
Transcriptomic Analysis in Multiple Myeloma and Primary Plasma Cell Leukemia with t(11;14) Reveals Different Expression Patterns with Biological Implications in Venetoclax Sensitivity

 , , , , , ,
, , , , , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Gene Expression Profiling
2.3. Quantitative Real-Rime PCR (qRT-PCR)
3. Results
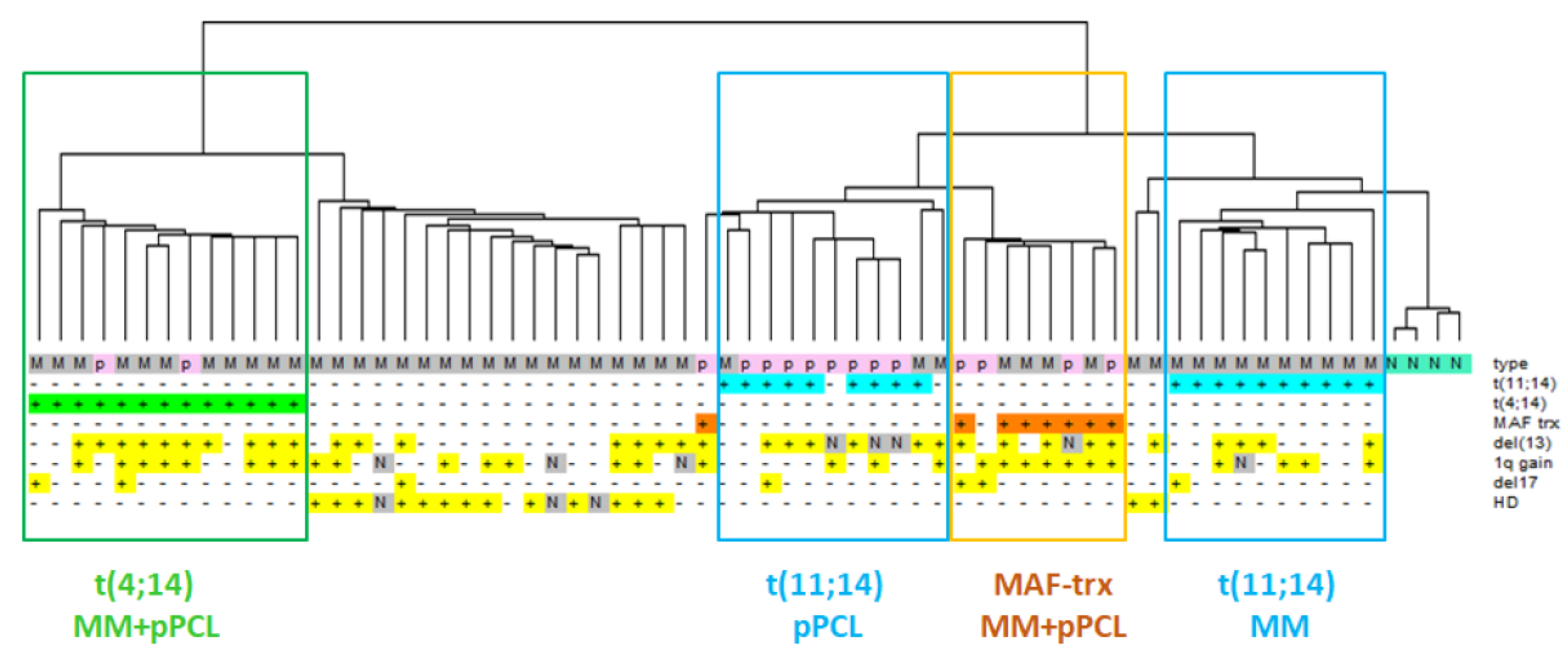
3.1. Distinct Transcriptomic Signature of MM and pPCL Samples with t(11;14)
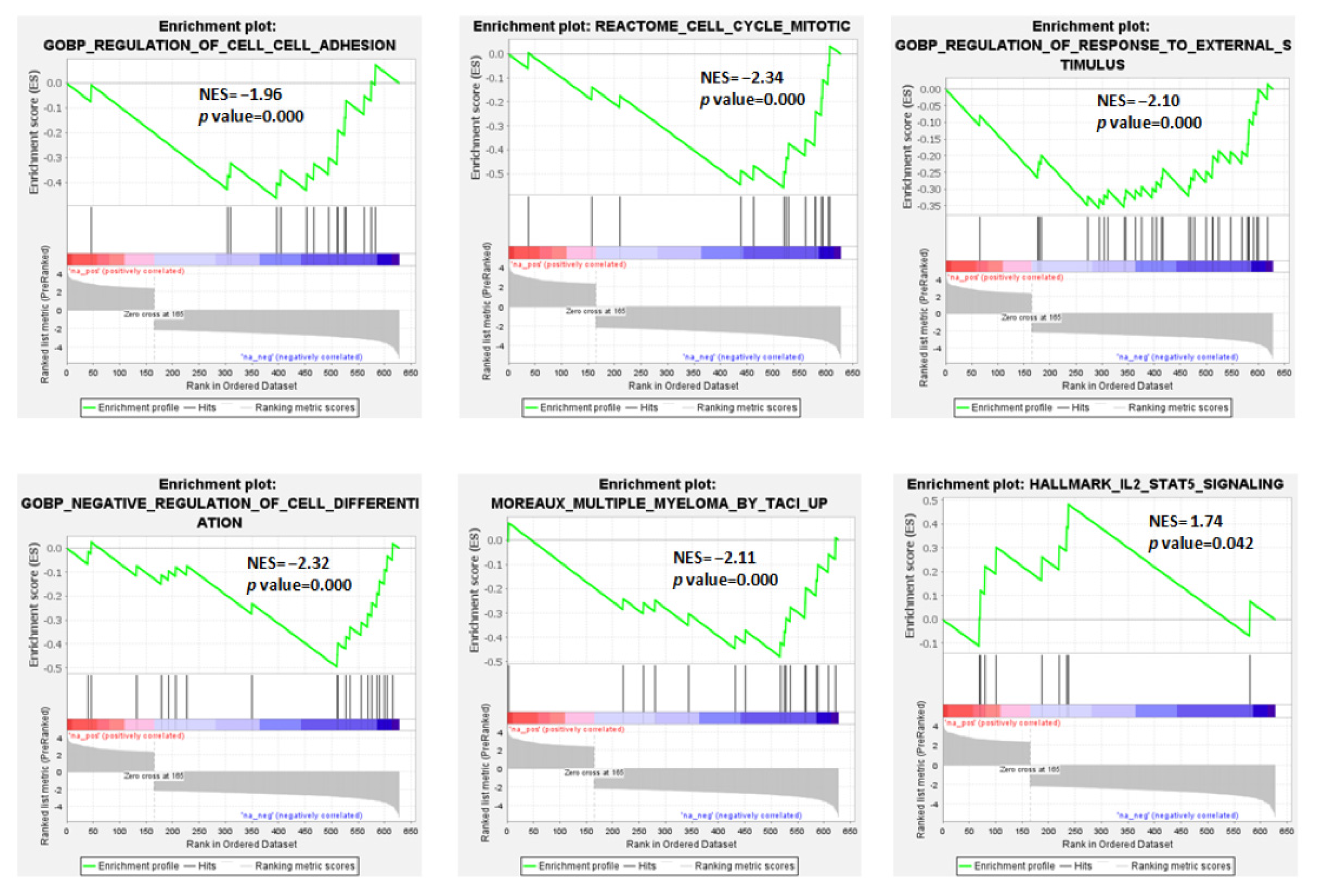
3.2. Protein-Coding Genes: Molecular Pathways and Gene Sets Modulated in MM and pPCL in Association with t(11;14)
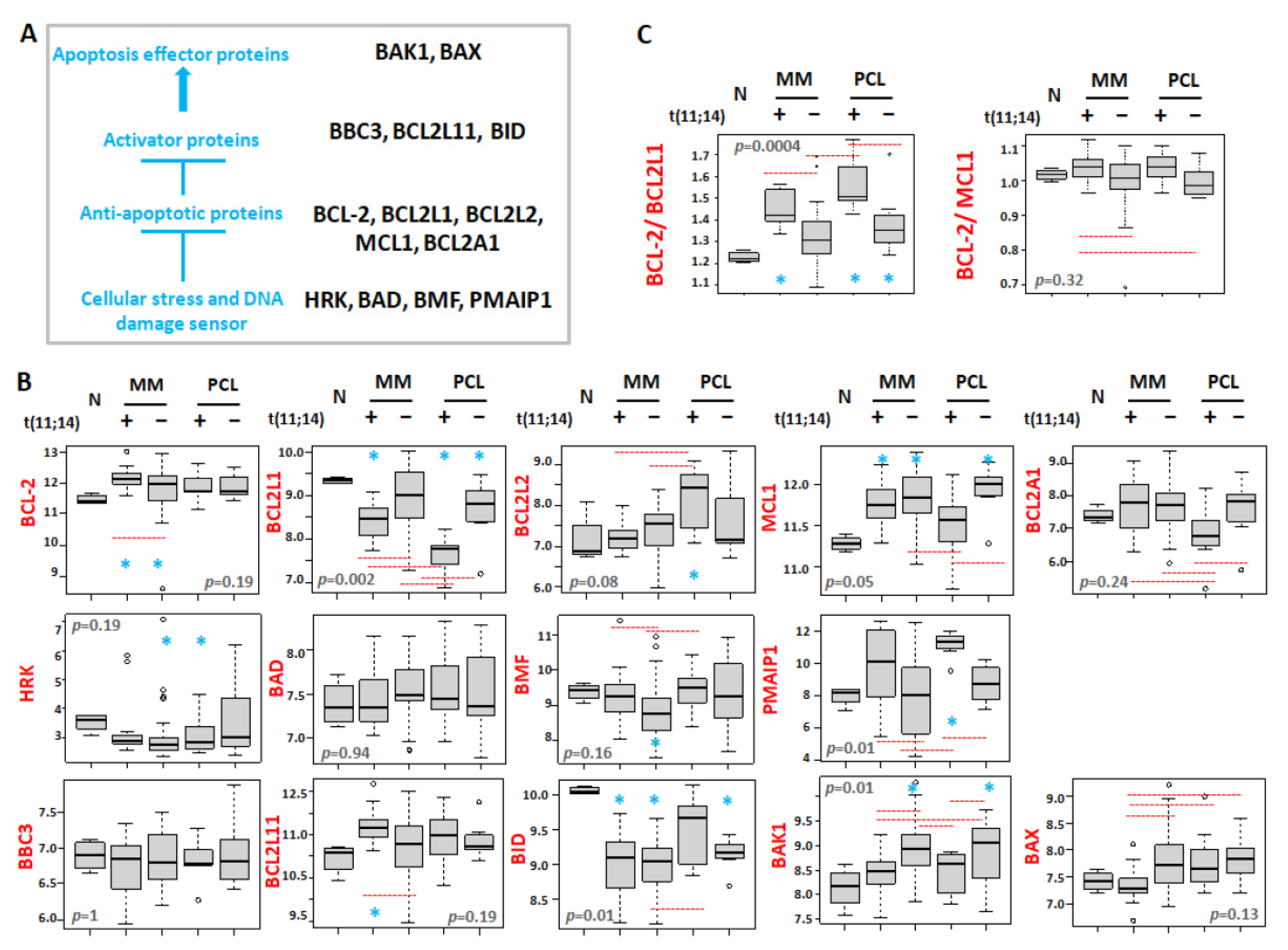
3.3. Differential Expression of BCL2 Gene Family Members in Association with t(11;14)
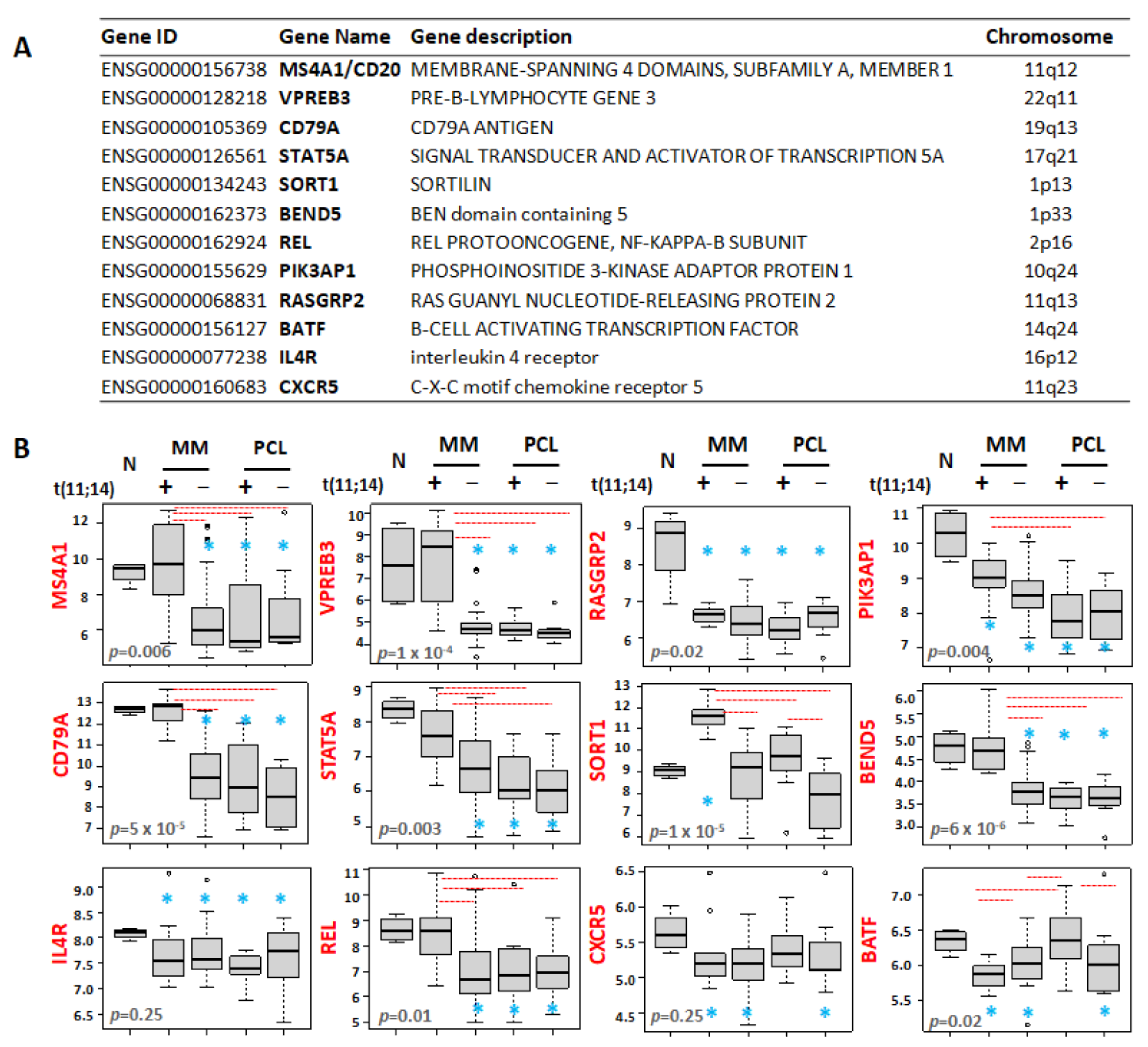
3.4. Differential Expression Patterns of B-Cell-Associated Genes
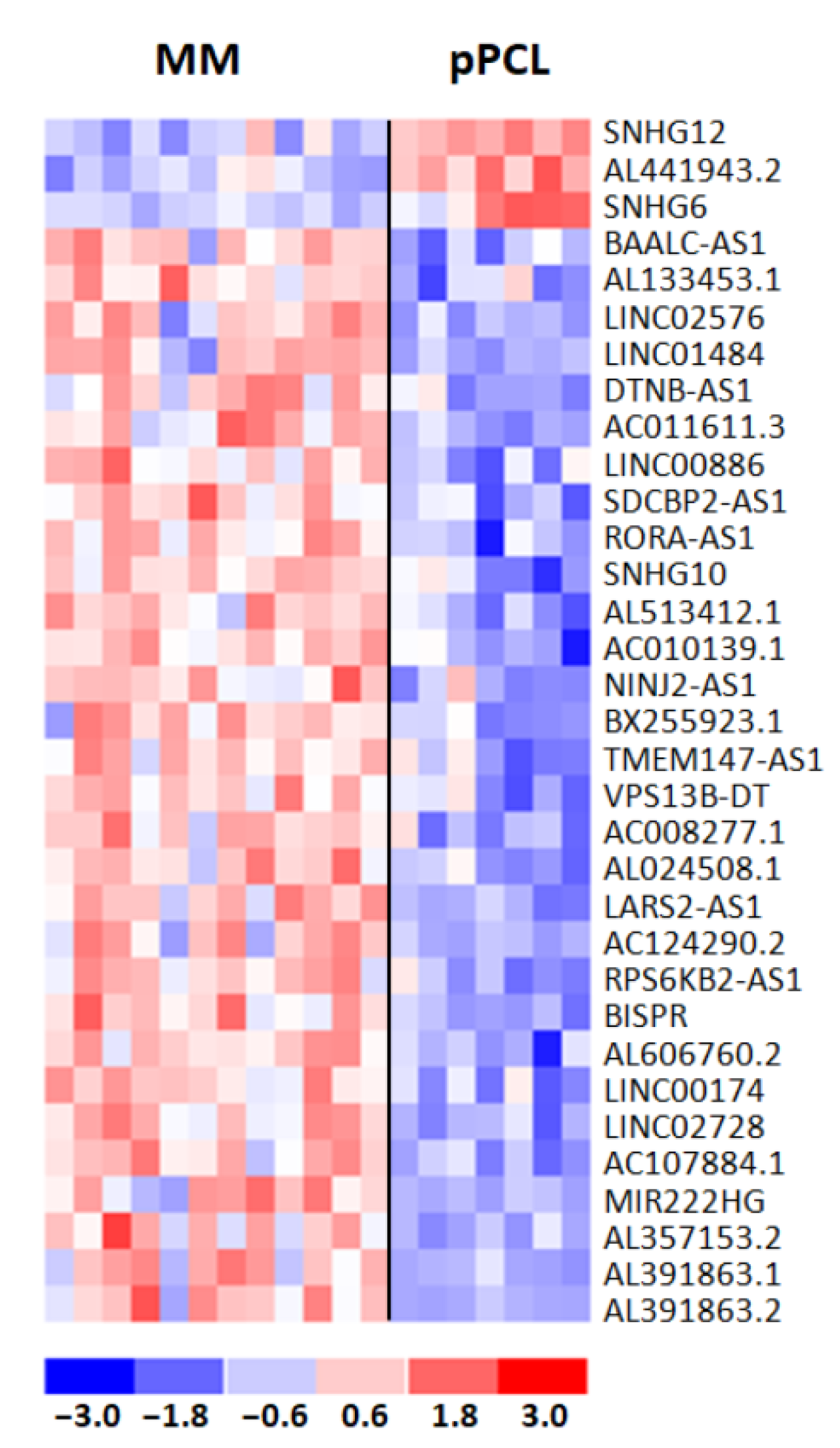
3.5. Differential Expression Patterns of lncRNAs Associated with t(11;14)
3.6. Clinical Relevance of lncRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Albarracin, F.; Fonseca, R. Plasma cell leukemia. Blood Rev. 2011, 25, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Gavriatopoulou, M.; Musto, P.; Caers, J.; Merlini, G.; Kastritis, E.; van de Donk, N.; Gay, F.; Hegenbart, U.; Hajek, R.; Zweegman, S.; et al. European myeloma network recommendations on diagnosis and management of patients with rare plasma cell dyscrasias. Leukemia 2018, 32, 1883–1898. [Google Scholar] [CrossRef] [PubMed]
- Musto, P. Progress in the Treatment of Primary Plasma Cell Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2082–2084. [Google Scholar] [CrossRef]
- Musto, P.; Statuto, T.; Valvano, L.; Grieco, V.; Nozza, F.; Vona, G.; Bochicchio, G.B.; La Rocca, F.; D’Auria, F. An update on biology, diagnosis and treatment of primary plasma cell leukemia. Expert Rev. Hematol. 2019, 12, 245–253. [Google Scholar] [CrossRef]
- Cifola, I.; Lionetti, M.; Pinatel, E.; Todoerti, K.; Mangano, E.; Pietrelli, A.; Fabris, S.; Mosca, L.; Simeon, V.; Petrucci, M.T.; et al. Whole-exome sequencing of primary plasma cell leukemia discloses heterogeneous mutational patterns. Oncotarget 2015, 6, 17543–17558. [Google Scholar] [CrossRef]
- Lionetti, M.; Musto, P.; Di Martino, M.T.; Fabris, S.; Agnelli, L.; Todoerti, K.; Tuana, G.; Mosca, L.; Gallo Cantafio, M.E.; Grieco, V.; et al. Biological and clinical relevance of miRNA expression signatures in primary plasma cell leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3130–3142. [Google Scholar] [CrossRef]
- Mattioli, M.; Agnelli, L.; Fabris, S.; Baldini, L.; Morabito, F.; Bicciato, S.; Verdelli, D.; Intini, D.; Nobili, L.; Cro, L.; et al. Gene expression profiling of plasma cell dyscrasias reveals molecular patterns associated with distinct IGH translocations in multiple myeloma. Oncogene 2005, 24, 2461–2473. [Google Scholar] [CrossRef]
- Rojas, E.A.; Corchete, L.A.; Mateos, M.V.; Garcia-Sanz, R.; Misiewicz-Krzeminska, I.; Gutierrez, N.C. Transcriptome analysis reveals significant differences between primary plasma cell leukemia and multiple myeloma even when sharing a similar genetic background. Blood Cancer J. 2019, 9, 90. [Google Scholar] [CrossRef]
- Schinke, C.; Boyle, E.M.; Ashby, C.; Wang, Y.; Lyzogubov, V.; Wardell, C.; Qu, P.; Hoering, A.; Deshpande, S.; Ryan, K.; et al. Genomic analysis of primary plasma cell leukemia reveals complex structural alterations and high-risk mutational patterns. Blood Cancer J. 2020, 10, 70. [Google Scholar] [CrossRef]
- Todoerti, K.; Agnelli, L.; Fabris, S.; Lionetti, M.; Tuana, G.; Mosca, L.; Lombardi, L.; Grieco, V.; Bianchino, G.; D’Auria, F.; et al. Transcriptional characterization of a prospective series of primary plasma cell leukemia revealed signatures associated with tumor progression and poorer outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3247–3258. [Google Scholar] [CrossRef]
- Todoerti, K.; Calice, G.; Trino, S.; Simeon, V.; Lionetti, M.; Manzoni, M.; Fabris, S.; Barbieri, M.; Pompa, A.; Baldini, L.; et al. Global methylation patterns in primary plasma cell leukemia. Leukemia Res. 2018, 73, 95–102. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Nair, B.; Qu, P.; Hansen, E.; Zhang, Q.; Petty, N.; Waheed, S.; Shaughnessy, J.D., Jr.; Alsayed, Y.; Heuck, C.J.; et al. Primary plasma cell leukemia: Clinical and laboratory presentation, gene-expression profiling and clinical outcome with Total Therapy protocols. Leukemia 2012, 26, 2398–2405. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Daviet, A.; Brigaudeau, C.; Callet-Bauchu, E.; Terre, C.; Lafage-Pochitaloff, M.; Desangles, F.; Ramond, S.; Talmant, P.; Bataille, R. Cytogenetic, interphase, and multicolor fluorescence in situ hybridization analyses in primary plasma cell leukemia: A study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myelome and the Groupe Francais de Cytogenetique Hematologique. Blood 2001, 97, 822–825. [Google Scholar] [CrossRef]
- Gutierrez, N.C.; Hernandez, J.M.; Garcia, J.L.; Canizo, M.C.; Gonzalez, M.; Hernandez, J.; Gonzalez, M.B.; Garcia-Marcos, M.A.; San Miguel, J.F. Differences in genetic changes between multiple myeloma and plasma cell leukemia demonstrated by comparative genomic hybridization. Leukemia 2001, 15, 840–845. [Google Scholar] [CrossRef][Green Version]
- Mosca, L.; Musto, P.; Todoerti, K.; Barbieri, M.; Agnelli, L.; Fabris, S.; Tuana, G.; Lionetti, M.; Bonaparte, E.; Sirchia, S.M.; et al. Genome-wide analysis of primary plasma cell leukemia identifies recurrent imbalances associated with changes in transcriptional profiles. Am. J. Hematol. 2013, 88, 16–23. [Google Scholar] [CrossRef]
- Touzeau, C.; Maciag, P.; Amiot, M.; Moreau, P. Targeting Bcl-2 for the treatment of multiple myeloma. Leukemia 2018, 32, 1899–1907. [Google Scholar] [CrossRef]
- Gupta, V.A.; Ackley, J.; Kaufman, J.L.; Boise, L.H. BCL2 Family Inhibitors in the Biology and Treatment of Multiple Myeloma. Blood Lymphat. Cancer Targets Ther. 2021, 11, 11–24. [Google Scholar] [CrossRef]
- Gupta, V.A.; Barwick, B.G.; Matulis, S.M.; Shirasaki, R.; Jaye, D.L.; Keats, J.; Oberlton, B.; Joseph, N.; Hofmeister, C.; Heffner, L.T.; et al. Venetoclax sensitivity in multiple myeloma is associated with B cell gene expression. Blood 2021, 137, 3604–3615. [Google Scholar] [CrossRef]
- Ronchetti, D.; Agnelli, L.; Pietrelli, A.; Todoerti, K.; Manzoni, M.; Taiana, E.; Neri, A. A compendium of long non-coding RNAs transcriptional fingerprint in multiple myeloma. Sci. Rep. 2018, 8, 6557. [Google Scholar] [CrossRef]
- Ronchetti, D.; Agnelli, L.; Taiana, E.; Galletti, S.; Manzoni, M.; Todoerti, K.; Musto, P.; Strozzi, F.; Neri, A. Distinct lncRNA transcriptional fingerprints characterize progressive stages of multiple myeloma. Oncotarget 2016, 7, 14814–14830. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Taiana, E.; Ronchetti, D.; Favasuli, V.; Todoerti, K.; Manzoni, M.; Amodio, N.; Tassone, P.; Agnelli, L.; Neri, A. Long non-coding RNA NEAT1 shows high expression unrelated to molecular features and clinical outcome in multiple myeloma. Haematologica 2019, 104, e72–e76. [Google Scholar] [CrossRef]
- Ronchetti, D.; Todoerti, K.; Vinci, C.; Favasuli, V.; Agnelli, L.; Manzoni, M.; Pelizzoni, F.; Chiaramonte, R.; Platonova, N.; Giuliani, N.; et al. Expression Pattern and Biological Significance of the lncRNA ST3GAL6-AS1 in Multiple Myeloma. Cancers 2020, 12, 782. [Google Scholar] [CrossRef]
- Moreaux, J.; Cremer, F.W.; Reme, T.; Raab, M.; Mahtouk, K.; Kaukel, P.; Pantesco, V.; De Vos, J.; Jourdan, E.; Jauch, A.; et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood 2005, 106, 1021–1030. [Google Scholar] [CrossRef]
- Jelinek, T.; Mihalyova, J.; Kascak, M.; Duras, J.; Popkova, T.; Benkova, K.; Richterova, P.; Plonkova, H.; Zuchnicka, J.; Broskevicova, L.; et al. Single-agent venetoclax induces MRD-negative response in relapsed primary plasma cell leukemia with t(11;14). Am. J. Hematol. 2019, 94, E35–E37. [Google Scholar] [CrossRef]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef]
- Sigova, A.A.; Mullen, A.C.; Molinie, B.; Gupta, S.; Orlando, D.A.; Guenther, M.G.; Almada, A.E.; Lin, C.; Sharp, P.A.; Giallourakis, C.C.; et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.Y.; Smith, A.A.T.; da Silva, M.F.; Matthey-Doret, C.; Rueedi, R.; Sönmez, R.; Ding, D.; Kutalik, Z.; Bergmann, S.; Marques, A.C. cis-Acting Complex-Trait-Associated lincRNA Expression Correlates with Modulation of Chromosomal Architecture. Cell Rep. 2017, 18, 2280–2288. [Google Scholar] [CrossRef] [PubMed]
- Trinklein, N.D.; Aldred, S.F.; Hartman, S.J.; Schroeder, D.I.; Otillar, R.P.; Myers, R.M. An abundance of bidirectional promoters in the human genome. Genome Res. 2004, 14, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-X.; Leonard, W.J. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 2000, 19, 2566–2576. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef]
- Matulis, S.M.; Gupta, V.A.; Neri, P.; Bahlis, N.J.; Maciag, P.; Leverson, J.D.; Heffner, L.T., Jr.; Lonial, S.; Nooka, A.K.; Kaufman, J.L.; et al. Functional profiling of venetoclax sensitivity can predict clinical response in multiple myeloma. Leukemia 2019, 33, 1291–1296. [Google Scholar] [CrossRef]
- Touzeau, C.; Dousset, C.; Le Gouill, S.; Sampath, D.; Leverson, J.D.; Souers, A.J.; Maiga, S.; Bene, M.C.; Moreau, P.; Pellat-Deceunynck, C.; et al. The Bcl-2 specific BH3 mimetic ABT-199: A promising targeted therapy for t(11;14) multiple myeloma. Leukemia 2014, 28, 210–212. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Buadi, F.K.; Kumar, S.K. Combination therapy incorporating Bcl-2 inhibition with Venetoclax for the treatment of refractory primary plasma cell leukemia with t (11;14). Eur. J. Haematol. 2018, 100, 215–217. [Google Scholar] [CrossRef]
- Nalghranyan, S.; Singh, A.P.; Schinke, C. The combination of venetoclax, daratumumab and dexamethasone for the treatment of refractory primary plasma cell leukemia. Am. J. Hematol. 2020, 95, E34–E35. [Google Scholar] [CrossRef]
- Valliani, S.; Ali, M.; Mahmoo, O.; Hinduja, S.; Chen, C.K.; Damon, L.; Abdulhaq, H. Efficacy of Venetoclax and Dexamethasone in Refractory IgM Primary Plasma Cell Leukemia with t(11;14) and TP53 Mutation: A Case Report and Literature Review. Case Rep. Hematol. 2020, 2020, 8823877. [Google Scholar] [CrossRef]
- Gomez-Bougie, P.; Maiga, S.; Tessoulin, B.; Bourcier, J.; Bonnet, A.; Rodriguez, M.S.; Le Gouill, S.; Touzeau, C.; Moreau, P.; Pellat-Deceunynck, C.; et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood 2018, 132, 2656–2669. [Google Scholar] [CrossRef]
- Touzeau, C.; Ryan, J.; Guerriero, J.; Moreau, P.; Chonghaile, T.N.; Le Gouill, S.; Richardson, P.; Anderson, K.; Amiot, M.; Letai, A. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 2016, 30, 761–764. [Google Scholar] [CrossRef]
- Murakami, K.; Kikugawa, S.; Kobayashi, Y.; Uehara, S.; Suzuki, T.; Kato, H.; Udagawa, N.; Nakamura, Y. Olfactomedin-like protein OLFML1 inhibits Hippo signaling and mineralization in osteoblasts. Biochem. Biophys. Res. Commun. 2018, 505, 419–425. [Google Scholar] [CrossRef]
- Worby, C.A.; Dixon, J.E. Sorting out the cellular functions of sorting nexins. Rev. Mol. Cell Biol. 2002, 3, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Mostoslavsky, G.; Sattler, M.; Tai, Y.T.; Hayashi, T.; Catley, L.P.; Hideshima, T.; Mulligan, R.C.; Chauhan, D.; Anderson, K.C. Critical role for hematopoietic cell kinase (Hck)-mediated phosphorylation of Gab1 and Gab2 docking proteins in interleukin 6-induced proliferation and survival of multiple myeloma cells. J. Biol. Chem. 2004, 279, 21658–21665. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Zhang, T.; Zhang, D.; Xie, L.; Zou, X.; Lei, L.; Wu, D.; Liu, L. The long non-coding RNA, SNHG6-003, functions as a competing endogenous RNA to promote the progression of hepatocellular carcinoma. Oncogene 2017, 36, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Yuan, Y.; Li, C.; Guo, T.; Qi, H.; Xiao, Y.; Dong, X.; Liu, Z.; Liu, Q. Upregulation of SNHG6 regulates ZEB1 expression by competitively binding miR-101-3p and interacting with UPF1 in hepatocellular carcinoma. Cancer Lett. 2016, 383, 183–194. [Google Scholar] [CrossRef]
- Zhu, X.; Yang, G.; Xu, J.; Zhang, C. Silencing of SNHG6 induced cell autophagy by targeting miR-26a-5p/ULK1 signaling pathway in human osteosarcoma. Cancer Cell Int. 2019, 19, 82. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | MM | pPCL | ||||
|---|---|---|---|---|---|---|
| Median age, years (range) | 67 (39–78) | 67 (39–78) | ||||
| Male gender (%) | 23 (46%) | 5 (33%) | ||||
| ISS, I–II–III | 13–28–9 | |||||
| Sample Features | Positive | Negative | NA | Positive | Negative | NA |
| HD | 15 | 32 | 3 | 0 | 15 | 0 |
| t(11;14) | 12 | 38 | 0 | 7 | 8 | 0 |
| t(4;14) | 12 | 38 | 0 | 2 | 13 | 0 |
| MAF-trx | 5 | 45 | 0 | 4 | 11 | 0 |
| none | 3 | 47 | 2 | 13 | ||
| del(17p) | 4 | 46 | 0 | 3 | 12 | 0 |
| del(13q) | 27 | 23 | 0 | 9 | 2 | 4 |
| 1q gain | 25 | 21 | 4 | 7 | 8 | 0 |
| lncRNA Gene ID | lncRNA Gene Name | Overlapping/Nearby Gene ID | Overlapping/Nearby Gene Name | Pearson’s Correlation |
|---|---|---|---|---|
| ENSG00000223642 | AC008277.1 | ENSG00000123636 | BAZ2B | r = 0.53, q-value = 1.4 × 10−57 |
| ENSG00000224220 | DTNB-AS1 | ENSG00000138101 | DTNB | r = 0.89, q-value = 4.3 × 10−272 |
| ENSG00000229502 | AL391863.1 | ENSG00000130340 | SNX9 | r = 0.65, q-value = 3.5 × 10−95 |
| ENSG00000234361 | AL391863.2 | ENSG00000130340 | SNX9 | r = 0.58, q-value = 2.4 × 10−71 |
| ENSG00000234684 | SDCBP2-AS1 | ENSG00000088832 | FKBP1A | r = 0.56, q-value = 1.8 × 10−64 |
| ENSG00000245534 | RORA-AS1 | ENSG00000128915 | ICE2 | r = 0.51, q-value = 1.5 × 10−38 |
| ENSG00000247081 | BAALC-AS1 | ENSG00000164930 | FZD6 | r = 0.84, q-value = 1.5 × 10−209 |
| ENSG00000251364 | AC107884.1 | ENSG00000183801 | OLFML1 | r = 0.61, q-value = 2.2 × 10−79 |
| ENSG00000257453 | AC011611.3 | ENSG00000139289 | PHLDA1 | r = 0.87, q-value = 1.2 × 10−233 |
| ENSG00000272189 | AL024508.1 | ENSG00000135525 | MAP7 | r = 0.55, q-value = 1.6 × 10−63 |
| OS Univariate Cox Analysis | PFS Univariate Cox Analysis | ||||
|---|---|---|---|---|---|
| Molecular Lesion | N (%) High Level | HR (95% CI) | p-Value | HR (95% CI) | p-Value |
| Linc00886.median | 240 (48%) | 0.65 (0.45–0.94) | 0.0213 | 0.73 (0.57–0.95) | 0.017 |
| NINJ2-AS1.median | 246 (49%) | 0.71 (0.49–1.03) | 0.0691 | 0.76 (0.59–0.99) | 0.0386 |
| AL513412.1.median | 233 (47%) | 0.77 (0.53–1.12) | 0.173 | 0.78 (0.60–1.01) | 0.0641 |
| Linc02728.median | 263 (53%) | 0.79 (0.55–1.13) | 0.195 | 0.76 (0.59–0.98) | 0.0343 |
| SNHG6.median | 262 (53%) | 1.45 (1.00–2.09) | 0.0478 | 1.11 (0.86–1.43) | 0.437 |
| del1p.CDKN2C | 143 (29%) | 1.61 (1.10–2.35) | 0.0148 | 1.30 (0.98–1.71) | 0.0646 |
| del13q.RB1 | 258 (52%) | 2.11 (1.44–3.09) | 0.000119 | 1.71 (1.32–2.21) | 4.71 × 10−5 |
| HD | 281 (57%) | 0.64 (0.44–0.92) | 0.0149 | 0.67 (0.52–0.86) | 0.00166 |
| TP53.alt.1q.gain.amp | 19 (4%) | 3.63 (1.89–6.97) | 0.000103 | 2.46 (1.38–4.42) | 0.00243 |
| TP53.alt | 40 (8%) | 1.05 (0.53–2.07) | 0.892 | 0.85 (0.52–1.39) | 0.511 |
| gain.amp.1q | 164 (33%) | 1.68 (1.17–2.43) | 0.0051 | 1.52 (1.17–1.98) | 0.00151 |
| DIS3mut | 50 (10%) | 1.63 (1.06–2.50) | 0.0264 | 1.62 (1.18–2.22) | 0.00308 |
| NRASmut | 117 (24%) | 0.90 (0.62–1.32) | 0.596 | 1.01 (0.79–1.29) | 0.924 |
| KRASmut | 121 (24%) | 1.07 (0.77–1.49) | 0.694 | 1.03 (0.80–1.31) | 0.827 |
| BRAF.mut | 36 (7%) | 1.18 (0.60–2.31) | 0.628 | 0.88 (0.52–1.48) | 0.626 |
| FAM46C.mut | 49 (10%) | 0.78 (0.42–1.43) | 0.419 | 1.07 (0.74–1.53) | 0.728 |
| TRAF3.mut | 38 (8%) | 0.42 (0.17–1.06) | 0.0668 | 0.80 (0.52–1.22) | 0.297 |
| t11.14 | 102 (21%) | 0.92 (0.58–1.46) | 0.726 | 0.94 (0.68–1.30) | 0.694 |
| t4.14 | 69 (14%) | 1.63 (1.04–2.55) | 0.0335 | 1.60 (1.14–2.23) | 0.00612 |
| trx.MAF | 33 (7%) | 1.96 (1.08–3.57) | 0.0276 | 1.63 (1.03–2.57) | 0.0376 |
| trx.MYC | 20 (4%) | 1.92 (0.94–3.94) | 0.0748 | 1.94 (1.13–3.34) | 0.0163 |
| ISS I | 187 (38%) | 0.31 (0.19–0.50) | 1.86 × 10−6 | 0.46 (0.34–0.61) | 1.20 × 10−7 |
| ISS II | 171 (34%) | 1.13 (0.78–1.64) | 0.524 | 1.18 (0.91–1.54) | 0.213 |
| ISS III | 139 (28%) | 2.45 (1.70–3.53) | 1.62 × 10−6 | 1.92 (1.48–2.50) | 1.14 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todoerti, K.; Taiana, E.; Puccio, N.; Favasuli, V.; Lionetti, M.; Silvestris, I.; Gentile, M.; Musto, P.; Morabito, F.; Gianelli, U.; et al. Transcriptomic Analysis in Multiple Myeloma and Primary Plasma Cell Leukemia with t(11;14) Reveals Different Expression Patterns with Biological Implications in Venetoclax Sensitivity. Cancers 2021, 13, 4898. https://doi.org/10.3390/cancers13194898
Todoerti K, Taiana E, Puccio N, Favasuli V, Lionetti M, Silvestris I, Gentile M, Musto P, Morabito F, Gianelli U, et al. Transcriptomic Analysis in Multiple Myeloma and Primary Plasma Cell Leukemia with t(11;14) Reveals Different Expression Patterns with Biological Implications in Venetoclax Sensitivity. Cancers. 2021; 13(19):4898. https://doi.org/10.3390/cancers13194898
Chicago/Turabian StyleTodoerti, Katia, Elisa Taiana, Noemi Puccio, Vanessa Favasuli, Marta Lionetti, Ilaria Silvestris, Massimo Gentile, Pellegrino Musto, Fortunato Morabito, Umberto Gianelli, and et al. 2021. "Transcriptomic Analysis in Multiple Myeloma and Primary Plasma Cell Leukemia with t(11;14) Reveals Different Expression Patterns with Biological Implications in Venetoclax Sensitivity" Cancers 13, no. 19: 4898. https://doi.org/10.3390/cancers13194898
APA StyleTodoerti, K., Taiana, E., Puccio, N., Favasuli, V., Lionetti, M., Silvestris, I., Gentile, M., Musto, P., Morabito, F., Gianelli, U., Bolli, N., Baldini, L., Neri, A., & Ronchetti, D. (2021). Transcriptomic Analysis in Multiple Myeloma and Primary Plasma Cell Leukemia with t(11;14) Reveals Different Expression Patterns with Biological Implications in Venetoclax Sensitivity. Cancers, 13(19), 4898. https://doi.org/10.3390/cancers13194898