
Evaluation of Comprehensive Gene Expression and NK Cell-Mediated Killing in Glioblastoma Cell Line-Derived Spheroids

 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Method
2.1. Gene Expression Based on The Cancer Genome Atlas (TCGA) Data Set
2.2. Cell Lines
2.3. Spheroid Culture
2.4. Expansion of Primary Human NK Cells and Lymphocyte-Activated Killer Cells
2.5. CFSE-Based Cytotoxic Assay
2.6. Microarray Gene Expression Assay
2.7. Statistical Analysis
3. Results
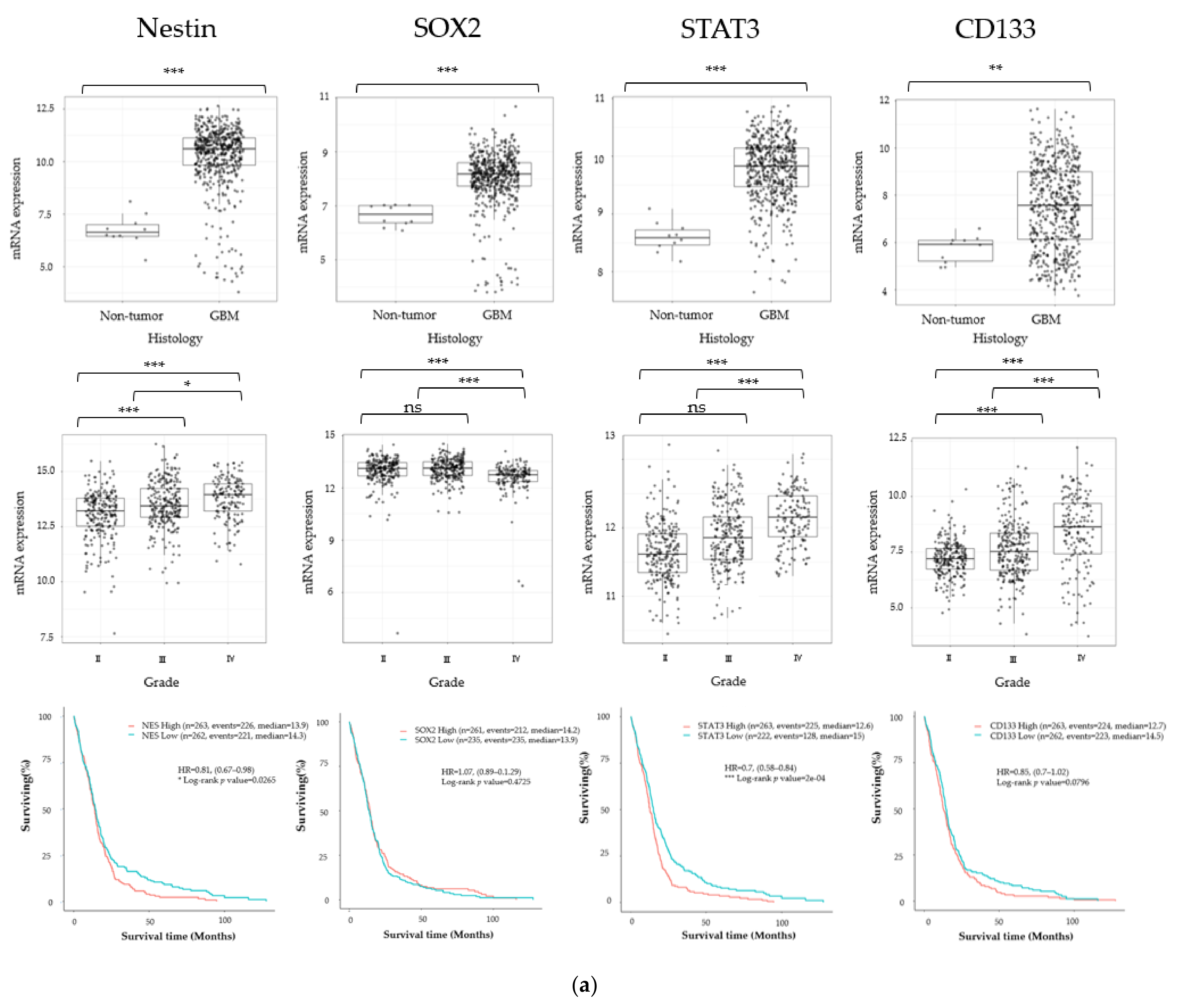
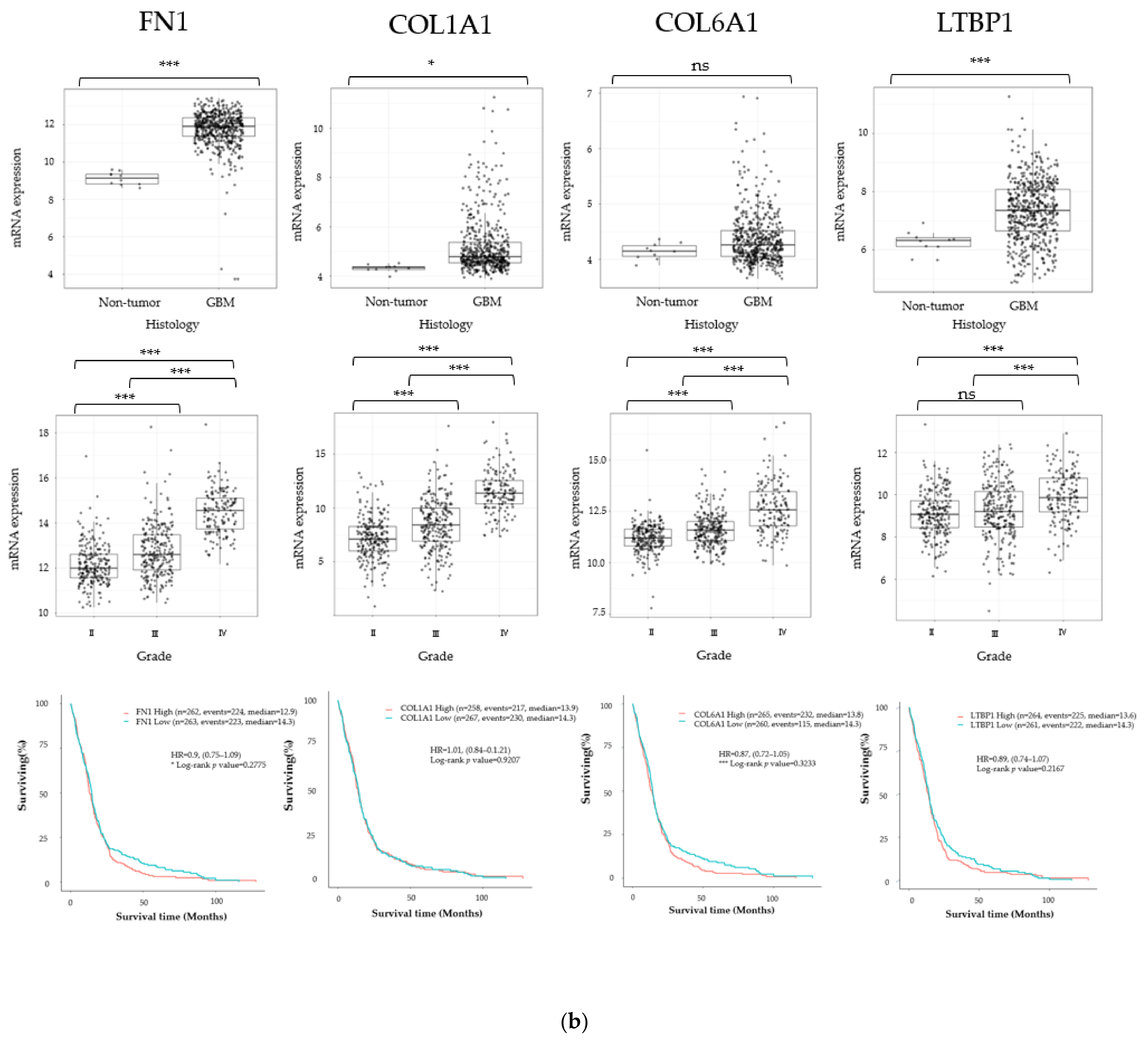
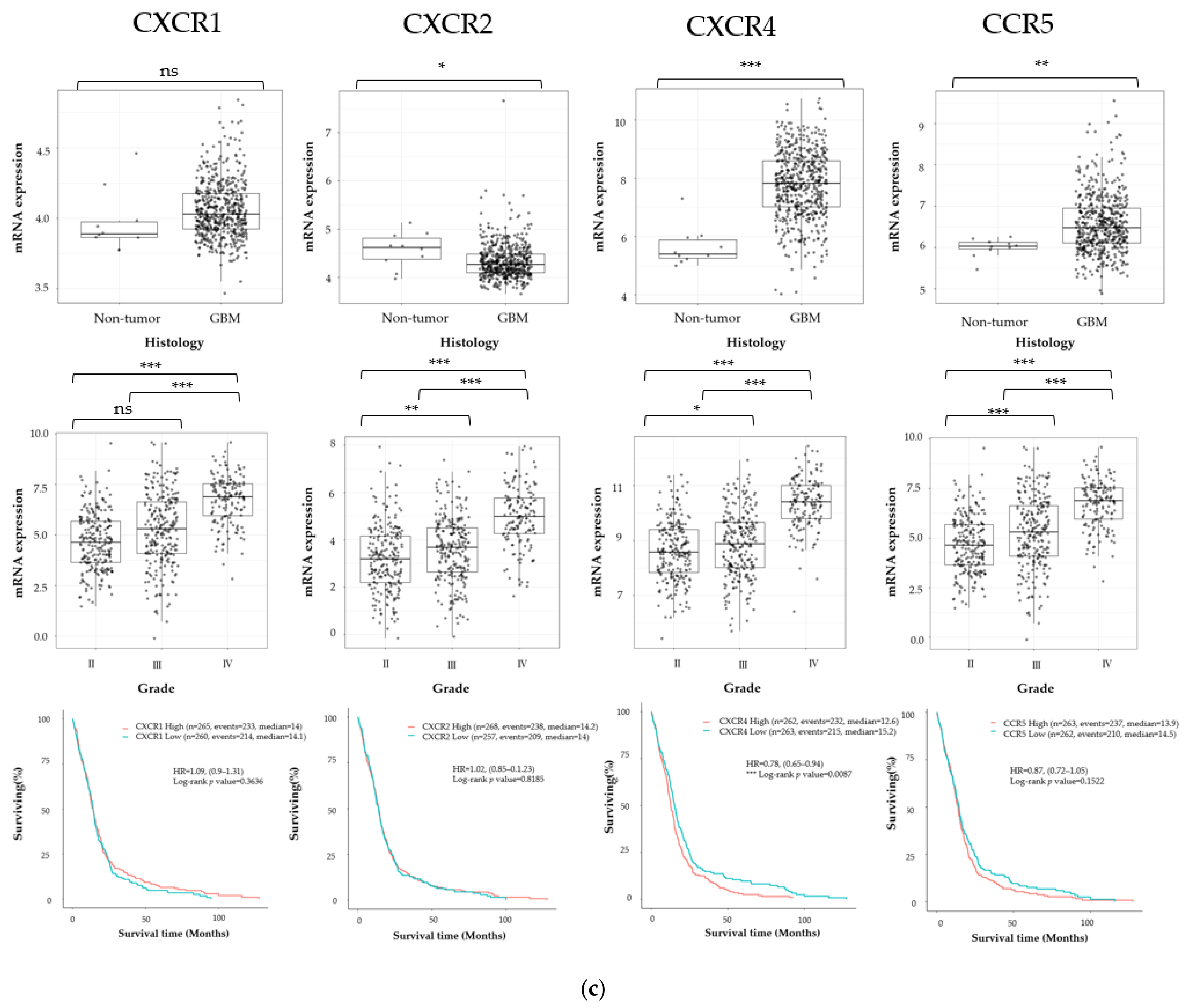
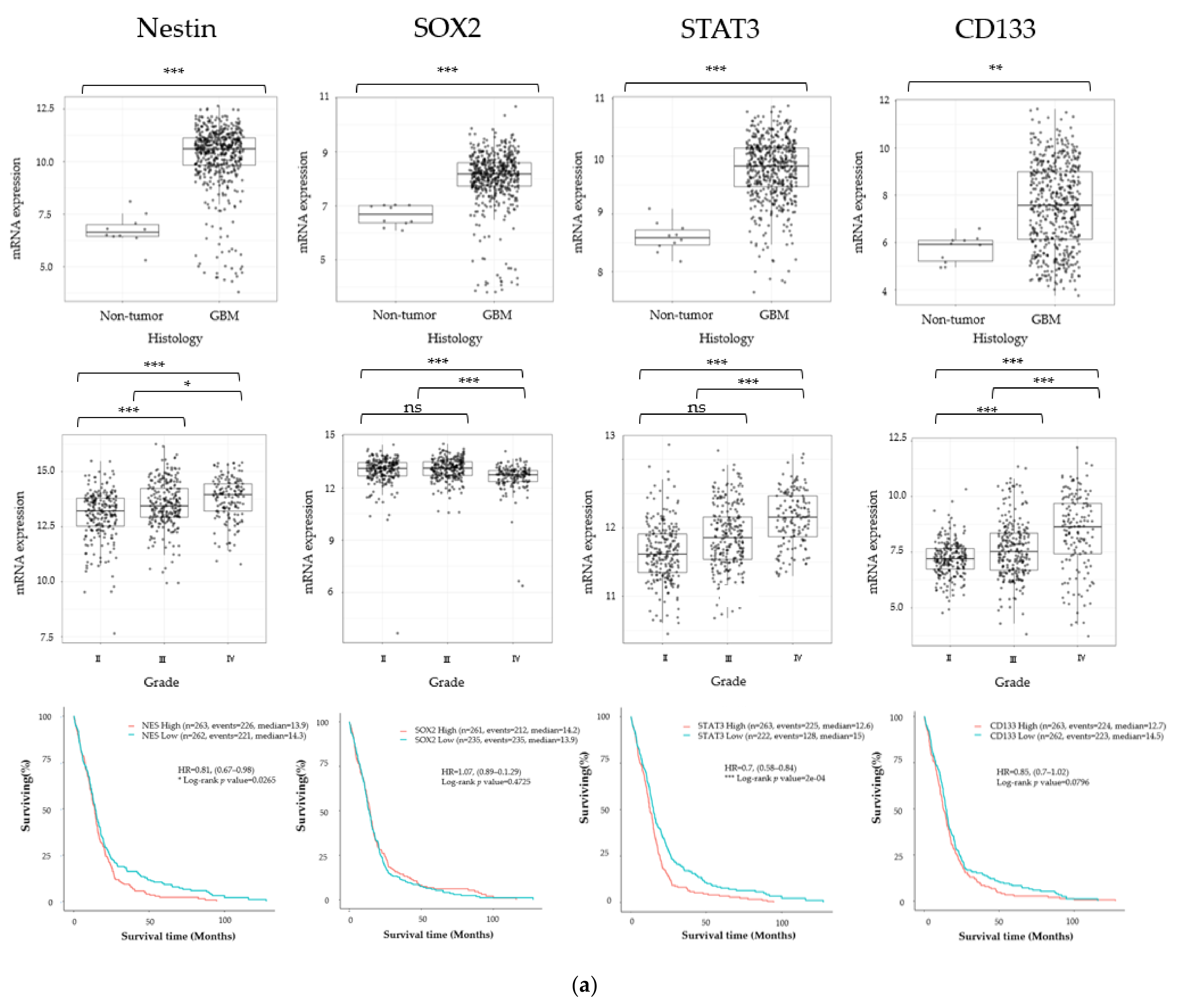
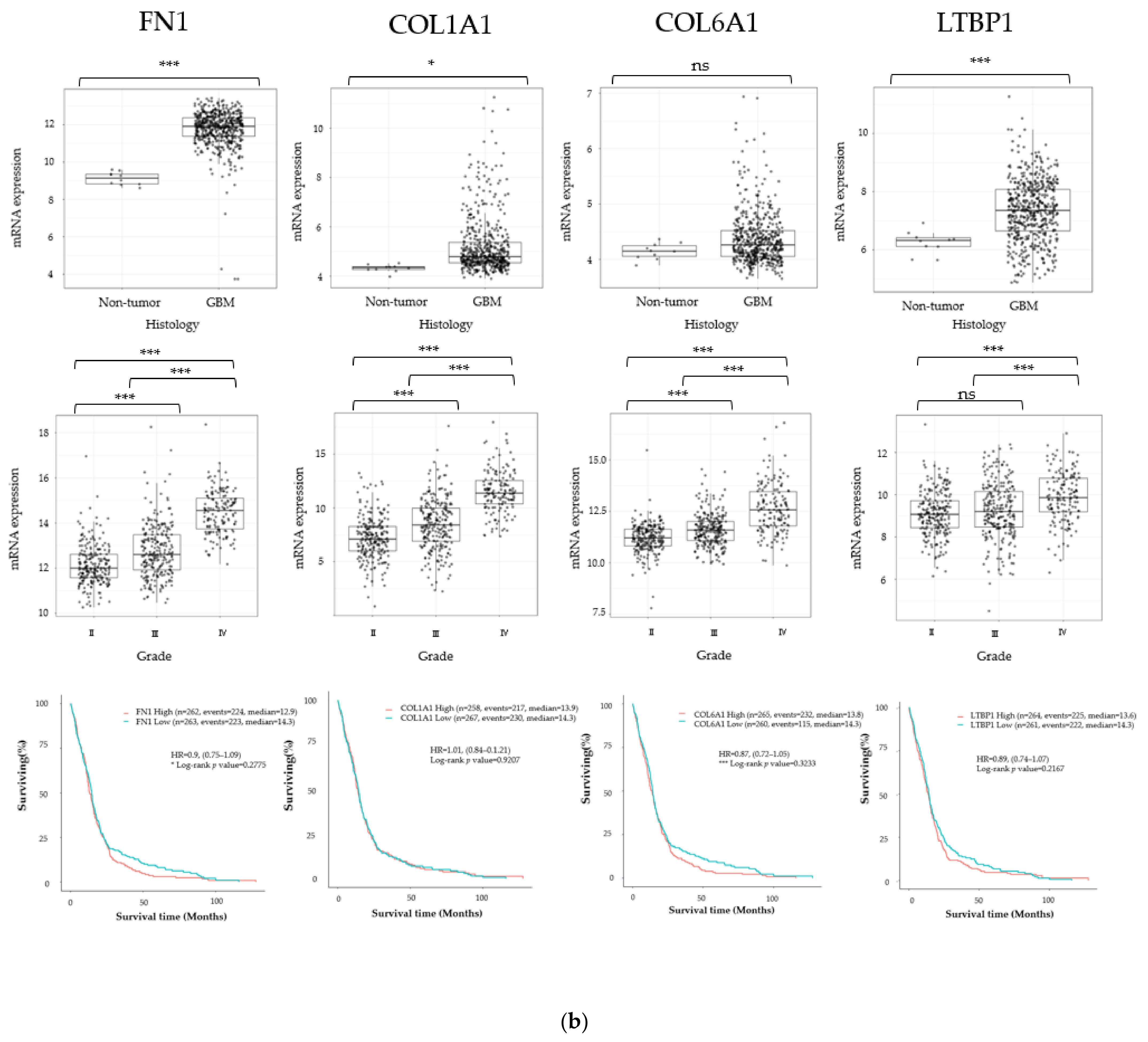
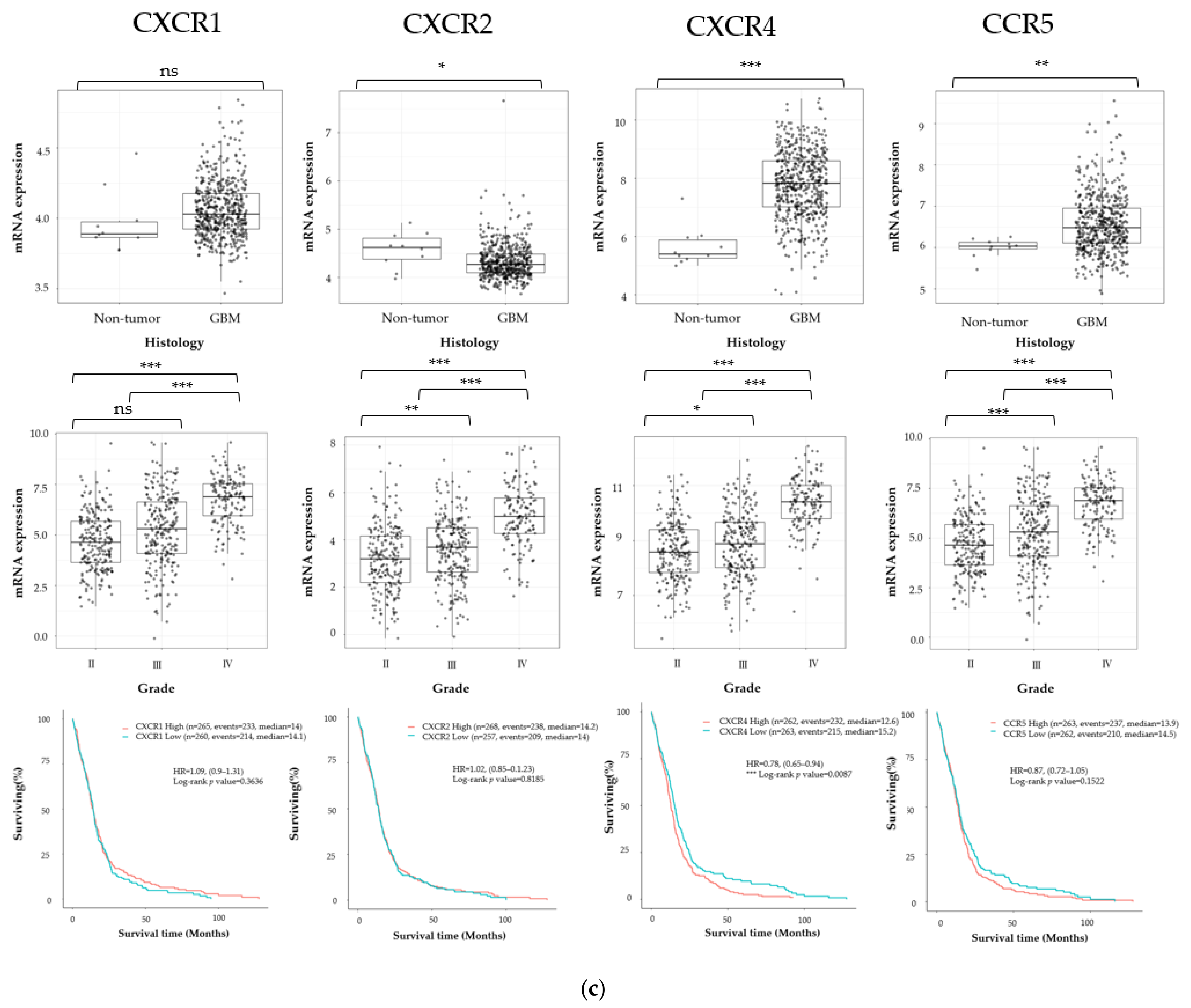
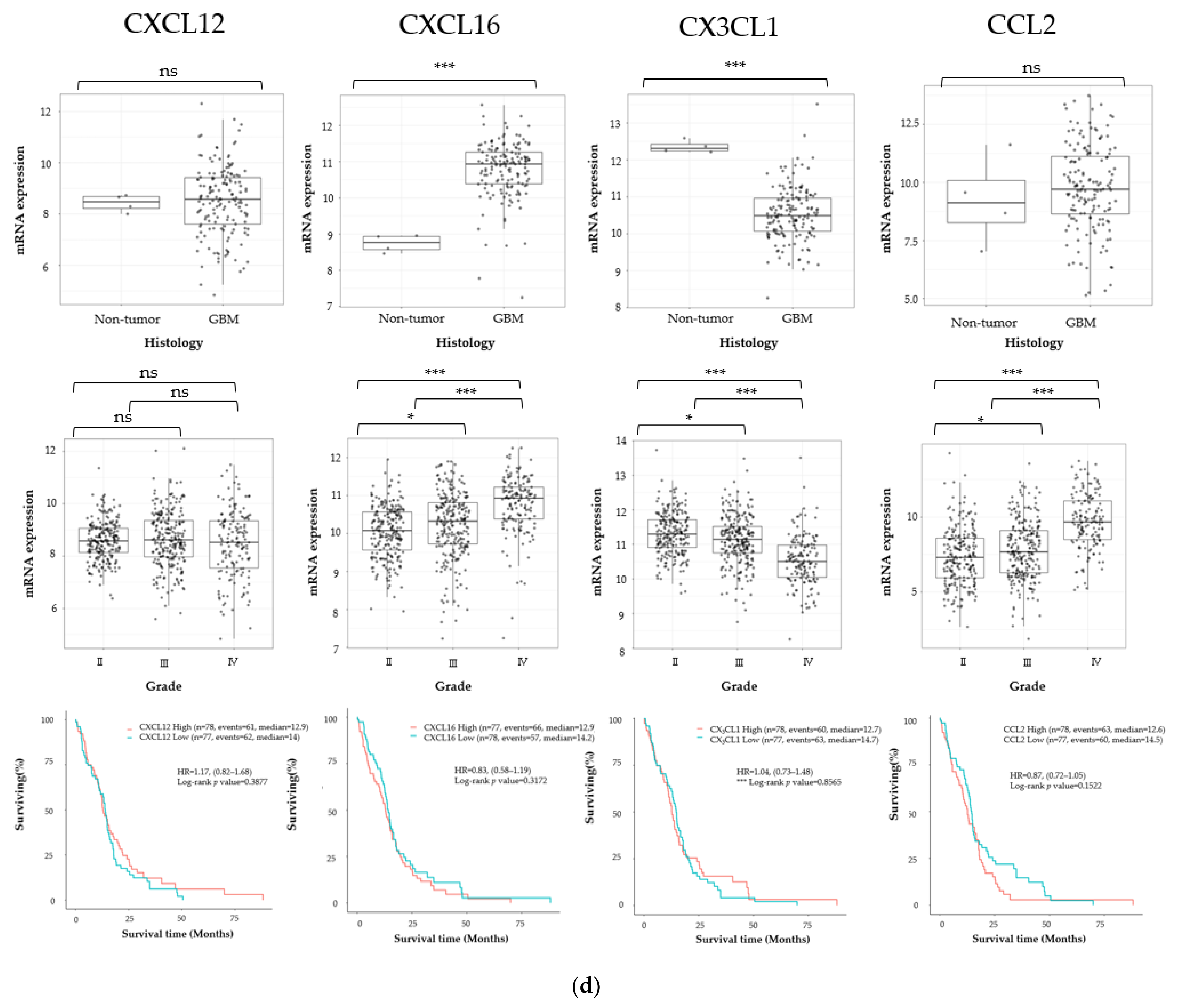
3.1. Cancer Stem Cell Marker, ECM Marker, and Chemokine/Chemokine Receptor Expression in Human Glioma
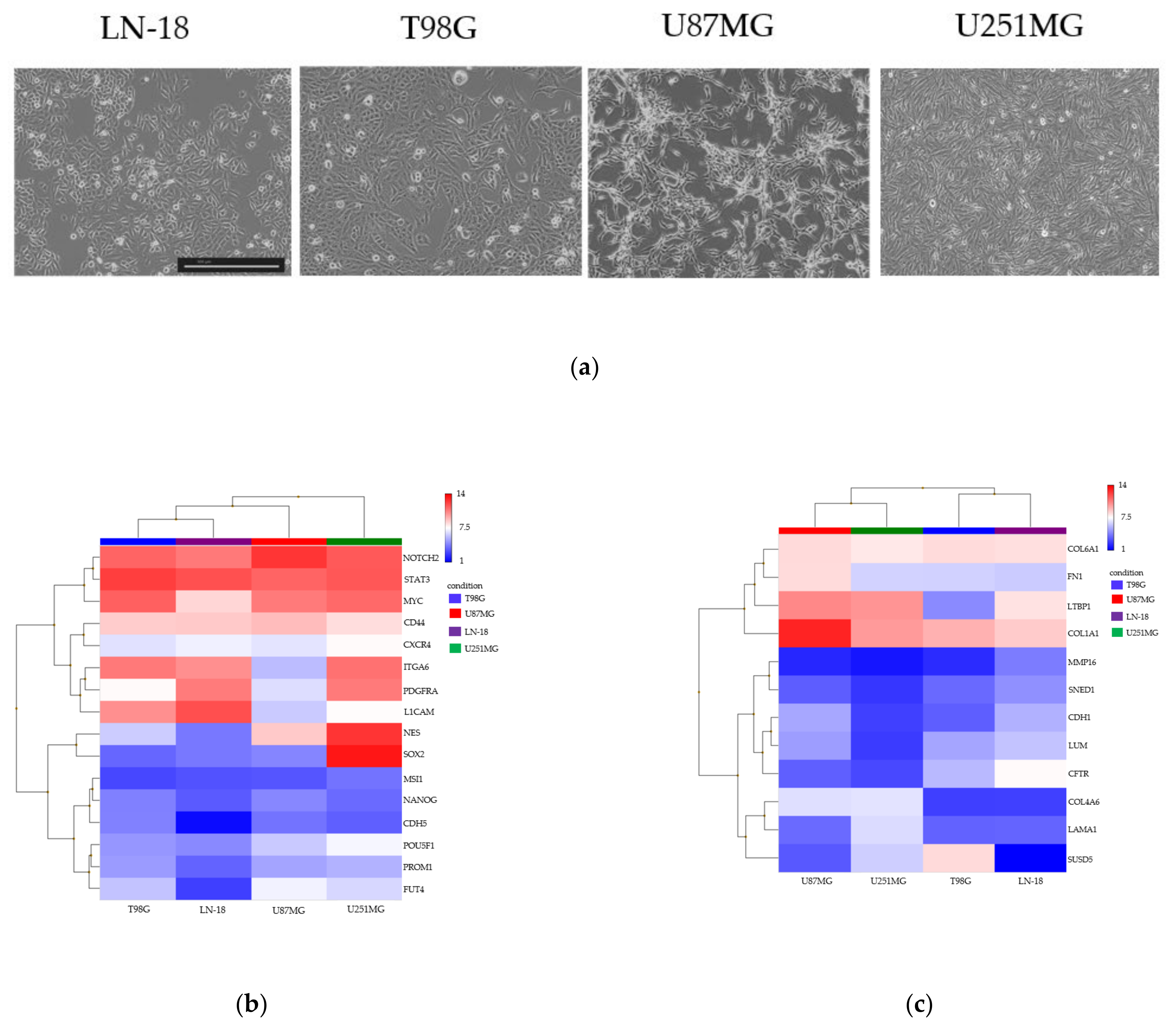
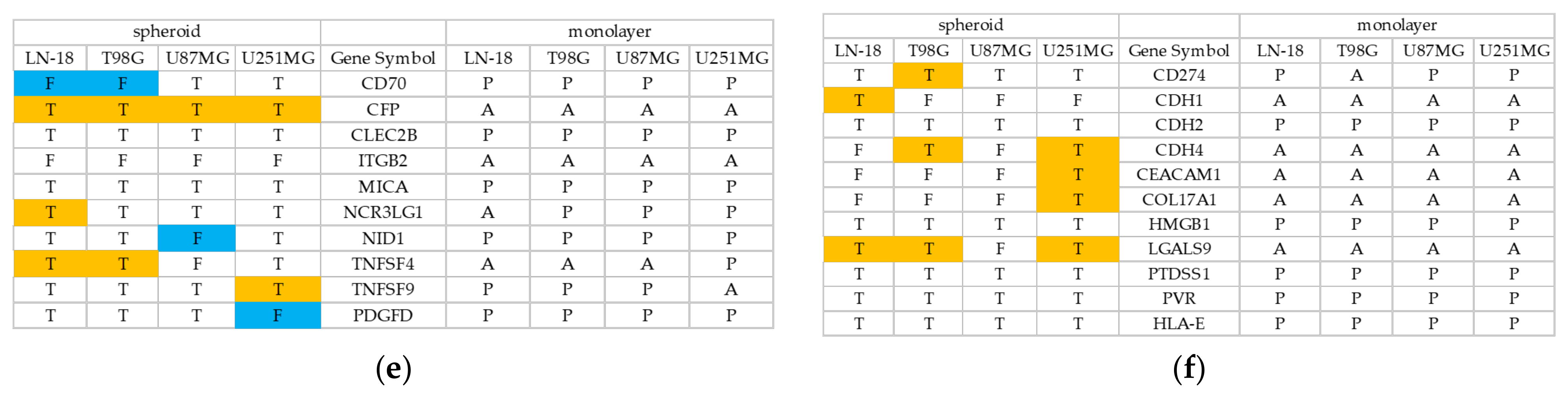
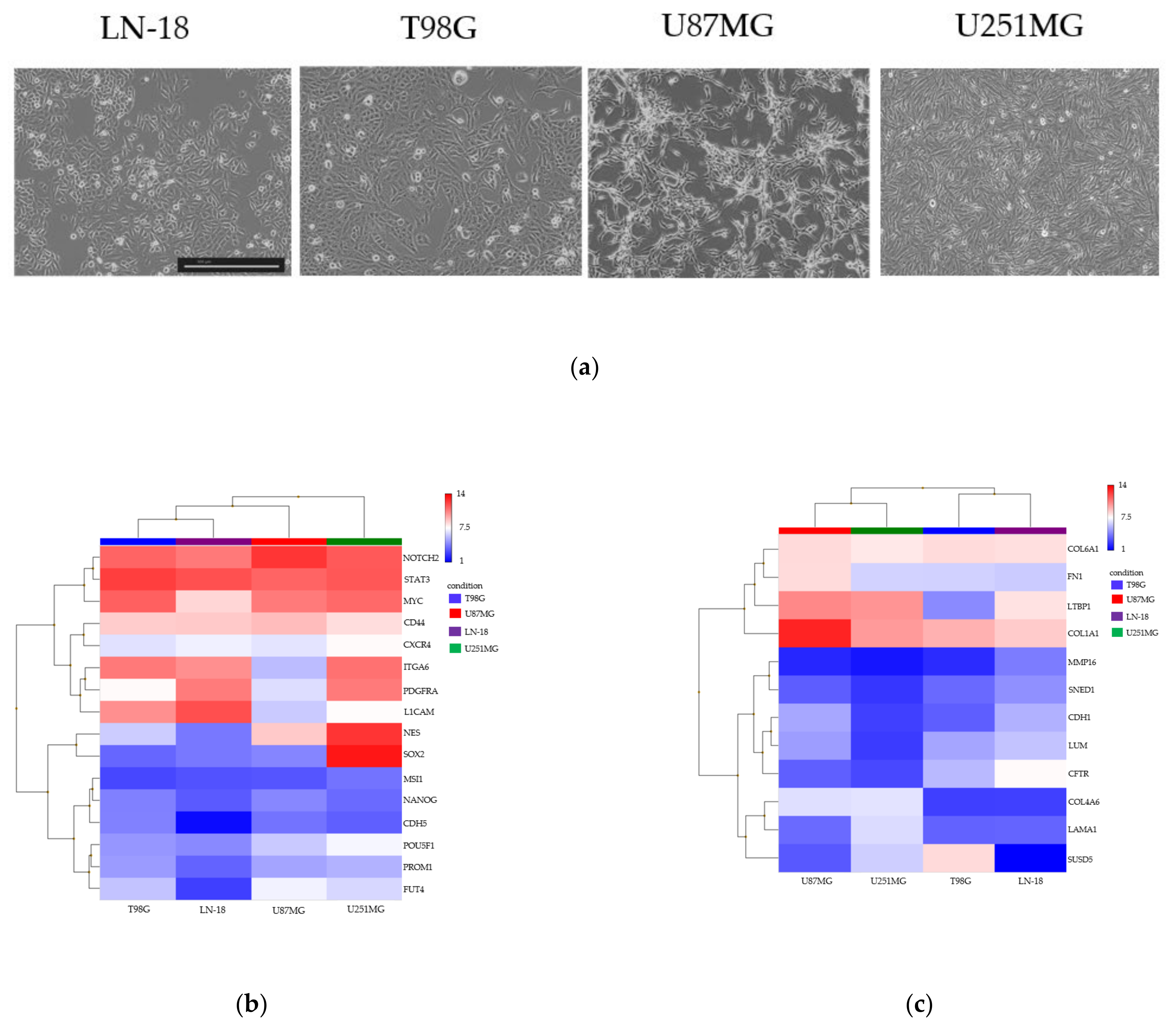
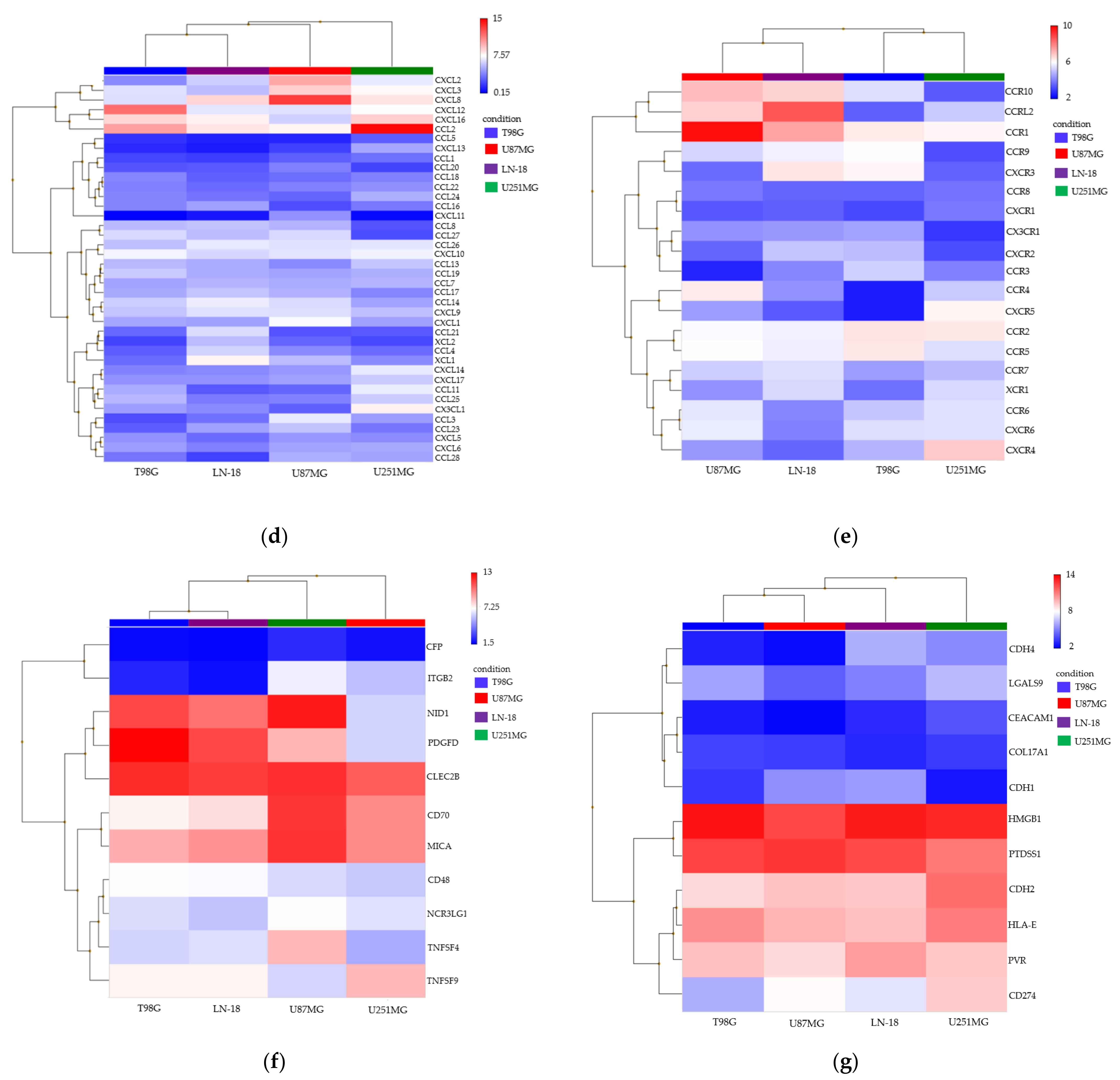
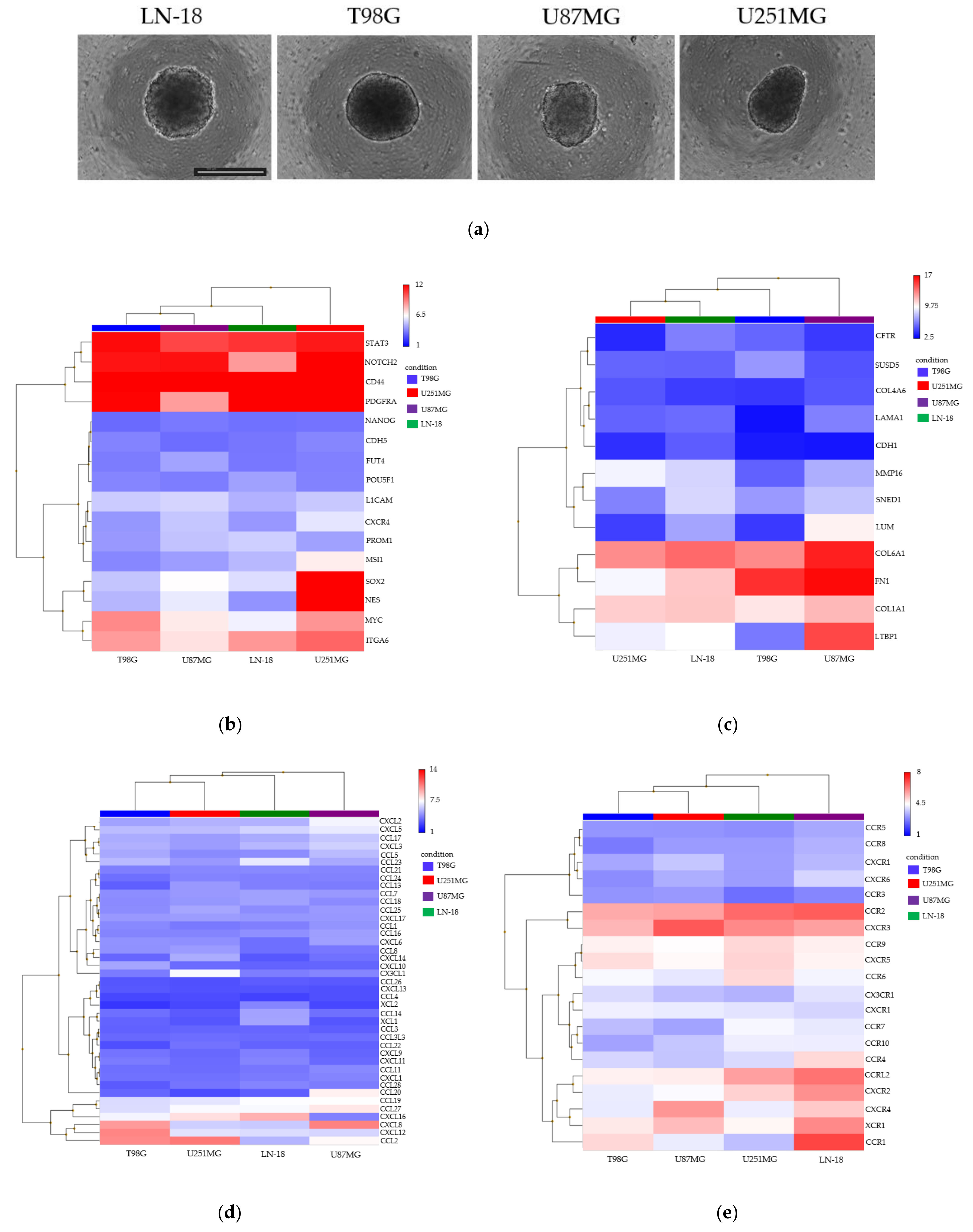
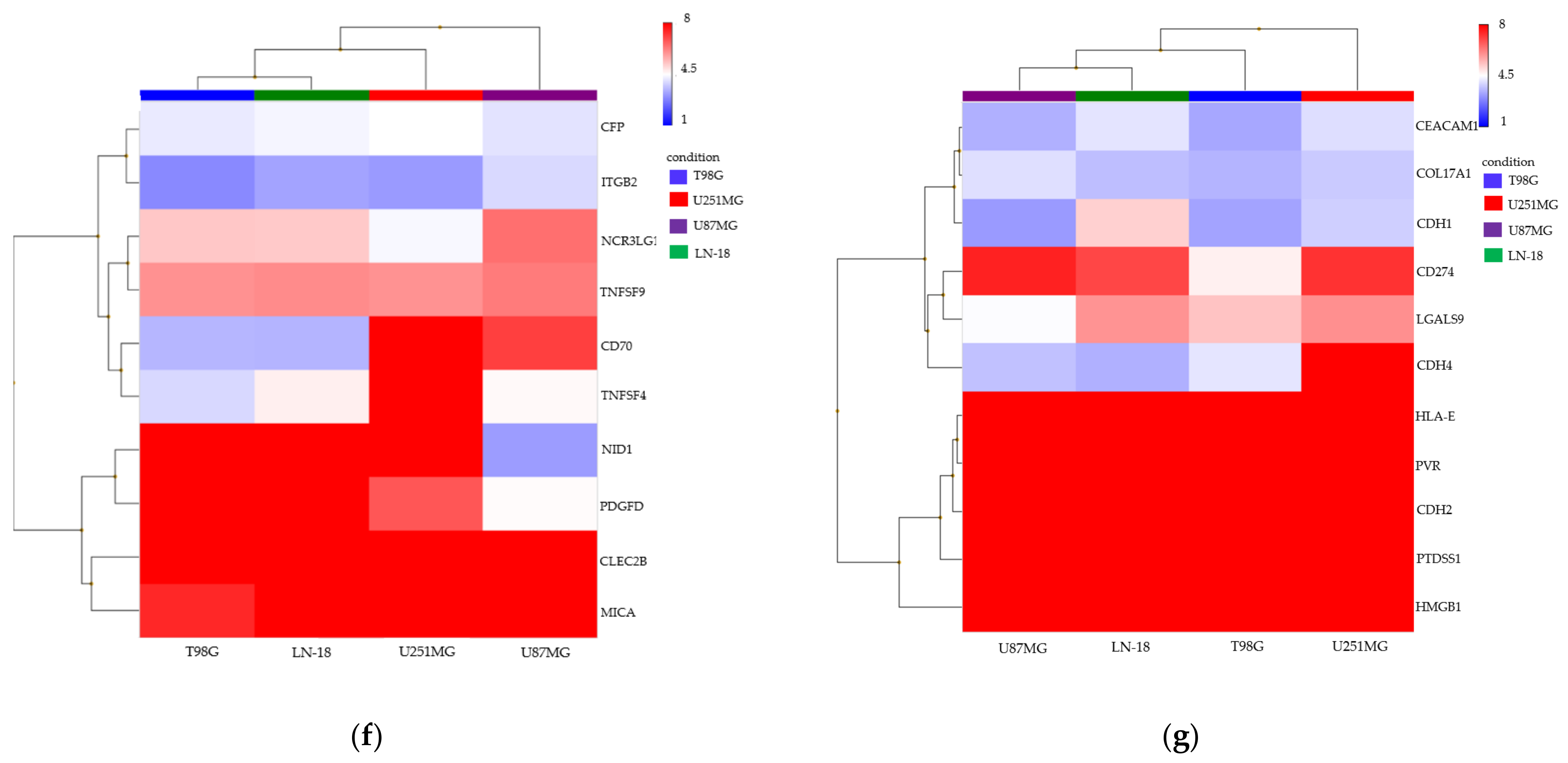
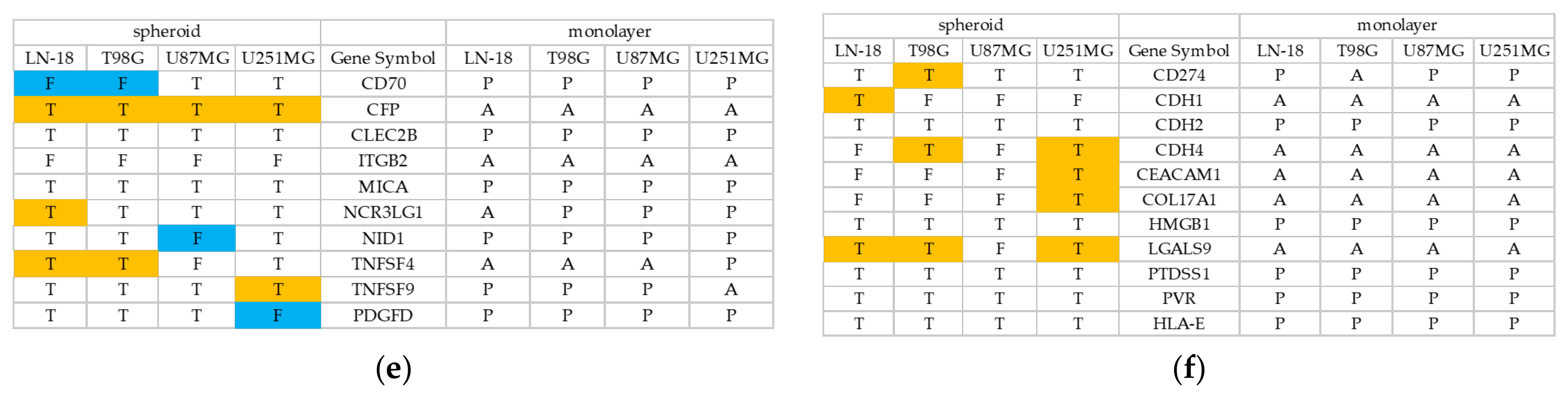
3.2. Spheroid Formation and Comprehensive Gene Expression Analysis of 2D Adherent Cells vs. 3D Spheroids Derived from GBM Cell Lines
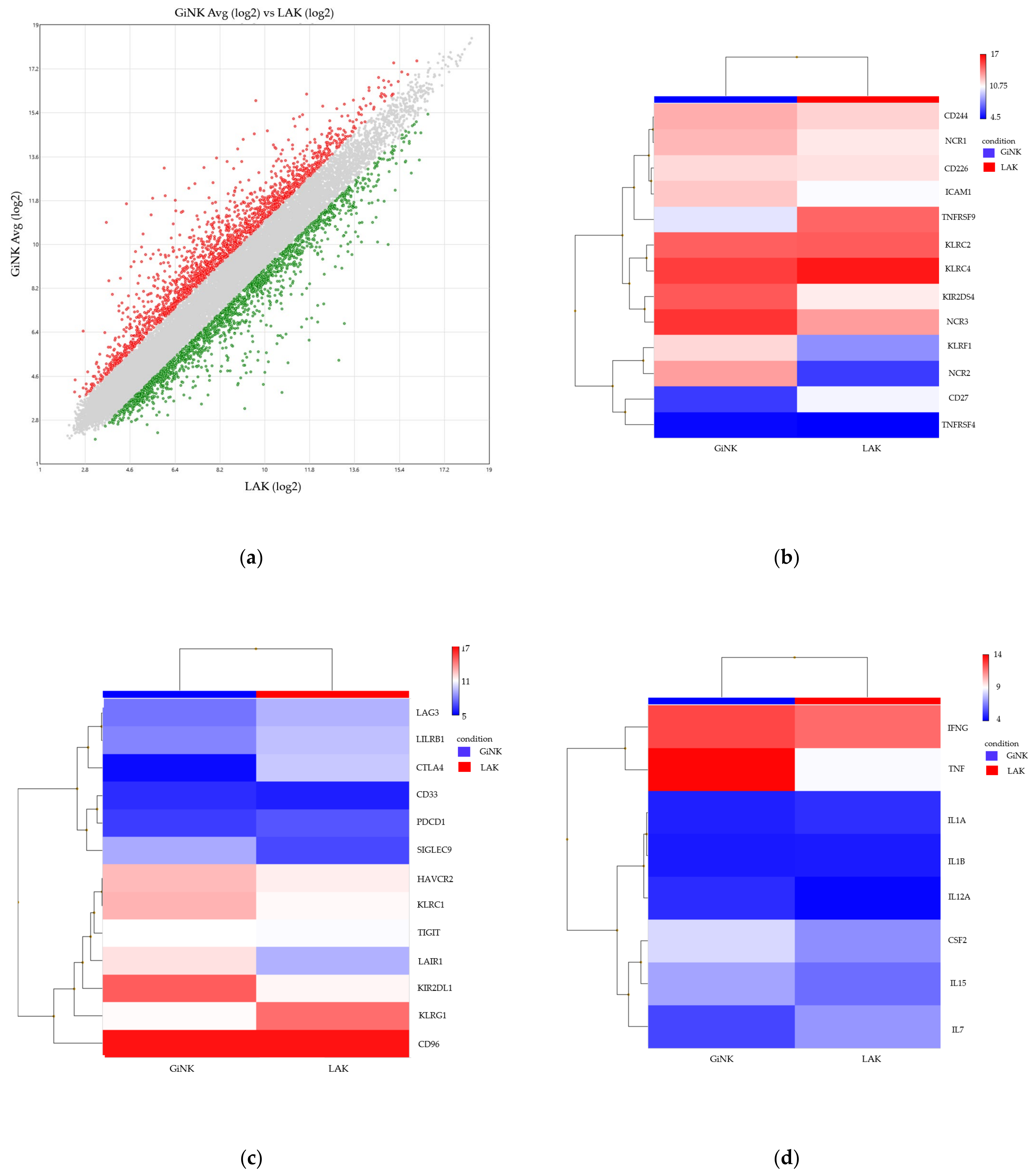
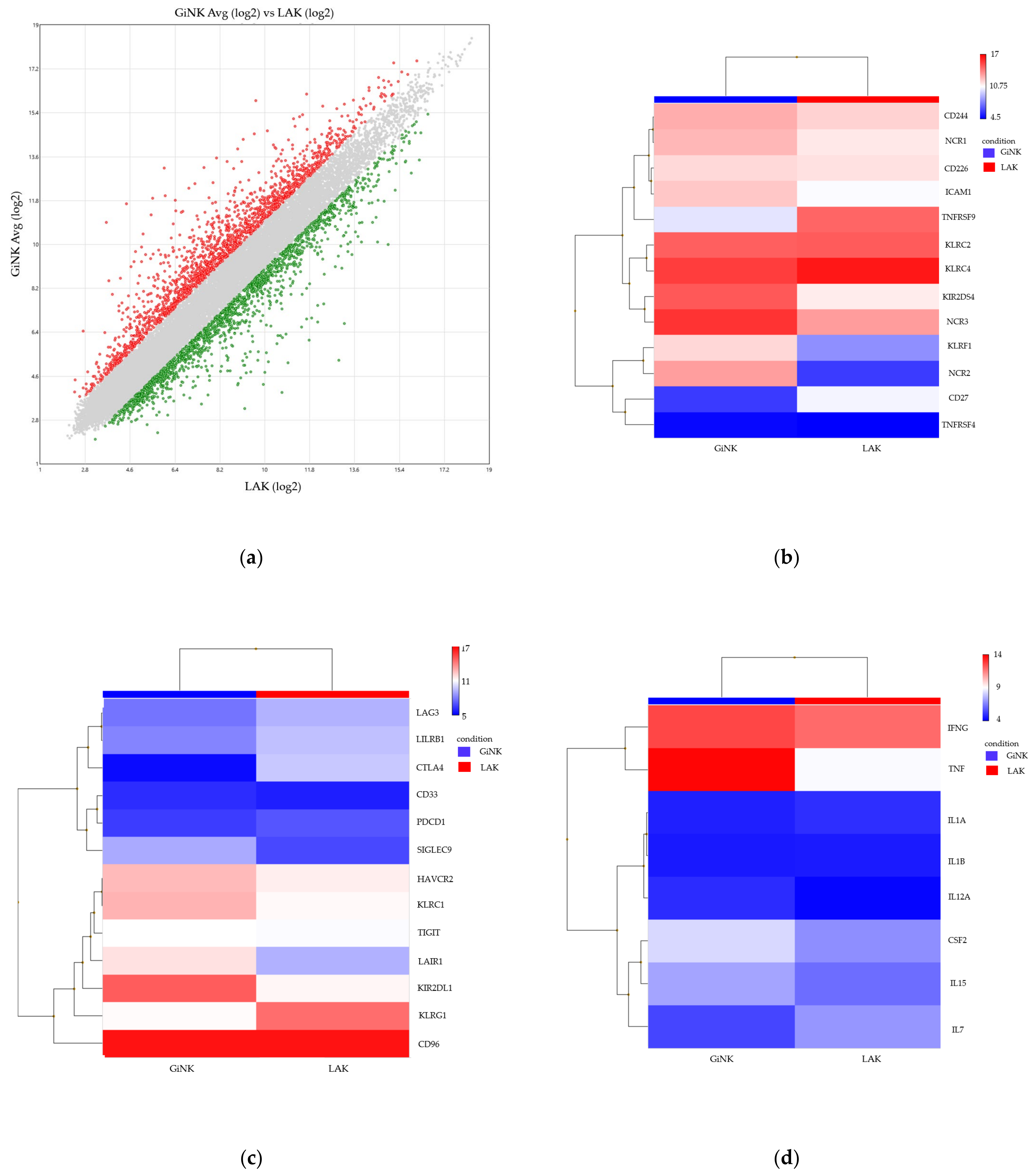
3.3. Comprehensive Gene Expression Analysis of the Expanded NK Cells as Compared to Conventional LAK Cells
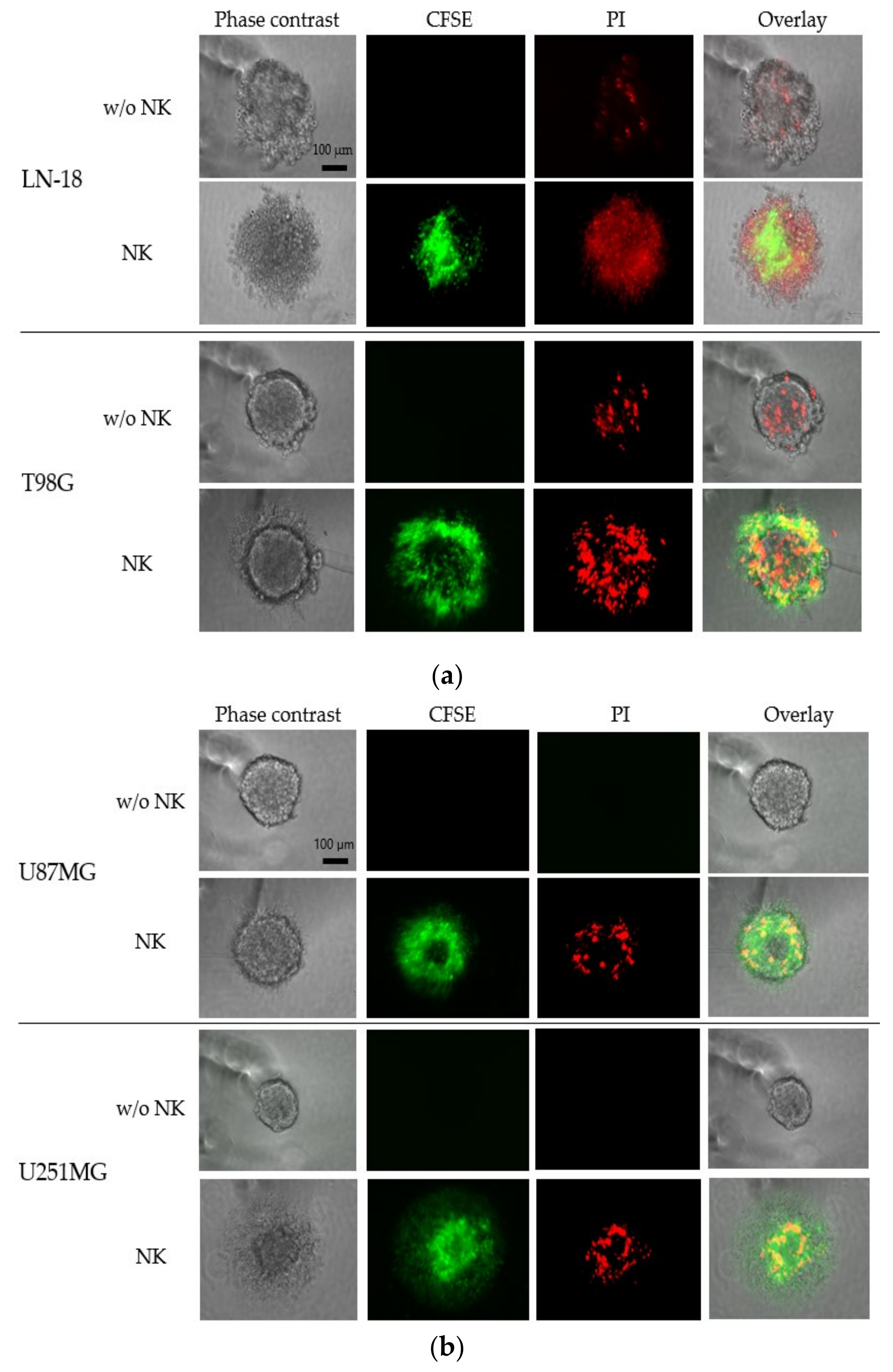
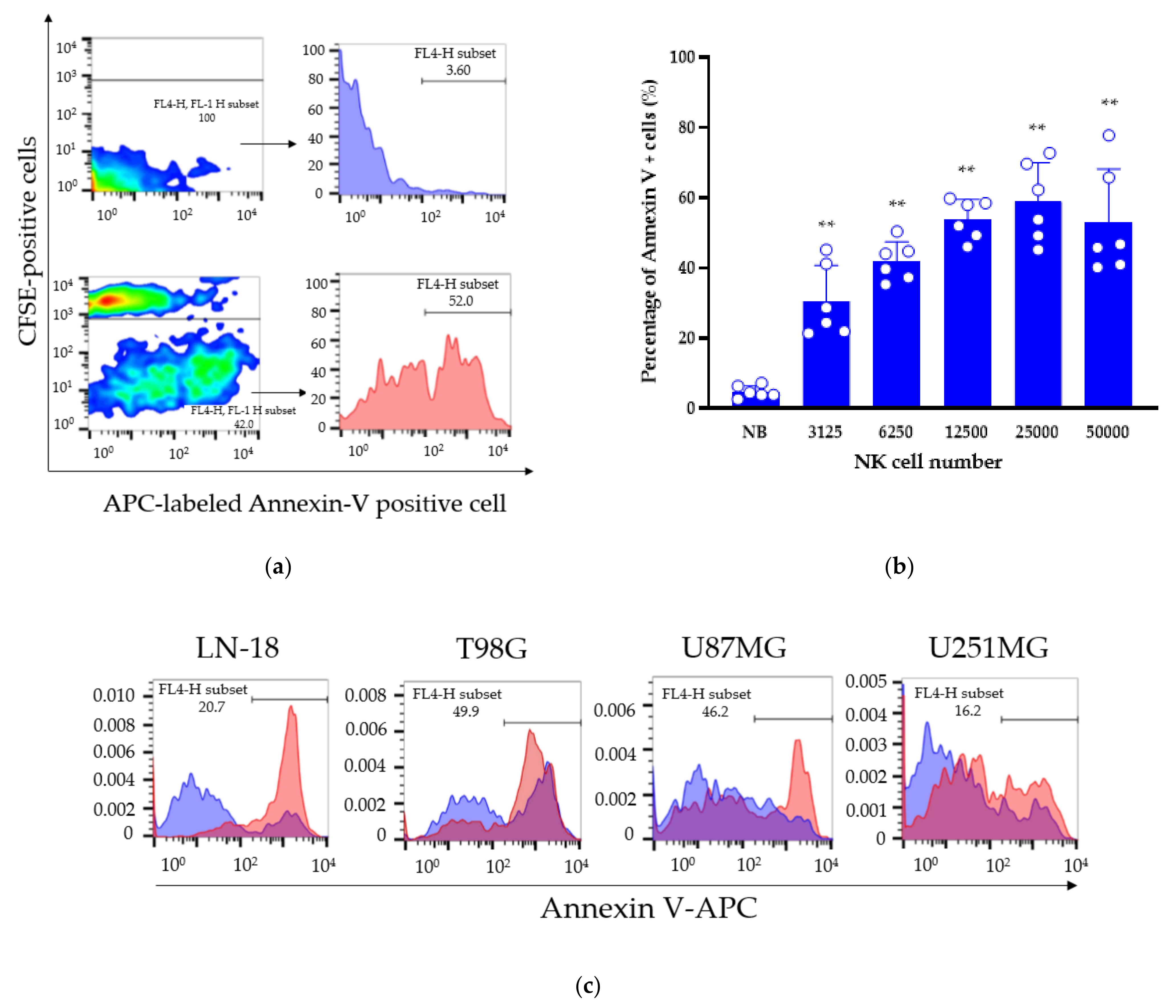
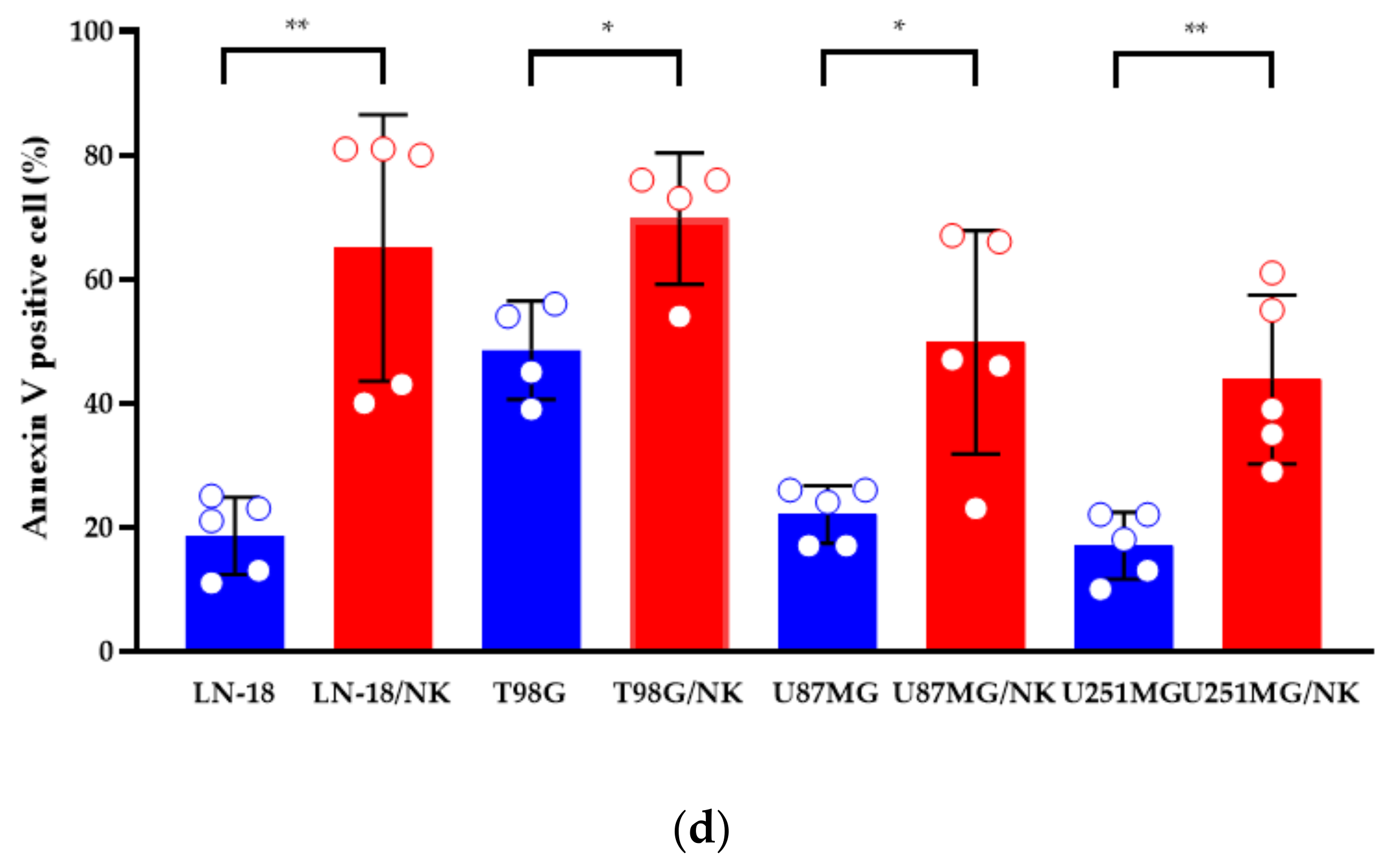
3.4. Cytotoxicity Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 1–9. [Google Scholar]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, R. Tumor immunotherapy: New aspects of natural killer cells. Chin. J. Cancer Res. 2018, 30, 173–196. [Google Scholar] [CrossRef]
- Rautela, J.; Souza-Fonseca-Guimaraes, F.; Hediyeh-Zadeh, S.; Delconte, R.B.; Davis, M.J.; Huntington, N.D. Molecular insight into targeting the NK cell immune response to cancer. Immunol. Cell Biol. 2018, 96, 477–484. [Google Scholar] [CrossRef]
- Huntington, N.D.; Legrand, N.; Alves, N.L.; Jaron, B.; Weijer, K.; Plet, A.; Corcuff, E.; Mortier, E.; Jacques, Y.; Spits, H.; et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J. Exp. Med. 2009, 206, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Puthalakath, H.; Gunn, P.; Naik, E.; Michalak, E.M.; Smyth, M.J.; Tabarias, H.; Degli-Esposti, M.A.; Dewson, G.; Willis, S.N.; et al. Interleukin 15–mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat. Immunol. 2007, 8, 856–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walzer, T.; Dalod, M.; Robbins, S.H.; Zitvogel, L.; Vivier, E. Natural-killer cells and dendritic cells: “l’union fait la force”. Blood 2005, 106, 2252–2258. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Arias, D.A.; Campbell, K.S. Protein Kinase C Regulates Expression and Function of Inhibitory Killer Cell Ig-Like Receptors in NK Cells. J. Immunol. 2007, 179, 5281–5290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.-G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L.; Yu, G.; Phillips, J.H. Co-association of CD3ζ with a receptor (CD16) for IgG Fc on human natural killer cells. Nature 1989, 342, 803–805. [Google Scholar] [CrossRef]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Friebel, E.; Kapolou, K.; Unger, S.; Nunez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e1620. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e1617. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirschl, C.J.; Drake, C.G. Molecular Pathways: Coexpression of Immune Checkpoint Molecules: Signaling Pathways and Implications for Cancer Immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [Green Version]
- Richards, J.O.; Chang, X.; Blaser, B.W.; Caligiuri, M.A.; Zheng, P.; Liu, Y. Tumor growth impedes natural-killer-cell maturation in the bone marrow. Blood 2006, 108, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Mundy-Bosse, B.L.; Scoville, S.D.; Chen, L.; McConnell, K.; Mao, H.C.; Ahmed, E.H.; Zorko, N.; Harvey, S.; Cole, J.; Zhang, X.; et al. MicroRNA-29b mediates altered innate immune development in acute leukemia. J. Clin. Investig. 2016, 126, 4404–4416. [Google Scholar] [CrossRef] [Green Version]
- Pasero, C.; Gravis, G.; Guerin, M.; Granjeaud, S.; Thomassin-Piana, J.; Rocchi, P.; Paciencia-Gros, M.; Poizat, F.; Bentobji, M.; Azario-Cheillan, F.; et al. Inherent and Tumor-Driven Immune Tolerance in the Prostate Microenvironment Impairs Natural Killer Cell Antitumor Activity. Cancer Res. 2016, 76, 2153–2165. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-Cell Atlas Reveals Complexity of the Immunosuppressive Microenvironment of Initial and Recurrent Glioblastoma. Front. Immunol. 2020, 11, 835. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nakazawa, T.; Nakamura, M.; Nishimura, F.; Matsuda, R.; Omoto, K.; Shida, Y.; Murakami, T.; Nakagawa, I.; Motoyama, Y.; et al. Ex vivo-expanded highly purified natural killer cells in combination with temozolomide induce antitumor effects in human glioblastoma cells in vitro. PLoS ONE 2019, 14, e0212455. [Google Scholar] [CrossRef]
- Klein, E.; Hau, A.-C.; Oudin, A.; Golebiewska, A.; Niclou, S.P. Glioblastoma Organoids: Pre-Clinical Applications and Challenges in the Context of Immunotherapy. Front. Oncol. 2020, 10, 2755. [Google Scholar] [CrossRef]
- Joo, K.M.; Kim, J.; Jin, J.; Kim, M.; Seol, H.J.; Muradov, J.; Yang, H.; Choi, Y.L.; Park, W.Y.; Kong, D.S.; et al. Patient-Specific Orthotopic Glioblastoma Xenograft Models Recapitulate the Histopathology and Biology of Human Glioblastomas In Situ. Cell Rep. 2013, 3, 260–273. [Google Scholar] [CrossRef] [Green Version]
- Shu, Q.; Wong, K.K.; Su, J.M.; Adesina, A.M.; Yu, L.T.; Tsang, Y.T.M.; Antalffy, B.C.; Baxter, P.; Perlaky, L.; Yang, J.; et al. Direct Orthotopic Transplantation of Fresh Surgical Specimen Preserves CD133+Tumor Cells in Clinically Relevant Mouse Models of Medulloblastoma and Glioma. Stem Cells 2008, 26, 1414–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oraiopoulou, M.-E.; Tampakaki, M.; Tzamali, E.; Tamiolakis, T.; Makatounakis, V.; Vakis, A.F.; Zacharakis, G.; Sakkalis, V.; Papamatheakis, J. A 3D tumor spheroid model for the T98G Glioblastoma cell line phenotypic characterization. Tissue Cell 2019, 59, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.C.; Moreira, A.F.; de Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: An overview on the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef] [PubMed]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, X.; Chedid, K.; Kalkanis, S.N. Glioblastoma cell line-derived spheres in serum-containing medium versus serum-free medium: A comparison of cancer stem cell properties. Int. J. Oncol. 2012, 41, 1693–1700. [Google Scholar] [CrossRef] [Green Version]
- Christofides, A.; Kosmopoulos, M.; Piperi, C. Pathophysiological mechanisms regulated by cytokines in gliomas. Cytokine 2015, 71, 377–384. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.M.; Mei, R.; Di, X.; Ryder, T.B.; Hubbell, E.; Dee, S.; Webster, T.A.; Harrington, C.A.; Ho, M.H.; Baid, J.; et al. Analysis of high density expression microarrays with signed-rank call algorithms. Bioinformatics 2002, 18, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Kuang, Y.; Xing, X.; Simpson, R.J.; Huang, H.; Yang, T.; Chen, J.; Yang, L.; Liu, E.; He, W.; et al. Proteomic Comparison of 3D and 2D Glioma Models Reveals Increased HLA-E Expression in 3D Models is Associated with Resistance to NK Cell-Mediated Cytotoxicity. J. Proteome Res. 2014, 13, 2272–2281. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 1–17. [Google Scholar] [CrossRef]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Qiao, B.; Johnson, N.W.; Chen, X.; Li, R.; Tao, Q.; Gao, J. Disclosure of a stem cell phenotype in an oral squamous cell carcinoma cell line induced by BMP-4 via an epithelial-mesenchymal transition. Oncol. Rep. 2011, 26, 455–461. [Google Scholar]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer Stem Cells: Models and Concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Venere, M.; Fine, H.A.; Dirks, P.B.; Rich, J.N. Cancer stem cells in gliomas: Identifying and understanding the apex cell in cancer’s hierarchy. Glia 2011, 59, 1148–1154. [Google Scholar] [CrossRef] [Green Version]
- Man, J.; Shoemake, J.; Zhou, W.; Fang, X.; Wu, Q.; Rizzo, A.; Prayson, R.; Bao, S.; Rich, J.N.; Yu, J.S. Sema3C Promotes the Survival and Tumorigenicity of Glioma Stem Cells through Rac1 Activation. Cell Rep. 2014, 9, 1812–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.-M.; Maciaczyk, J.; et al. Notch Pathway Blockade Depletes CD133-Positive Glioblastoma Cells and Inhibits Growth of Tumor Neurospheres and Xenografts. Stem Cells 2009, 28, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Lawrenson, K.; Grun, B.; Gayther, S.A. Heterotypic three-dimensional in vitro modeling of stromal-epithelial interactions during ovarian cancer initiation and progression. J. Vis. Exp. 2012, 66, e4206. [Google Scholar] [CrossRef] [Green Version]
- Bell, H.S.; Whittle, I.R.; Walker, M.; Leaver, H.A.; Wharton, S.B. The development of necrosis and apoptosis in glioma: Experimental findings using spheroid culture systems. Neuropathol. Appl. Neurobiol. 2001, 27, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mou, L.; Pan, Y.; Feng, C.; Zhang, J.; Li, J. CXCL8 Promotes Glioma Progression by Activating The JAK/STAT1/HIF-1α/Snail Signaling Axis. OncoTargets Ther. 2019, 12, 8125–8138. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.D.; Sweeney, C.J.; Sledge, G.W. Can tumor angiogenesis be inhibited without resistance? In Mechanisms of Angiogenesis; Springer: Berlin/Heidelberg, Germany, 2005; pp. 95–112. [Google Scholar]
- Angara, K.; Borin, T.F.; Rashid, M.H.; Lebedyeva, I.; Ara, R.; Lin, P.-C.; Iskander, A.; Bollag, R.J.; Achyut, B.R.; Arbab, A.S. CXCR2-Expressing Tumor Cells Drive Vascular Mimicry in Antiangiogenic Therapy–Resistant Glioblastoma. Neoplasia 2018, 20, 1070–1082. [Google Scholar] [CrossRef]
- Rempel, S.A.; Dudas, S.; Ge, S.; Gutiérrez, J.A. Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin. Cancer Res. 2000, 6, 102–111. [Google Scholar]
- Ping, Y.-F.; Yao, X.-H.; Jiang, J.-Y.; Zhao, L.-T.; Yu, S.-C.; Jiang, T.; Lin, M.C.; Chen, J.-H.; Wang, B.; Zhang, R.; et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Carvalho Da Fonseca, A.C.; Badie, B. Microglia and Macrophages in Malignant Gliomas: Recent Discoveries and Implications for Promising Therapies. Clin. Dev. Immunol. 2013, 2013, 1–5. [Google Scholar] [CrossRef]
- Kumar, R.; De Mooij, T.; Peterson, T.E.; Kaptzan, T.; Johnson, A.J.; Daniels, D.J.; Parney, I.F. Modulating glioma-mediated myeloid-derived suppressor cell development with sulforaphane. PLoS ONE 2017, 12, e0179012. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Lei, Z.; Zhao, J.; Gong, W.; Liu, J.; Chen, Z.; Liu, Y.; Li, D.; Yuan, Y.; Zhang, G.-M.; et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007, 252, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Groblewska, M.; Litman-Zawadzka, A.; Mroczko, B. The Role of Selected Chemokines and Their Receptors in the Development of Gliomas. Int. J. Mol. Sci. 2020, 21, 3704. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitprez, F.; Meylan, M.; de Reyniès, A.; Sautès-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [Google Scholar] [CrossRef]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131, e142116. [Google Scholar] [CrossRef]
- Close, H.J.; Stead, L.F.; Nsengimana, J.; Reilly, K.A.; Droop, A.; Wurdak, H.; Mathew, R.K.; Corns, R.; Newton-Bishop, J.; Melcher, A.A.; et al. Expression profiling of single cells and patient cohorts identifies multiple immunosuppressive pathways and an altered NK cell phenotype in glioblastoma. Clin. Exp. Immunol. 2020, 200, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Torsvik, A.; Stieber, D.; Enger, P.Ø.; Golebiewska, A.; Molven, A.; Svendsen, A.; Westermark, B.; Niclou, S.P.; Olsen, T.K.; Chekenya Enger, M.; et al. U-251 revisited: Genetic drift and phenotypic consequences of long-term cultures of glioblastoma cells. Cancer Med. 2014, 3, 812–824. [Google Scholar] [CrossRef]
- De Witt Hamer, P.C.; Van Tilborg, A.A.G.; Eijk, P.P.; Sminia, P.; Troost, D.; Van Noorden, C.J.F.; Ylstra, B.; Leenstra, S. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene 2008, 27, 2091–2096. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.J.; Homer, N.; O’Connor, B.D.; Chen, Z.; Eskin, A.; Lee, H.; Merriman, B.; Nelson, S.F. U87MG Decoded: The Genomic Sequence of a Cytogenetically Aberrant Human Cancer Cell Line. PLoS Genet. 2010, 6, e1000832. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Ishikawa, T.; Kokura, S.; Okayama, T.; Oka, K.; Ideno, M.; Sakai, F.; Kato, A.; Tanabe, M.; Enoki, T.; et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J. Transl. Med. 2015, 13, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, L.L.; Jain, R.K. Vascular regulation of antitumor immunity. Science 2019, 365, 544–545. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morimoto, T.; Nakazawa, T.; Matsuda, R.; Nishimura, F.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Park, Y.-S.; Tsujimura, T.; Nakase, H. Evaluation of Comprehensive Gene Expression and NK Cell-Mediated Killing in Glioblastoma Cell Line-Derived Spheroids. Cancers 2021, 13, 4896. https://doi.org/10.3390/cancers13194896
Morimoto T, Nakazawa T, Matsuda R, Nishimura F, Nakamura M, Yamada S, Nakagawa I, Park Y-S, Tsujimura T, Nakase H. Evaluation of Comprehensive Gene Expression and NK Cell-Mediated Killing in Glioblastoma Cell Line-Derived Spheroids. Cancers. 2021; 13(19):4896. https://doi.org/10.3390/cancers13194896
Chicago/Turabian StyleMorimoto, Takayuki, Tsutomu Nakazawa, Ryosuke Matsuda, Fumihiko Nishimura, Mitsutoshi Nakamura, Shuichi Yamada, Ichiro Nakagawa, Young-Soo Park, Takahiro Tsujimura, and Hiroyuki Nakase. 2021. "Evaluation of Comprehensive Gene Expression and NK Cell-Mediated Killing in Glioblastoma Cell Line-Derived Spheroids" Cancers 13, no. 19: 4896. https://doi.org/10.3390/cancers13194896
APA StyleMorimoto, T., Nakazawa, T., Matsuda, R., Nishimura, F., Nakamura, M., Yamada, S., Nakagawa, I., Park, Y.-S., Tsujimura, T., & Nakase, H. (2021). Evaluation of Comprehensive Gene Expression and NK Cell-Mediated Killing in Glioblastoma Cell Line-Derived Spheroids. Cancers, 13(19), 4896. https://doi.org/10.3390/cancers13194896