
Heterogeneity of Circulating Tumor Cell Neoplastic Subpopulations Outlined by Single-Cell Transcriptomics


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
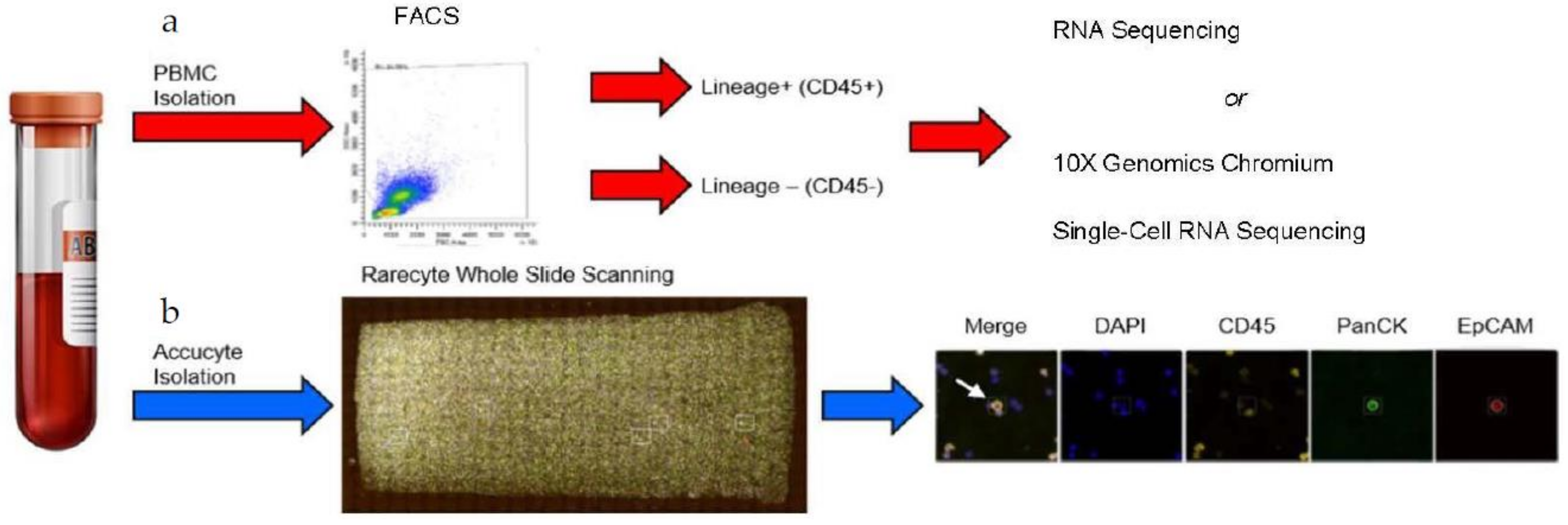
2.1. Patients’ Blood Collection and Analyses
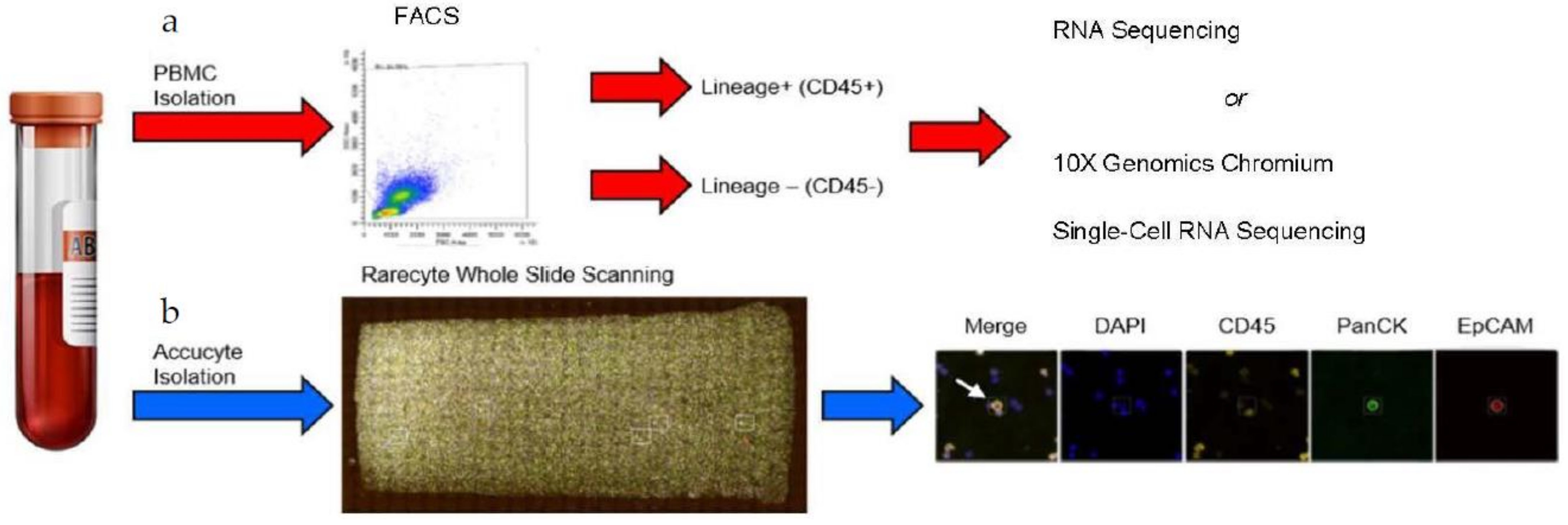
2.2. Fluorescence Activated Cell Sorting (FACS)
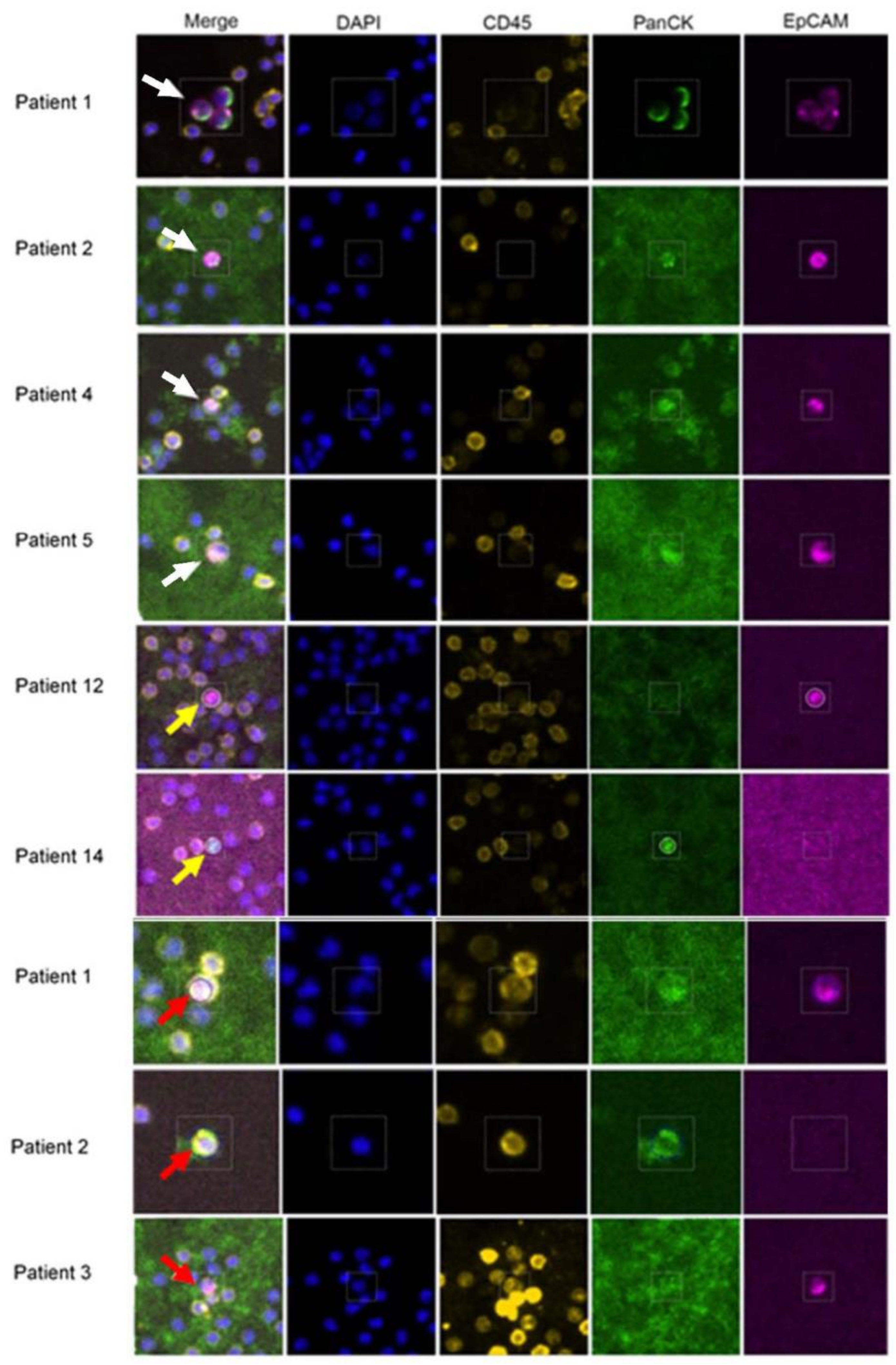
2.3. RareCyte Cytefinder II™ Analysis
2.4. RNA Isolation and Sequencing
2.5. RNA-Seq Analyses
2.6. 10x Genomics Chromium Single-Cell RNA Sequencing
2.7. Single-Cell RNA Transcriptome Analyses
3. Results
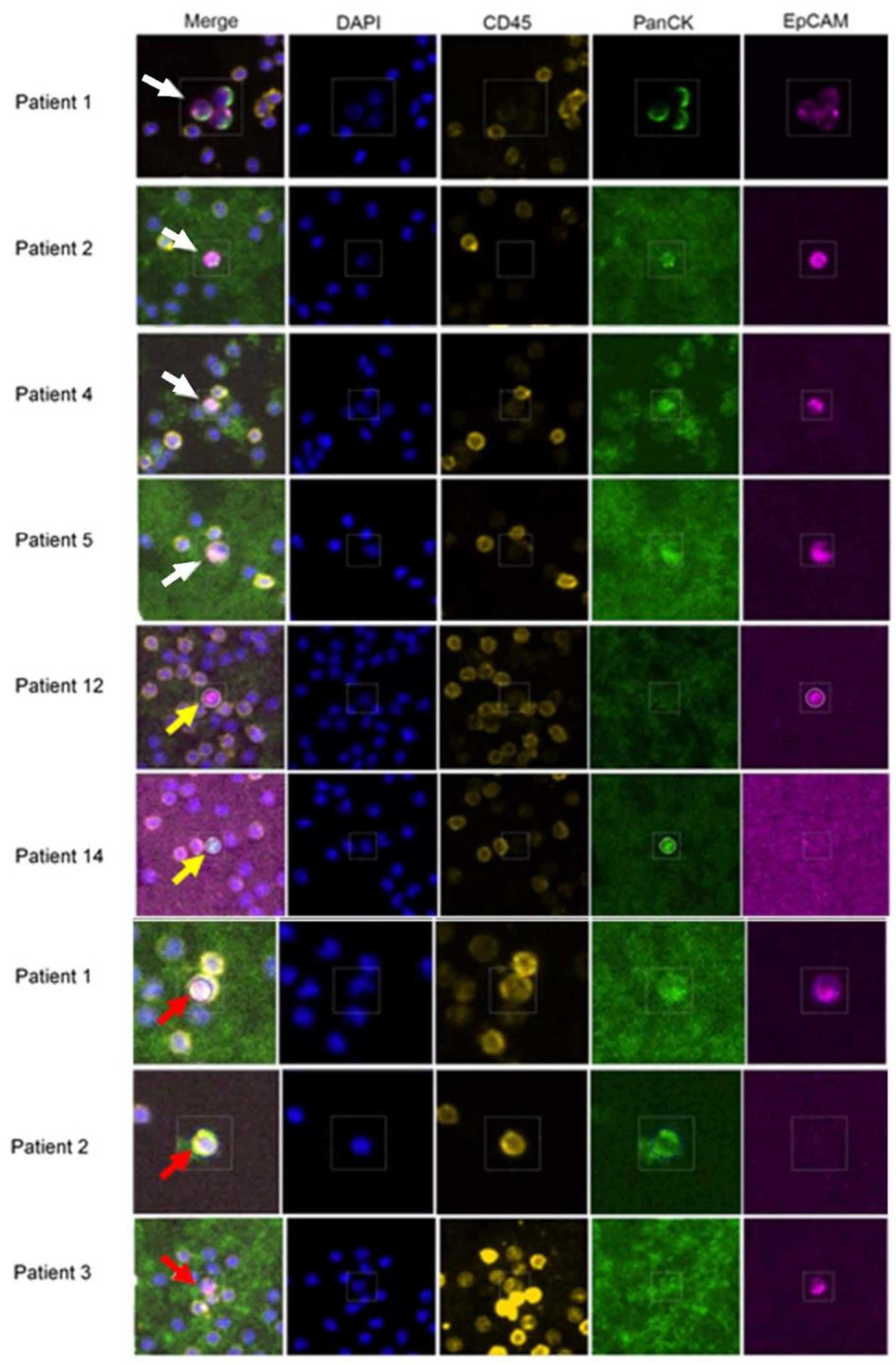
3.1. Isolation and Characterization of CTCs from mBC Patients by RareCyte Analyses
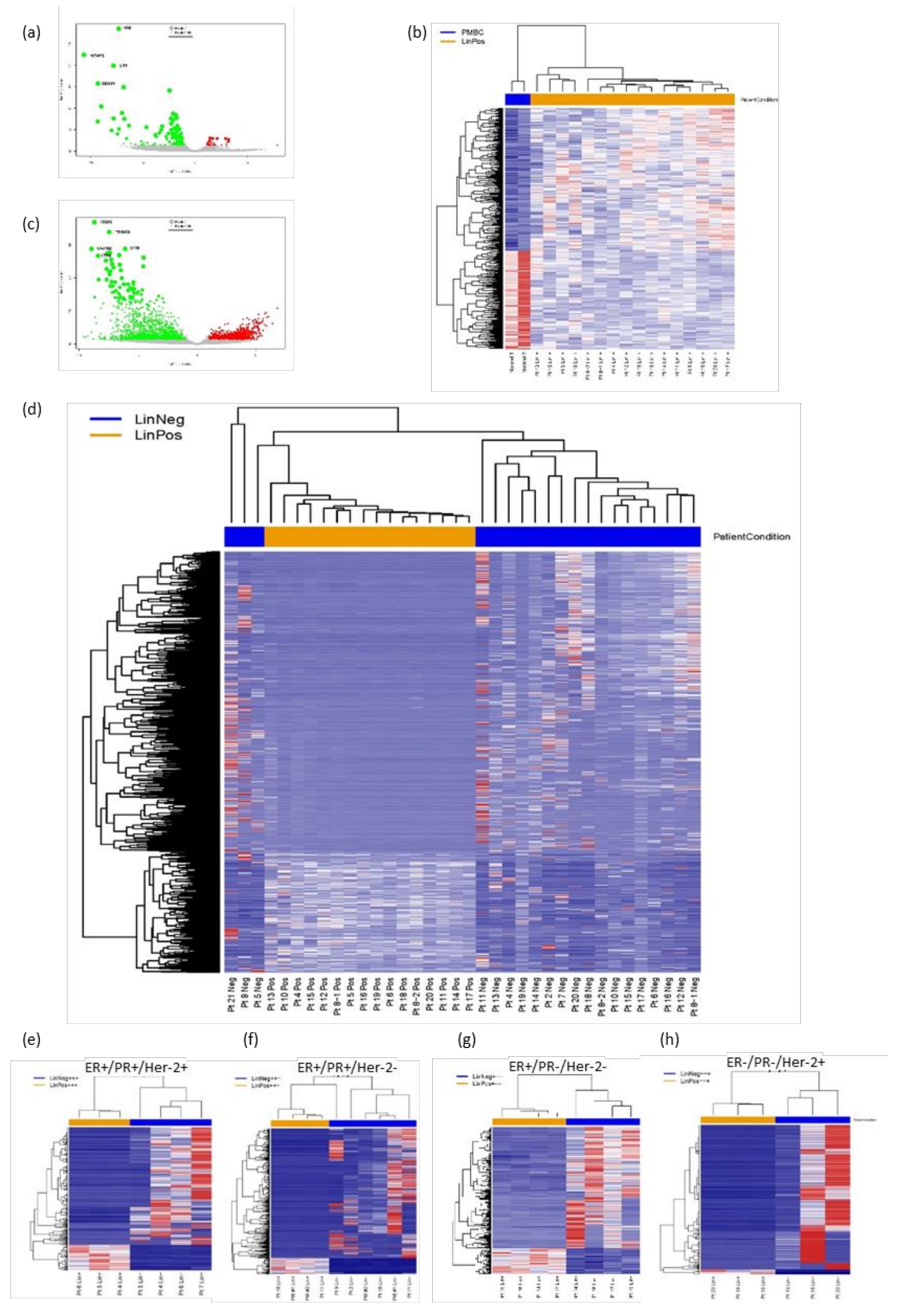
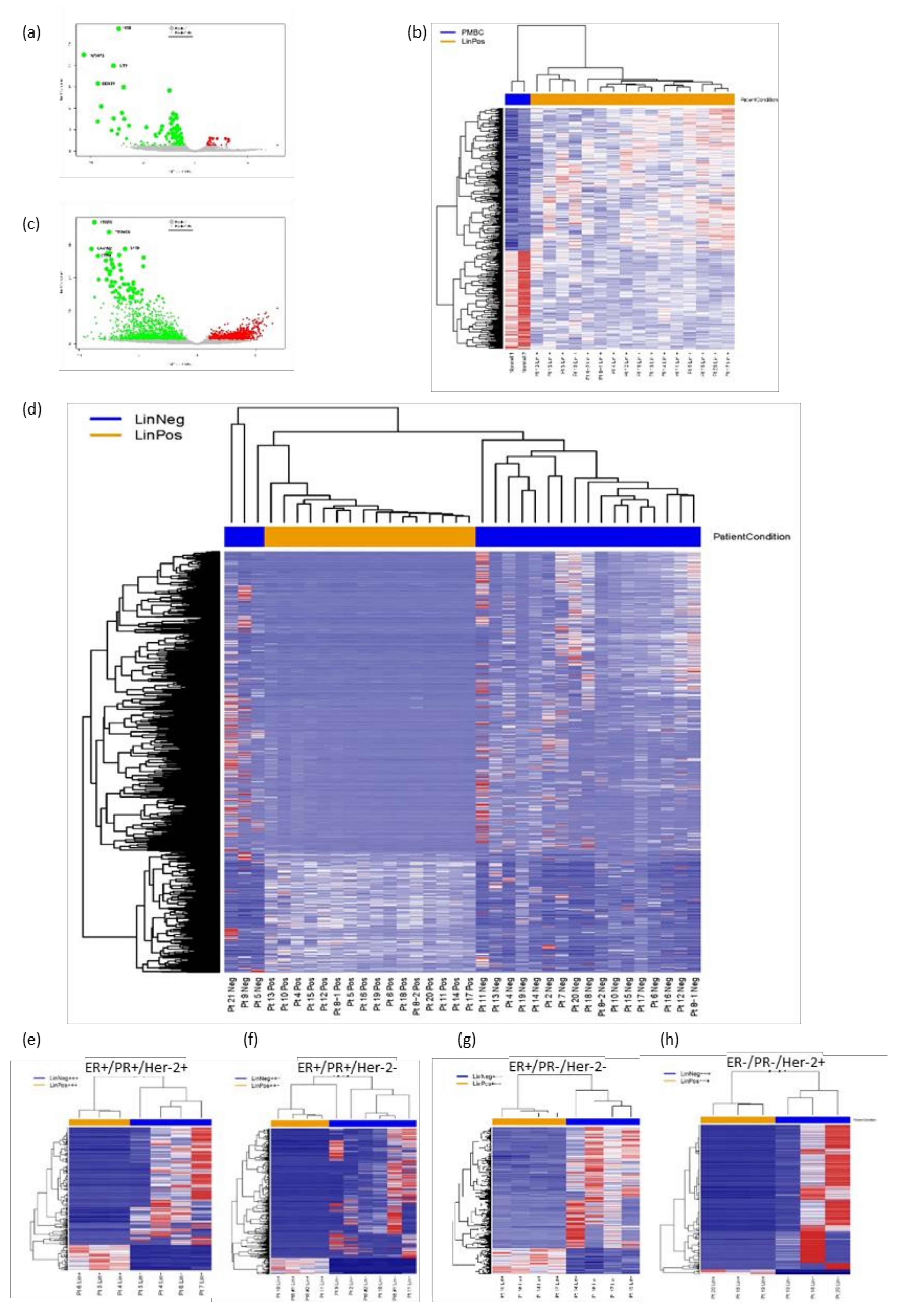
3.2. FACS Isolation of Patient Lin− and Lin+ Cell Populations and RNA-Seq Analyses
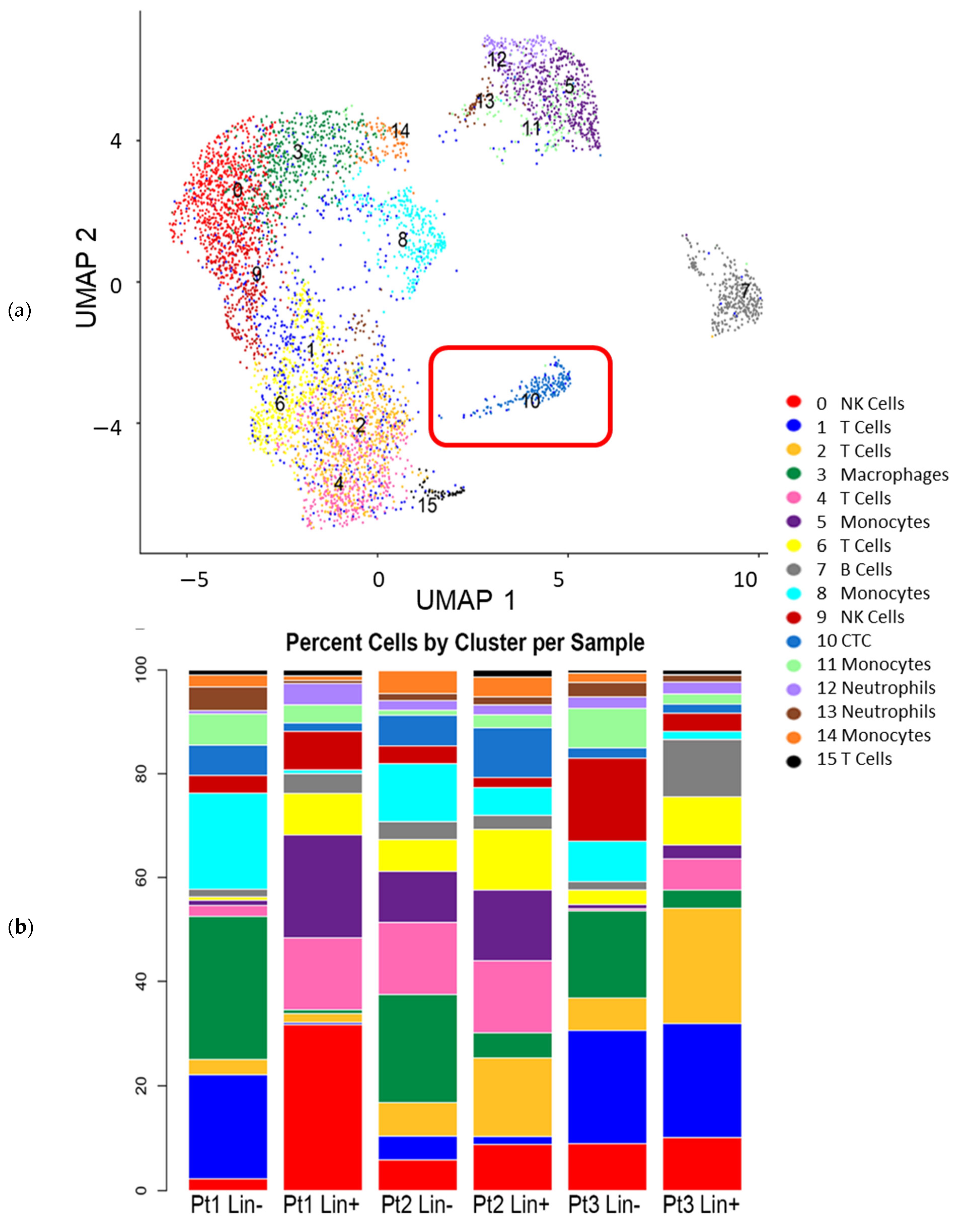
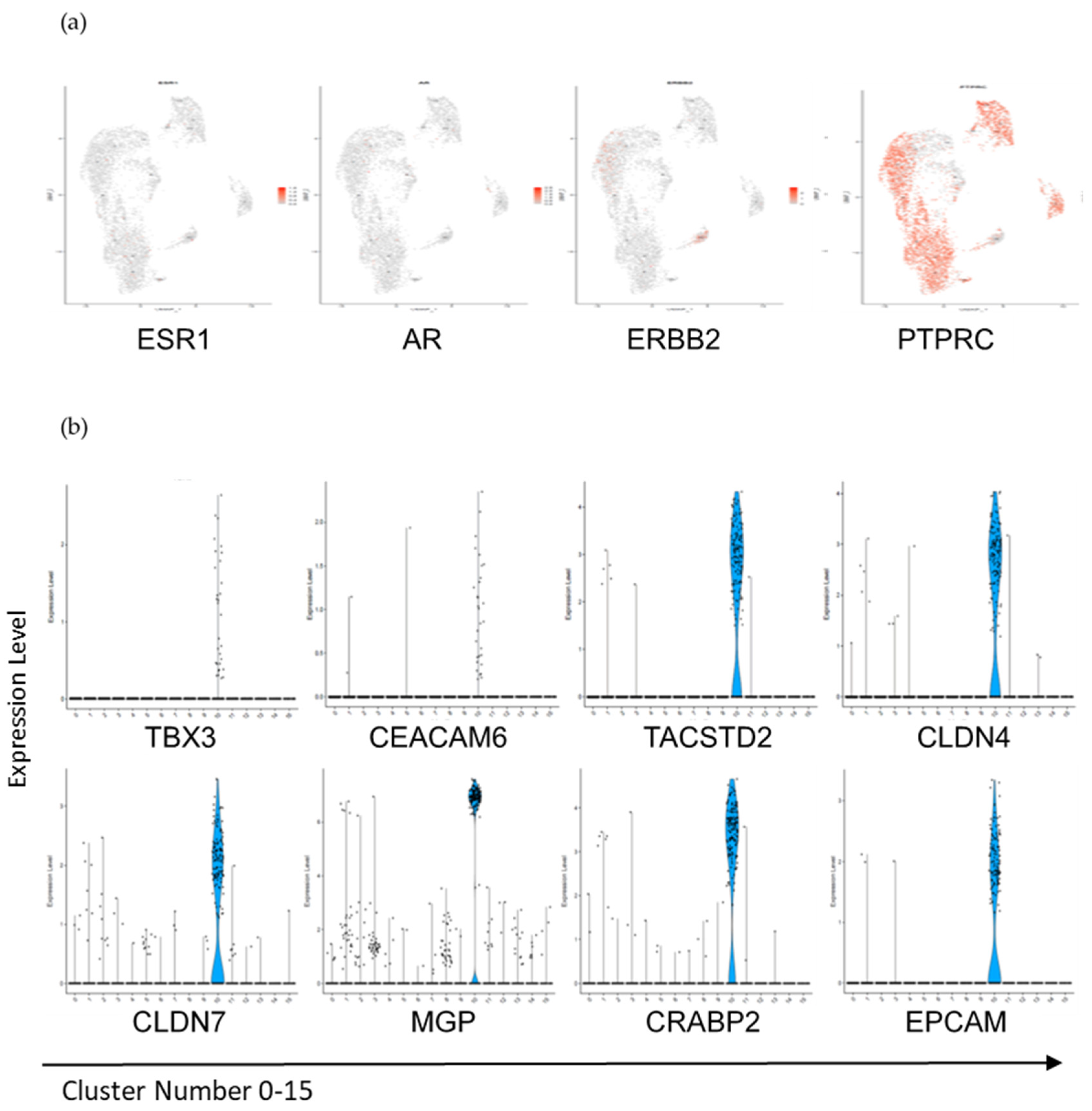
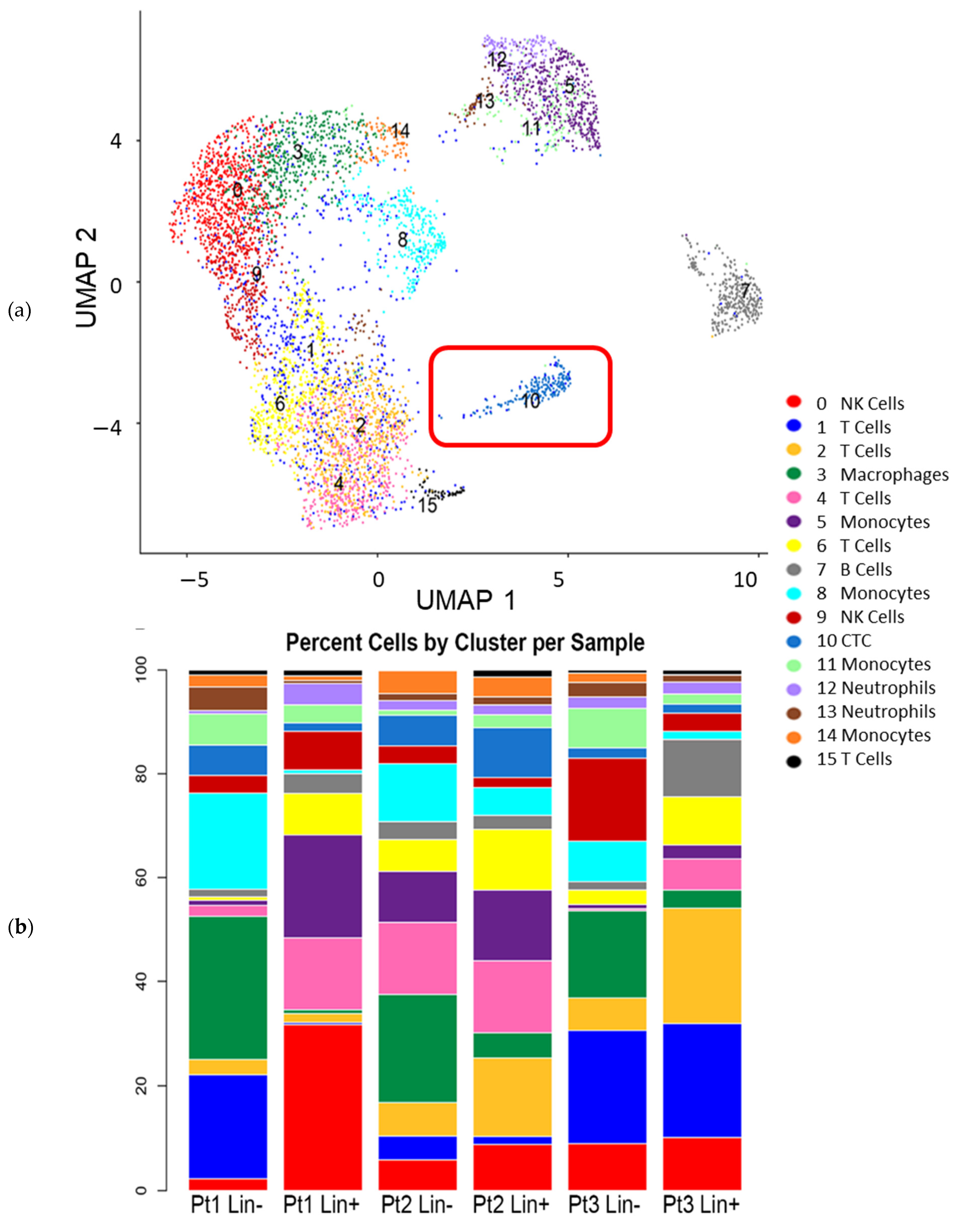
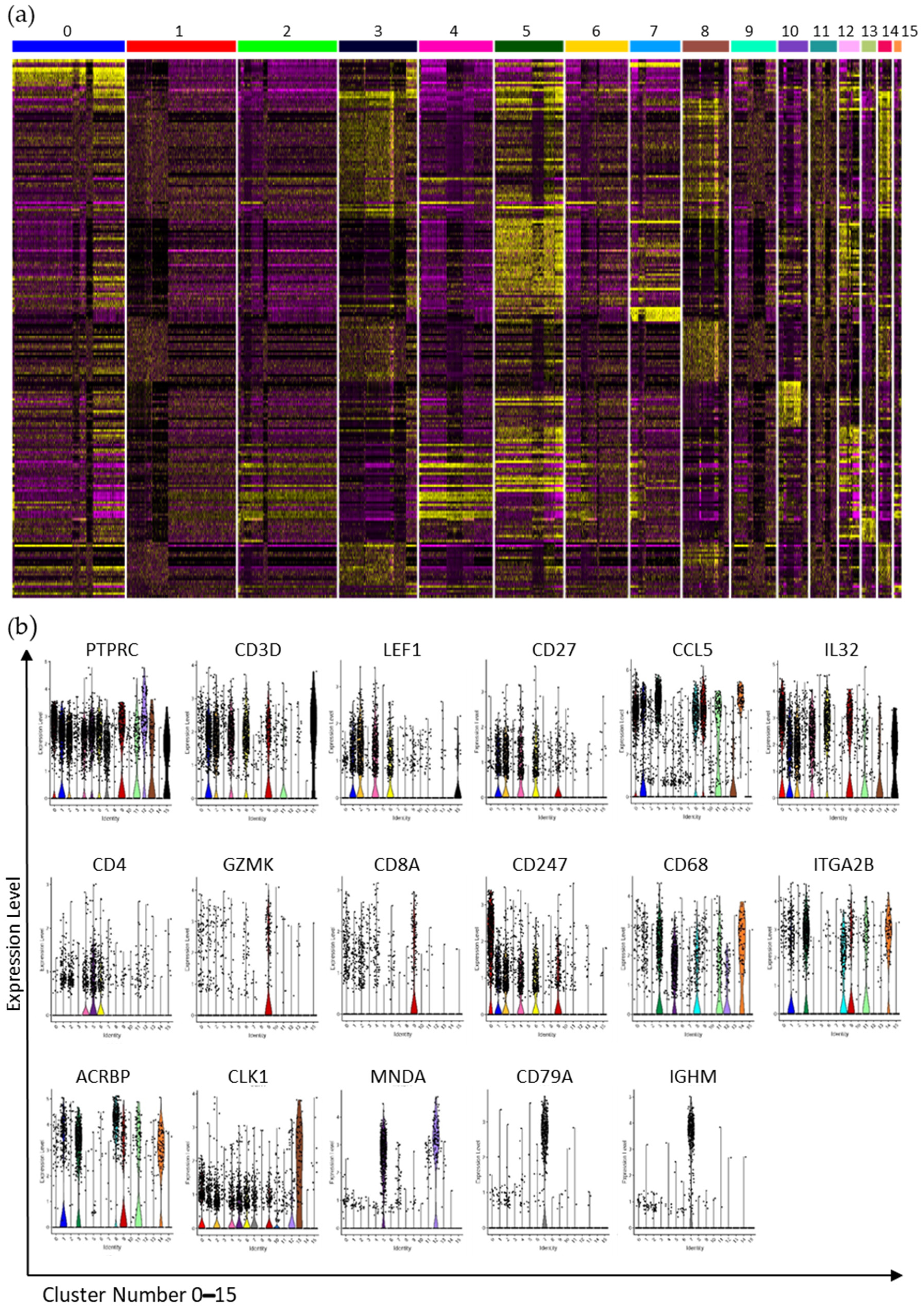
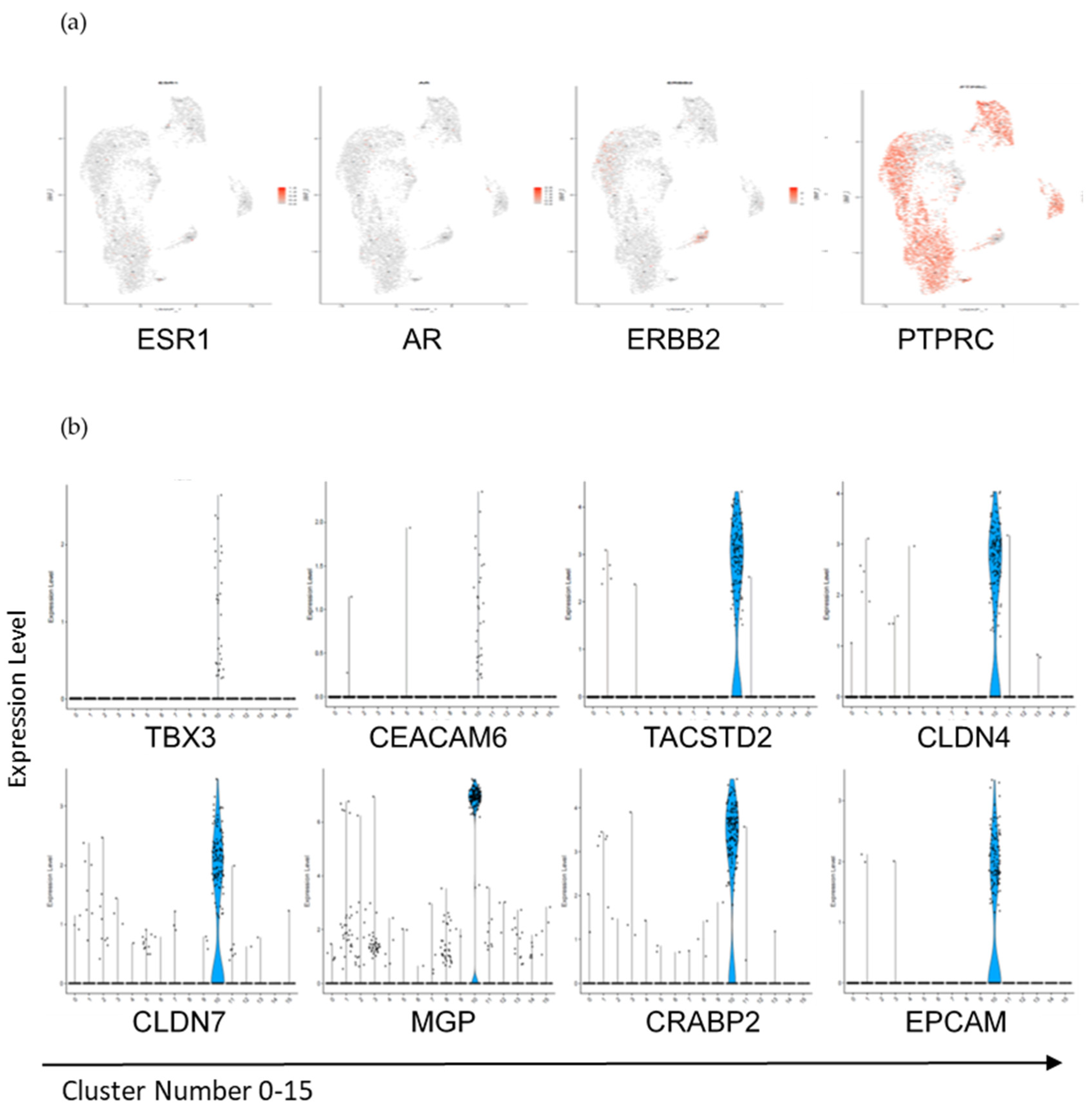
3.3. Interrogation of the Lin− and Lin+ Populations by Single-Cell RNA Sequencing (scRNA-Seq)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) Program Populations (1969–2019). 2019. Available online: www.seer.cancer.gov/popdata (accessed on 5 February 2021).
- Gupta, G.P.; Massague, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A Pooled Analysis of Bone Marrow Micrometastasis in Breast Cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Kiosses, W.B. Intratumoral Cancer Cell Intravasation Can Occur Independent of Invasion into the Adjacent Stroma. Cell Rep. 2017, 19, 601–616. [Google Scholar] [CrossRef] [Green Version]
- Rack, B.S.C.; Jückstock, J.; Andergassen, U.; Hepp, P.; Zwingers, T.; Friedl, T.W.; Lorenz, R.; Tesch, H.; Fasching, P.A.; Fehm, T.; et al. SUCCESS Study Group., Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl. Cancer Inst. 2014, 106, dju066. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Azizi, M.; Eslami-S, Z.; Cortés-Hernández, L.E.; Heidarifard, M.; Nouri, M.; Alix-Panabières, C. The Role of Circulating Tumor Cells in the Metastatic Cascade: Biology, Technical Challenges, and Clinical Relevance. Cancers 2020, 12, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, H.; Obradović, M.M.S.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, S.L.K.; Werno, C.; Ehrl, C.; Maneck, M.; et al. Early dissemination seeds metastasis in breast cancer. Nature 2016, 540, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Pantel, K. Tumor Cell Dissemination: Emerging Biological Insights from Animal Models and Cancer Patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Reduzzi, C.; Di Cosimo, S.; Gerratana, L.; Motta, R.; Martinetti, A.; Vingiani, A.; D’Amico, P.; Zhang, Y.; Vismara, M.; Depretto, C.; et al. Circulating Tumor Cell Clusters Are Frequently Detected in Women with Early-Stage Breast Cancer. Cancers 2021, 13, 2356. [Google Scholar] [CrossRef]
- Boral, D.; Vishnoi, M.; Liu, H.N.; Yin, W.; Sprouse, M.L.; Scamardo, A.; Hong, D.S.; Tan, T.Z.; Thiery, J.P.; Chang, J.C.; et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat. Commun. 2017, 8, 196. [Google Scholar] [CrossRef] [Green Version]
- Vishnoi, M.; Boral, D.; Liu, H.; Sprouse, M.L.; Yin, W.; Goswami-Sewell, D.; Tetzlaff, M.T.; Davies, M.A.; Oliva, I.C.G.; Marchetti, D. Targeting USP7 Identifies a Metastasis-Competent State within Bone Marrow–Resident Melanoma CTCs. Cancer Res. 2018, 78, 5349–5362. [Google Scholar] [CrossRef] [Green Version]
- Lang, J.E.; Ring, A.; Porras, T.; Kaur, P.; Forte, V.A.; Mineyev, N.; Tripathy, D.; Press, M.F.; Campo, D. RNA-Seq of Circulating Tumor Cells in Stage II–III Breast Cancer. Ann. Surg. Oncol. 2018, 25, 2261–2270. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef]
- Ramirez, A.B.; Bhat, R.; Sahay, D.; De Angelis, C.; Thangavel, H.; Hedayatpour, S.; Dobrolecki, L.E.; Nardone, A.; Giuliano, M.; Nagi, C.; et al. Circulating tumor cell investigation in breast cancer patient-derived xenograft models by automated immunofluorescence staining, image acquisition, and single cell retrieval and analysis. BMC Cancer 2019, 19, 220. [Google Scholar] [CrossRef]
- Lustberg, M.B.; Balasubramanian, P.; Miller, B.; Garcia-Villa, A.; Deighan, C.; Wu, Y.; Carothers, S.; Berger, M.; Ramaswamy, B.; Macrae, E.R.; et al. Heterogeneous atypical cell populations are present in blood of metastatic breast cancer patients. Breast Cancer Res. 2014, 16, R23. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The Identification and Characterization of Breast Cancer CTCs Competent for Brain Metastasis. Sci. Transl. Med. 2013, 5, 180ra48. [Google Scholar] [CrossRef] [Green Version]
- Vishnoi, M.; Peddibhotla, S.; Yin, W.; Scamardo, A.T.; George, G.C.; Hong, D.S.; Marchetti, D. The isolation and characterization of CTC subsets related to breast cancer dormancy. Sci. Rep. 2015, 5, 17533. [Google Scholar] [CrossRef] [Green Version]
- Boral, D.; Liu, H.N.; Kenney, S.R.; Marchetti, D. Molecular Interplay between Dormant Bone Marrow-Resident Cells (BMRCs) and CTCs in Breast Cancer. Cancers 2020, 12, 1626. [Google Scholar] [CrossRef]
- Sprouse, M.L.; Boral, W.T.D.; Liu, H.N.; Yin, W.; Vishnoi, M.; Goswami-Sewell, D.; Li, L.; Pei, G.; Jia, P.; Glitza-Oliva, I.C.; et al. PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling. Int. J. Mol. Sci. 2019, 20, 1916. [Google Scholar] [CrossRef] [Green Version]
- Frerich, C.A.; Brayer, K.J.; Painter, B.M.; Kang, H.; Mitani, Y.; El-Naggar, A.K.; Ness, S.A. Transcriptomes define distinct subgroups of salivary gland adenoid cystic carcinoma with different driver mutations and outcomes. Oncotarget 2017, 9, 7341–7358. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.B.; Madrid, N.J.; Suzuki, H.; Ness, S.A. Optimized approach for Ion Proton RNA sequencing reveals details of RNA splicing and editing features of the transcriptome. PLoS ONE 2017, 12, e0176675. [Google Scholar] [CrossRef] [Green Version]
- Brayer, K.J.; Frerich, C.A.; Kang, H.; Ness, S.A. Recurrent Fusions in MYB and MYBL1; Define a Common, Transcription Factor–Driven Oncogenic Pathway in Salivary Gland Adenoid Cystic Carcinoma. Cancer Discov. 2016, 6, 176. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J.R. topGO: Enrichment Analysis for Gene Ontology. R Package Version 2.42.0; Bioconductor: Buffalo, NY, USA, 2020. [Google Scholar]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., III; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Team, R Core. A Language and Environment for Statistical Computing; Team R Core: Vienna, Austria, 2020. [Google Scholar]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337.e4. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Mabbott, A.N.; Baillie, J.K.; Brown, H.; Freeman, T.C.; Hume, A.D. An expression atlas of human primary cells: Inference of gene function from coexpression networks. BMC Genom. 2013, 14, 632. [Google Scholar] [CrossRef] [Green Version]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [Green Version]
- Giraddi, R.; Chung, C.-Y.; Heinz, R.E.; Balcioglu, O.; Novotny, M.; Trejo, C.L.; Dravis, C.; Hagos, B.M.; Mehrabad, E.M.; Rodewald, L.W.; et al. Single-Cell Transcriptomes Distinguish Stem Cell State Changes and Lineage Specification Programs in Early Mammary Gland Development. Cell Rep. 2018, 24, 1653–1666.e7. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Baeuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Krol, I.; Schwab, F.D.; Carbone, R.; Ritter, M.; Picocci, S.; De Marni, M.L.; Stepien, G.; Franchi, G.M.; Zanardi, A.; Rissoglio, M.D.; et al. Detection of clustered circulating tumour cells in early breast cancer. Br. J. Cancer 2021, 125, 23–27. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The human transcription factors. Cell 2018, 172, 650–665. [Google Scholar]
- Chakravarty, D.G.J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; Chang, M.T.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; Hollander, P.D.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, 2102050118. [Google Scholar] [CrossRef]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.-J.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef]
- Pece, S.; Tosoni, D.; Confalonieri, S.; Mazzarol, G.; Vecchi, M.; Ronzoni, S.; Bernard, L.; Viale, G.; Pelicci, P.G.; Di Fiore, P.P. Biological and Molecular Heterogeneity of Breast Cancers Correlates with Their Cancer Stem Cell Content. Cell 2010, 140, 62–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Behbod, F.; Atkinson, R.L.; Landis, M.D.; Kittrell, F.; Edwards, D.; Medina, D.; Tsimelzon, A.; Hilsenbeck, S.; Green, J.E.; et al. Identification of Tumor-Initiating Cells in a p53-Null Mouse Model of Breast Cancer. Cancer Res. 2008, 68, 4674–4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.B.; Onder, T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolsky, Y.; Sviridov, E.; Yao, J.; Dosymbekov, D.; Ustyansky, V.; Kaznacheev, V.; Dezso, Z.; Mulvey, L.; MacConaill, L.E.; Winckler, W.; et al. Genome-Wide Functional Synergy between Amplified and Mutated Genes in Human Breast Cancer. Cancer Res. 2008, 68, 9532–9540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blau, C.A.; Arturo, B.R.; Sibel, B.; Colin, C.P.; Michael, O.D.; Stephen, C.S.; Timothy, J.M.; Elisabeth, M.M.; Kimberly, A.B.; Vitalina, M.K.; et al. A Distributed Network for Intensive Longitudinal Monitoring in Metastatic Triple-Negative Breast Cancer. Natl. Compr. Canc. Netw. 2016, 14, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.H.H.; Liu, H.; Jin, J.; Zhu, P.; Wang, S.; Shen, K.; Hu, Y.; Li, Z.; Zhan, P.; Zhu, S.; et al. RNA sequencing enables systematic identification of platelet transcriptomic alterations in NSCLC patients. Biomed Pharmacother. 2018, 105, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Da Huang, W.; Sherman, B.T.; RA, L. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pauken, C.M.; Kenney, S.R.; Brayer, K.J.; Guo, Y.; Brown-Glaberman, U.A.; Marchetti, D. Heterogeneity of Circulating Tumor Cell Neoplastic Subpopulations Outlined by Single-Cell Transcriptomics. Cancers 2021, 13, 4885. https://doi.org/10.3390/cancers13194885
Pauken CM, Kenney SR, Brayer KJ, Guo Y, Brown-Glaberman UA, Marchetti D. Heterogeneity of Circulating Tumor Cell Neoplastic Subpopulations Outlined by Single-Cell Transcriptomics. Cancers. 2021; 13(19):4885. https://doi.org/10.3390/cancers13194885
Chicago/Turabian StylePauken, Christine M., Shelby Ray Kenney, Kathryn J. Brayer, Yan Guo, Ursa A. Brown-Glaberman, and Dario Marchetti. 2021. "Heterogeneity of Circulating Tumor Cell Neoplastic Subpopulations Outlined by Single-Cell Transcriptomics" Cancers 13, no. 19: 4885. https://doi.org/10.3390/cancers13194885
APA StylePauken, C. M., Kenney, S. R., Brayer, K. J., Guo, Y., Brown-Glaberman, U. A., & Marchetti, D. (2021). Heterogeneity of Circulating Tumor Cell Neoplastic Subpopulations Outlined by Single-Cell Transcriptomics. Cancers, 13(19), 4885. https://doi.org/10.3390/cancers13194885