagtagggacgggcggtgtg 3′
- Reverse—5′ cagccacccgagattgagca 3′
2.5. Western Blotting
2.6. Cell Cycle Analysis
2.7. Annexin V Assay
2.8. Cell Migration and Invasion Assay
2.9. Statistics
3. Results
3.1. Genetic Knockdown of HuR Inhibited Cell Growth in Melanoma
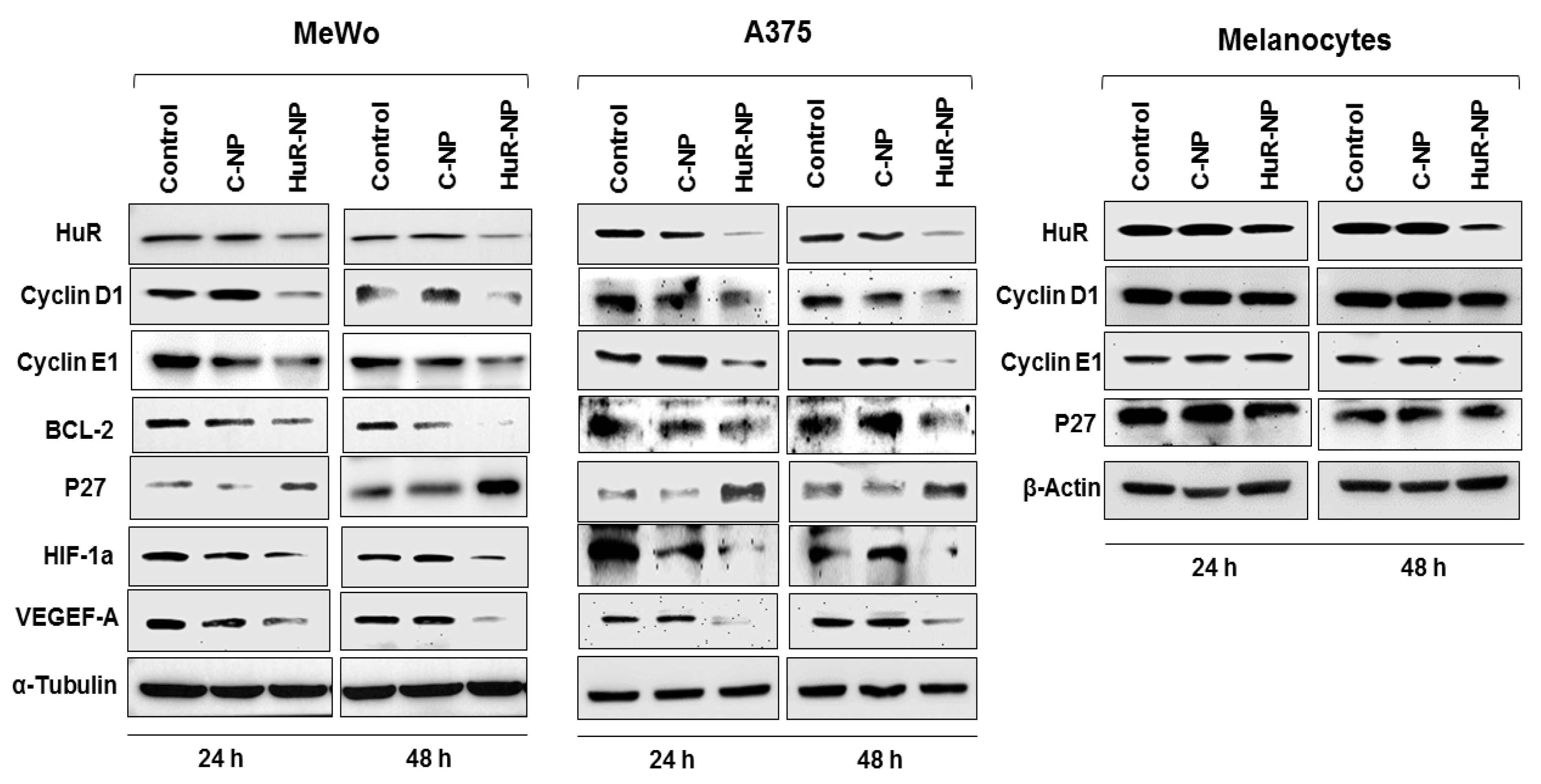
3.2. Genetic Knockdown of HuR Using HuR-NP Reduced the Expression of HuR and HuR-Regulated Oncoproteins in Melanoma Cell Lines but Not in Melanocytes
3.3. Genetic Knockdown of HuR Induced G1 Cell Cycle Arrest in Melanoma
3.4. Genetic Knockdown of HuR in Melanoma Cells Activated the Apoptosis Cascade
3.5. Genetic Knockdown of HuR Inhibited Melanoma Cell Migration and Invasion
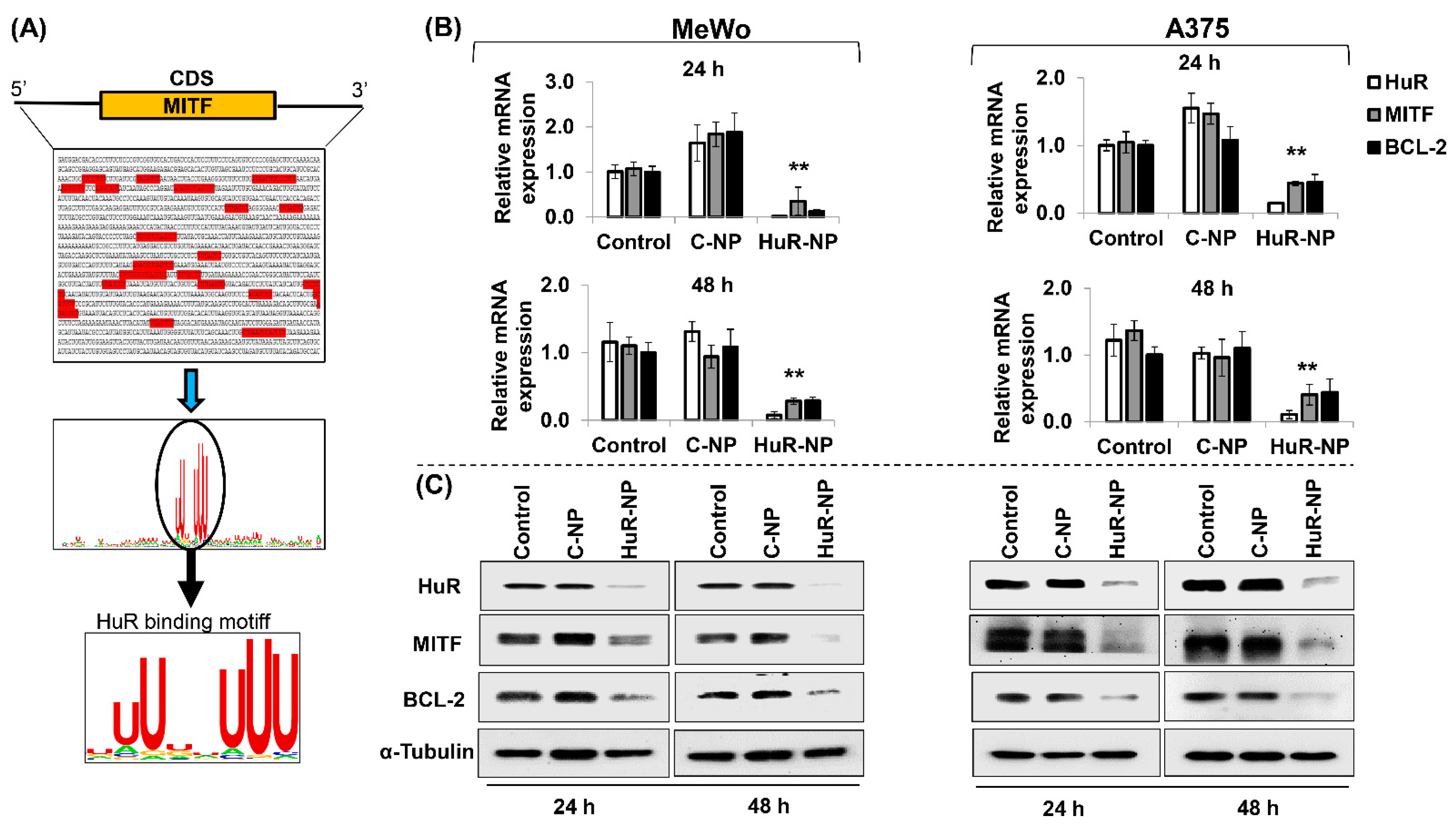
3.6. HuR Inhibition Reduced MITF in Melanoma Cells
3.7. HuR-NP Reduces U0126 Induced MITF in Melanoma Cells
3.8. HuR-NP Suppresses the Cell Viability of MITF Overexpressing Melanoma Cell Line
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wyant, T.; Alteri, R.; Kalidas, M.; Ogoro, C.; Lubejko, B.; Eidsmoe, K.; McDowell, S.; Greene, B.; Delfin-Davis, R.; Cance, G.W.; et al. Melanoma Skin Cancer Causes, RiskFactors, and Prevention. American Cancer Society. 2020. Available online: https://www.cancer.org/content/dam/CRC/PDF/Public/8824.00.pdf (accessed on 20 October 2020).
- Kakavand, H.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Targeted therapies and immune checkpoint inhibitors in the treatment of metastatic melanoma patients: A guide and update for pathologists. Pathology 2016, 48, 194–202. [Google Scholar] [CrossRef]
- Wong, D.J.; Ribas, A. Targeted Therapy for Melanoma. Cancer Treat. Res. 2016, 167, 251–262. [Google Scholar] [CrossRef]
- Kee, D.; McArthur, G. Targeted therapies for cutaneous melanoma. Hematol. Oncol. Clin. N. Am. 2014, 28, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Manzano, J.L.; Layos, L.; Buges, C.; de Los Llanos Gil, M.; Vila, L.; Martinez-Balibrea, E.; Martinez-Cardus, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Ogilvie, L.; Hedley, D.; Karasarides, M.; Martin, J.; Niculescu-Duvaz, D.; Springer, C.J.; Marais, R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004, 64, 2338–2342. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef]
- Hodi, F.S.; Friedlander, P.; Corless, C.L.; Heinrich, M.C.; Mac Rae, S.; Kruse, A.; Jagannathan, J.; Van den Abbeele, A.D.; Velazquez, E.F.; Demetri, G.D.; et al. Major response to imatinib mesylate in KIT-mutated melanoma. J. Clin. Oncol. 2008, 26, 2046–2051. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Hamid, O.; Antonescu, C.R. Selecting patients for KIT inhibition in melanoma. Methods Mol. Biol. 2014, 1102, 137–162. [Google Scholar] [CrossRef]
- Woodman, S.E.; Trent, J.C.; Stemke-Hale, K.; Lazar, A.J.; Pricl, S.; Pavan, G.M.; Fermeglia, M.; Gopal, Y.N.; Yang, D.; Podoloff, D.A.; et al. Activity of dasatinib against L576P KIT mutant melanoma: Molecular, cellular, and clinical correlates. Mol. Cancer Ther. 2009, 8, 2079–2085. [Google Scholar] [CrossRef]
- Busca, R.; Bertolotto, C.; Ortonne, J.P.; Ballotti, R. Inhibition of the phosphatidylinositol 3-kinase/p70(S6)-kinase pathway induces B16 melanoma cell differentiation. J. Biol. Chem. 1996, 271, 31824–31830. [Google Scholar] [CrossRef] [PubMed]
- Trojaniello, C.; Festino, L.; Vanella, V.; Ascierto, P.A. Encorafenib in combination with binimetinib for unresectable or metastatic melanoma with BRAF mutations. Expert. Rev. Clin. Pharmacol. 2019, 12, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, F.; Ghiorzo, P.; Orgiano, L.; Pastorino, L.; Picasso, V.; Tornari, E.; Ottaviano, V.; Queirolo, P. BRAF-mutant melanoma: Treatment approaches, resistance mechanisms, and diagnostic strategies. Onco Targets Ther. 2015, 8, 157–168. [Google Scholar] [CrossRef]
- Simeone, E.; Grimaldi, A.M.; Festino, L.; Vanella, V.; Palla, M.; Ascierto, P.A. Combination Treatment of Patients with BRAF-Mutant Melanoma: A New Standard of Care. BioDrugs 2017, 31, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Achkar, T.; Tarhini, A.A. The use of immunotherapy in the treatment of melanoma. J. Hematol. Oncol. 2017, 10, 88. [Google Scholar] [CrossRef]
- Karlsson, A.K.; Saleh, S.N. Checkpoint inhibitors for malignant melanoma: A systematic review and meta-analysis. Clin. Cosmet. Investig. Dermatol. 2017, 10, 325–339. [Google Scholar] [CrossRef]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Christiansen, S.A.; Khan, S.; Gibney, G.T. Targeted Therapies in Combination With Immune Therapies for the Treatment of Metastatic Melanoma. Cancer J. 2017, 23, 59–62. [Google Scholar] [CrossRef]
- Peng, S.S.; Chen, C.Y.; Xu, N.; Shyu, A.B. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 1998, 17, 3461–3470. [Google Scholar] [CrossRef]
- Ripin, N.; Boudet, J.; Duszczyk, M.M.; Hinniger, A.; Faller, M.; Krepl, M.; Gadi, A.; Schneider, R.J.; Sponer, J.; Meisner-Kober, N.C.; et al. Molecular basis for AU-rich element recognition and dimerization by the HuR C-terminal RRM. Proc. Natl. Acad. Sci. USA 2019, 116, 2935–2944. [Google Scholar] [CrossRef]
- Wang, J.; Guo, Y.; Chu, H.; Guan, Y.; Bi, J.; Wang, B. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int. J. Mol. Sci. 2013, 14, 10015–10041. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, M.; Bono, P.; Narko, K.; Chang, S.H.; Lundin, J.; Joensuu, H.; Furneaux, H.; Hla, T.; Haglund, C.; Ristimaki, A. Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. 2005, 65, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Weichert, W.; Pest, S.; Koch, I.; Licht, D.; Kobel, M.; Reles, A.; Sehouli, J.; Dietel, M.; Hauptmann, S. Overexpression of the embryonic-lethal abnormal vision-like protein HuR in ovarian carcinoma is a prognostic factor and is associated with increased cyclooxygenase 2 expression. Cancer Res. 2004, 64, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Mrena, J.; Wiksten, J.P.; Thiel, A.; Kokkola, A.; Pohjola, L.; Lundin, J.; Nordling, S.; Ristimaki, A.; Haglund, C. Cyclooxygenase-2 is an independent prognostic factor in gastric cancer and its expression is regulated by the messenger RNA stability factor HuR. Clin. Cancer Res. 2005, 11, 7362–7368. [Google Scholar] [CrossRef]
- Niesporek, S.; Kristiansen, G.; Thoma, A.; Weichert, W.; Noske, A.; Buckendahl, A.C.; Jung, K.; Stephan, C.; Dietel, M.; Denkert, C. Expression of the ELAV-like protein HuR in human prostate carcinoma is an indicator of disease relapse and linked to COX-2 expression. Int. J. Oncol. 2008, 32, 341–347. [Google Scholar] [CrossRef][Green Version]
- Stoppoloni, D.; Cardillo, I.; Verdina, A.; Vincenzi, B.; Menegozzo, S.; Santini, M.; Sacchi, A.; Baldi, A.; Galati, R. Expression of the embryonic lethal abnormal vision-like protein HuR in human mesothelioma: Association with cyclooxygenase-2 and prognosis. Cancer 2008, 113, 2761–2769. [Google Scholar] [CrossRef]
- Liaudet, N.; Fernandez, M.; Fontao, L.; Kaya, G.; Merat, R. Hu antigen R (HuR) heterogeneous expression quantification as a prognostic marker of melanoma. J. Cutan. Pathol. 2018, 45, 333–336. [Google Scholar] [CrossRef]
- Giaginis, C.; Alexandrou, P.; Tsoukalas, N.; Sfiniadakis, I.; Kavantzas, N.; Agapitos, E.; Patsouris, E.; Theocharis, S. Hu-antigen receptor (HuR) and cyclooxygenase-2 (COX-2) expression in human non-small-cell lung carcinoma: Associations with clinicopathological parameters, tumor proliferative capacity and patients’ survival. Tumour Biol. 2015, 36, 315–327. [Google Scholar] [CrossRef]
- Muralidharan, R.; Babu, A.; Amreddy, N.; Srivastava, A.; Chen, A.; Zhao, Y.D.; Kompella, U.B.; Munshi, A.; Ramesh, R. Tumor-targeted Nanoparticle Delivery of HuR siRNA Inhibits Lung Tumor Growth In Vitro and In Vivo By Disrupting the Oncogenic Activity of the RNA-binding Protein HuR. Mol. Cancer Ther. 2017, 16, 1470–1486. [Google Scholar] [CrossRef]
- Muralidharan, R.; Mehta, M.; Ahmed, R.; Roy, S.; Xu, L.; Aube, J.; Chen, A.; Zhao, Y.D.; Herman, T.; Ramesh, R.; et al. HuR-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci. Rep. 2017, 7, 9694. [Google Scholar] [CrossRef]
- Muralidharan, R.; Babu, A.; Amreddy, N.; Basalingappa, K.; Mehta, M.; Chen, A.; Zhao, Y.D.; Kompella, U.B.; Munshi, A.; Ramesh, R. Folate receptor-targeted nanoparticle delivery of HuR-RNAi suppresses lung cancer cell proliferation and migration. J. Nanobiotechnol. 2016, 14, 47. [Google Scholar] [CrossRef] [PubMed]
- Jimbo, M.; Blanco, F.F.; Huang, Y.H.; Telonis, A.G.; Screnci, B.A.; Cosma, G.L.; Alexeev, V.; Gonye, G.E.; Yeo, C.J.; Sawicki, J.A.; et al. Targeting the mRNA-binding protein HuR impairs malignant characteristics of pancreatic ductal adenocarcinoma cells. Oncotarget 2015, 6, 27312–27331. [Google Scholar] [CrossRef]
- Lang, M.; Berry, D.; Passecker, K.; Mesteri, I.; Bhuju, S.; Ebner, F.; Sedlyarov, V.; Evstatiev, R.; Dammann, K.; Loy, A.; et al. HuR Small-Molecule Inhibitor Elicits Differential Effects in Adenomatosis Polyposis and Colorectal Carcinogenesis. Cancer Res. 2017, 77, 2424–2438. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lan, L.; Wilson, D.M.; Marquez, R.T.; Tsao, W.C.; Gao, P.; Roy, A.; Turner, B.A.; McDonald, P.; Tunge, J.A.; et al. Identification and validation of novel small molecule disruptors of HuR-mRNA interaction. ACS Chem. Biol. 2015, 10, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.F.; Preet, R.; Aguado, A.; Vishwakarma, V.; Stevens, L.E.; Vyas, A.; Padhye, S.; Xu, L.; Weir, S.J.; Anant, S.; et al. Impact of HuR inhibition by the small molecule MS-444 on colorectal cancer cell tumorigenesis. Oncotarget 2016, 7, 74043–74058. [Google Scholar] [CrossRef] [PubMed]
- Romeo, C.; Weber, M.C.; Zarei, M.; DeCicco, D.; Chand, S.N.; Lobo, A.D.; Winter, J.M.; Sawicki, J.A.; Sachs, J.N.; Meisner-Kober, N.; et al. HuR Contributes to TRAIL Resistance by Restricting Death Receptor 4 Expression in Pancreatic Cancer Cells. Mol. Cancer Res. 2016, 14, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Basalingappa, K.; Griffith, J.N.; Andrade, D.; Babu, A.; Amreddy, N.; Muralidharan, R.; Gorospe, M.; Herman, T.; Ding, W.Q.; et al. HuR silencing elicits oxidative stress and DNA damage and sensitizes human triple-negative breast cancer cells to radiotherapy. Oncotarget 2016, 7, 64820–64835. [Google Scholar] [CrossRef] [PubMed]
- Amreddy, N.; Ahmed, R.A.; Munshi, A.; Ramesh, R. Tumor-Targeted Dendrimer Nanoparticles for Combinatorial Delivery of siRNA and Chemotherapy for Cancer Treatment. Methods Mol. Biol. 2020, 2059, 167–189. [Google Scholar] [CrossRef]
- Ramesh, R.; Saeki, T.; Templeton, N.S.; Ji, L.; Stephens, L.C.; Ito, I.; Wilson, D.R.; Wu, Z.; Branch, C.D.; Minna, J.D.; et al. Successful treatment of primary and disseminated human lung cancers by systemic delivery of tumor suppressor genes using an improved liposome vector. Mol. Ther. 2001, 3, 337–350. [Google Scholar] [CrossRef]
- Ahmed, R.; Amreddy, N.; Babu, A.; Munshi, A.; Ramesh, R. Combinatorial Nanoparticle Delivery of siRNA and Antineoplastics for Lung Cancer Treatment. Methods Mol. Biol. 2019, 1974, 265–290. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, J.; Srivastava, A.; Muralidharan, R.; Wang, Q.; Zheng, W.; Zhao, L.; Chen, A.; Zhao, Y.D.; Munshi, A.; Ramesh, R. IL-24 modulates the high mobility group (HMG) A1/miR222 /AKT signaling in lung cancer cells. Oncotarget 2016, 7, 70247–70263. [Google Scholar] [CrossRef] [PubMed]
- Andrade, D.; Mehta, M.; Griffith, J.; Oh, S.; Corbin, J.; Babu, A.; De, S.; Chen, A.; Zhao, Y.D.; Husain, S.; et al. HuR Reduces Radiation-Induced DNA Damage by Enhancing Expression of ARID1A. Cancers 2019, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Komina, A.V.; Palkina, N.V.; Aksenenko, M.B.; Lavrentev, S.N.; Moshev, A.V.; Savchenko, A.A.; Averchuk, A.S.; Rybnikov, Y.A.; Ruksha, T.G. Semaphorin-5A downregulation is associated with enhanced migration and invasion of BRAF-positive melanoma cells under vemurafenib treatment in melanomas with heterogeneous BRAF status. Melanoma Res. 2019, 29, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Glitza Oliva, I.; Tawbi, H.; Davies, M.A. Melanoma Brain Metastases: Current Areas of Investigation and Future Directions. Cancer J. 2017, 23, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Yajima, I.; Kumasaka, M.Y.; Thang, N.D.; Goto, Y.; Takeda, K.; Iida, M.; Ohgami, N.; Tamura, H.; Yamanoshita, O.; Kawamoto, Y.; et al. Molecular Network Associated with MITF in Skin Melanoma Development and Progression. J. Skin Cancer 2011, 2011, 730170. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.J.; Fisher, D.E. The roles of microphthalmia-associated transcription factor and pigmentation in melanoma. Arch. Biochem. Biophys. 2014, 563, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. Pro-survival role of MITF in melanoma. J. Investig. Dermatol. 2015, 135, 352–358. [Google Scholar] [CrossRef]
- Cheli, Y.; Giuliano, S.; Fenouille, N.; Allegra, M.; Hofman, V.; Hofman, P.; Bahadoran, P.; Lacour, J.P.; Tartare-Deckert, S.; Bertolotto, C.; et al. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene 2012, 31, 2461–2470. [Google Scholar] [CrossRef]
- Paz, I.; Kosti, I.; Ares, M., Jr.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014, 42, W361–W367. Available online: https://rbmap.technion.ac.il (accessed on 8 September 2018). [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. Available online: https://weblogo.berkeley.edu (accessed on 14 September 2018). [CrossRef] [PubMed]
- Sullivan, R.J.; Atkins, M.B. Molecular targeted therapy for patients with melanoma: The promise of MAPK pathway inhibition and beyond. Expert Opin. Investig. Drugs 2010, 19, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Goldinger, S.M.; Zimmer, L.; Schulz, C.; Ugurel, S.; Hoeller, C.; Kaehler, K.C.; Schadendorf, D.; Hassel, J.C.; Becker, J.; Hauschild, A.; et al. Upstream mitogen-activated protein kinase (MAPK) pathway inhibition: MEK inhibitor followed by a BRAF inhibitor in advanced melanoma patients. Eur. J. Cancer 2014, 50, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Czyz, M.; Sztiller-Sikorska, M.; Gajos-Michniewicz, A.; Osrodek, M.; Hartman, M.L. Plasticity of Drug-Naive and Vemurafenib- or Trametinib-Resistant Melanoma Cells in Execution of Differentiation/Pigmentation Program. J. Oncol. 2019, 2019, 1697913. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Long, G.V.; Weber, J.S.; Infante, J.R.; Kim, K.B.; Daud, A.; Gonzalez, R.; Sosman, J.A.; Hamid, O.; Schuchter, L.; Cebon, J.; et al. Overall Survival and Durable Responses in Patients With BRAF V600-Mutant Metastatic Melanoma Receiving Dabrafenib Combined With Trametinib. J. Clin. Oncol. 2016, 34, 871–878. [Google Scholar] [CrossRef]
- Pathria, G.; Garg, B.; Borgdorff, V.; Garg, K.; Wagner, C.; Superti-Furga, G.; Wagner, S.N. Overcoming MITF-conferred drug resistance through dual AURKA/MAPK targeting in human melanoma cells. Cell Death Dis. 2016, 7, e2135. [Google Scholar] [CrossRef]
- Muller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta. Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef]
- Busca, R.; Berra, E.; Gaggioli, C.; Khaled, M.; Bille, K.; Marchetti, B.; Thyss, R.; Fitsialos, G.; Larribere, L.; Bertolotto, C.; et al. Hypoxia-inducible factor 1{alpha} is a new target of microphthalmia-associated transcription factor (MITF) in melanoma cells. J. Cell Biol. 2005, 170, 49–59. [Google Scholar] [CrossRef]
- McGill, G.G.; Horstmann, M.; Widlund, H.R.; Du, J.; Motyckova, G.; Nishimura, E.K.; Lin, Y.L.; Ramaswamy, S.; Avery, W.; Ding, H.F.; et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 2002, 109, 707–718. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Zhao, F.; Evans, K.; Xiao, C.; DeVito, N.; Theivanthiran, B.; Holtzhausen, A.; Siska, P.J.; Blobe, G.C.; Hanks, B.A. Stromal Fibroblasts Mediate Anti-PD-1 Resistance via MMP-9 and Dictate TGFbeta Inhibitor Sequencing in Melanoma. Cancer Immunol. Res. 2018, 6, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.; Gill, J.; Kamra, S.; Chen, L.; Ahuja, A. Indirect treatment comparison of dabrafenib plus trametinib versus vemurafenib plus cobimetinib in previously untreated metastatic melanoma patients. J. Hematol. Oncol. 2017, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, D.; Ramalingam, S.; Sengupta, T.K.; Bandyopadhyay, S.; Dellis, S.; Tholanikunnel, B.G.; Fernandes, D.J.; Spicer, E.K. Regulation of Bcl-2 expression by HuR in HL60 leukemia cells and A431 carcinoma cells. Mol. Cancer Res. 2009, 7, 1354–1366. [Google Scholar] [CrossRef]
- Blanco, F.F.; Jimbo, M.; Wulfkuhle, J.; Gallagher, I.; Deng, J.; Enyenihi, L.; Meisner-Kober, N.; Londin, E.; Rigoutsos, I.; Sawicki, J.A.; et al. The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells. Oncogene 2016, 35, 2529–2541. [Google Scholar] [CrossRef]
- Urban, P.; Rabajdova, M.; Velika, B.; Spakova, I.; Bolerazska, B.; Marekova, M. The Importance of MITF Signaling Pathway in the Regulation of Proliferation and Invasiveness of Malignant Melanoma. Klin. Onkol. 2016, 29, 347–350. [Google Scholar] [CrossRef]
- Najem, A.; Krayem, M.; Sales, F.; Hussein, N.; Badran, B.; Robert, C.; Awada, A.; Journe, F.; Ghanem, G.E. P53 and MITF/Bcl-2 identified as key pathways in the acquired resistance of NRAS-mutant melanoma to MEK inhibition. Eur. J. Cancer 2017, 83, 154–165. [Google Scholar] [CrossRef]
- Ennen, M.; Keime, C.; Gambi, G.; Kieny, A.; Coassolo, S.; Thibault-Carpentier, C.; Margerin-Schaller, F.; Davidson, G.; Vagne, C.; Lipsker, D.; et al. MITF-High and MITF-Low Cells and a Novel Subpopulation Expressing Genes of Both Cell States Contribute to Intra- and Intertumoral Heterogeneity of Primary Melanoma. Clin. Cancer Res. 2017, 23, 7097–7107. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Arozarena, I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment. Cell Melanoma Res. 2015, 28, 390–406. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.; Rao, J.N.; Zou, T.; Liu, L.; Xiao, L.; Yu, T.X.; Turner, D.J.; Gorospe, M.; Wang, J.Y. Post-transcriptional regulation of MEK-1 by polyamines through the RNA-binding protein HuR modulating intestinal epithelial apoptosis. Biochem. J. 2010, 426, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Kim, T.K.; Brozyna, A.A.; Janjetovic, Z.; Brooks, D.L.; Schwab, L.P.; Skobowiat, C.; Jozwicki, W.; Seagroves, T.N. The role of melanogenesis in regulation of melanoma behavior: Melanogenesis leads to stimulation of HIF-1alpha expression and HIF-dependent attendant pathways. Arch. Biochem. Biophys. 2014, 563, 79–93. [Google Scholar] [CrossRef]
- Alam, M.B.; Ahmed, A.; Motin, M.A.; Kim, S.; Lee, S.H. Attenuation of melanogenesis by Nymphaea nouchali (Burm. f) flower extract through the regulation of cAMP/CREB/MAPKs/MITF and proteasomal degradation of tyrosinase. Sci. Rep. 2018, 8, 13928. [Google Scholar] [CrossRef]
- Jean, D.; Bar-Eli, M. Regulation of tumor growth and metastasis of human melanoma by the CREB transcription factor family. Mol. Cell. Biochem. 2000, 212, 19–28. [Google Scholar] [CrossRef]
- Padua, R.A.; Barrass, N.; Currie, G.A. A novel transforming gene in a human malignant melanoma cell line. Nature 1984, 311, 671–673. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Piulats, J.M.; Goebeler, M.; Galloway, I.; Lugowska, I.; Becker, J.C.; Vihinen, P.; Van Calster, J.; Hadjistilianou, T.; Proenca, R.; et al. Uveal Melanoma: A European Network to Face the Many Challenges of a Rare Cancer. Cancers 2019, 11, 817. [Google Scholar] [CrossRef]
- Eagle, R.C., Jr. Ocular tumors: Triumphs, challenges and controversies. Saudi. J. Ophthalmol. 2013, 27, 129–132. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, R.; Muralidharan, R.; Srivastava, A.; Johnston, S.E.; Zhao, Y.D.; Ekmekcioglu, S.; Munshi, A.; Ramesh, R. Molecular Targeting of HuR Oncoprotein Suppresses MITF and Induces Apoptosis in Melanoma Cells. Cancers 2021, 13, 166. https://doi.org/10.3390/cancers13020166
Ahmed R, Muralidharan R, Srivastava A, Johnston SE, Zhao YD, Ekmekcioglu S, Munshi A, Ramesh R. Molecular Targeting of HuR Oncoprotein Suppresses MITF and Induces Apoptosis in Melanoma Cells. Cancers. 2021; 13(2):166. https://doi.org/10.3390/cancers13020166
Chicago/Turabian StyleAhmed, Rebaz, Ranganayaki Muralidharan, Akhil Srivastava, Sarah E. Johnston, Yan D. Zhao, Suhendan Ekmekcioglu, Anupama Munshi, and Rajagopal Ramesh. 2021. "Molecular Targeting of HuR Oncoprotein Suppresses MITF and Induces Apoptosis in Melanoma Cells" Cancers 13, no. 2: 166. https://doi.org/10.3390/cancers13020166
APA StyleAhmed, R., Muralidharan, R., Srivastava, A., Johnston, S. E., Zhao, Y. D., Ekmekcioglu, S., Munshi, A., & Ramesh, R. (2021). Molecular Targeting of HuR Oncoprotein Suppresses MITF and Induces Apoptosis in Melanoma Cells. Cancers, 13(2), 166. https://doi.org/10.3390/cancers13020166