1. Introduction
Breast cancer is a major public health problem. It is the most frequent cancer in women and the second most common cancer overall, with over 2,000,000 newly diagnosed cases worldwide in 2020 (
https://www.who.int/news-room/fact-sheets/detail/breast-cancer accessed on 20 May 2021). It is highly heterogeneous in its pathological characteristics, with some cases showing slow growth rate with excellent prognosis, while others are very aggressive tumors. Expression of estrogen and progesterone receptors (ER and PR, respectively) usually suggests a better prognosis, based on lower aggressiveness and the possibility to use endocrine therapy (tamoxifen and aromatase inhibitors) [
1]. Conversely, overexpression of the HER2 protein, although allowing the use of specific drugs targeting this receptor (tyrosine kinase inhibitors or monoclonal antibodies), is usually associated with more aggressive tumors and dismal prognosis [
1]. Triple-negative breast cancer (TNBC) affects approximately 10 to 20 percent of patients and is characterized by tumors that do not express ER or PR at all, and do not overexpress HER2 [
1], all features making them insensitive to therapies targeting these proteins [
2]. Therefore, while endocrine therapy and HER2 “inhibitors” still represent the prototype of personalized approaches for several breast tumors, new actionable molecular targets need to be continuously discovered and validated to assist clinicians in their therapeutic decisions about a considerable number of cases [
3].
Cancer cells are dependent on the translational machinery due to their high demand for protein synthesis and mRNAs stabilization. DEAD-box helicases (so called because of their amino acid motif D-E-A-D, i.e., Asp-Glu-Ala-Asp) are intimately involved in these processes. They regulate RNA processing and metabolism through ATP-dependent unwinding of short double-stranded (ds) RNAs [
4]. Indeed, deregulation of expression, or function, of different members of the DEAD-box protein family has been shown to be involved in biological processes controlling cancer development [
5,
6,
7]. The
DEAD-box RNA helicase 3 X (
DDX3X) gene is located on the X-chromosome and is widely expressed in a broad range of organisms in which it controls pleiotropic physiological events in a variety of tissues. Specifically, DDX3X regulates different steps of RNA metabolism, including splicing, export, transcription, and translation initiation. As such, its activity has been shown to be involved in several cellular processes, from stress regulation to cell proliferation and apoptosis [
8]. Based on DDX3X’s involvement in many basic mechanisms controlling cell proliferation, it is unsurprising to observe aberrant expression and function of DDX3X in human tumors. Notably, DDX3X has been repeatedly proven to exert pro-tumorigenic effects in breast cancer [
8]. Specifically, knockdown of DDX3X in breast cancer cell lines decreased their proliferation and clonogenicity in vitro and reduced their ability to generate tumors and form metastases in vivo [
9]. Moreover, DDX3X is often overexpressed in breast cancers, and its expression is upregulated in distant breast cancer metastases, especially in the brain, with highly metastatic DDX3X protein expression correlated to a worse survival rate [
10]. Similarly, we have recently observed that women whose breast cancers have low
DDX3X gene expression had significantly better prognosis (Log-Rank test’s
p-value < 0.01) than the group with high expression (
Figure S1), thus supporting a specific prognostic role for
DDX3X in these tumor types. Therefore, based on all available data, DDX3X represents a promising potential target for pharmacological inhibition in breast cancers. However, none of the compounds identified that target the ATPase activity of DDX3X have progressed to clinical studies, yet. Taking advantage of in silico modeling approaches, a new class of molecules has been recently demonstrated to interact with the RNA-binding site of DDX3X [
11,
12], a mechanism distinct from the more conventional DDX3X inhibitors competing for the ATP binding sites. Here, we decided to study the antiproliferative activity of the FHP01 molecule [
13] against human breast cancer models. Indeed, FHP01 demonstrated very strong in vitro cytotoxic effects in a panel of breast cancer cell lines, including TNBC cellular models. Interestingly, this activity correlated with its ability to suppress the WNT/β-catenin signaling axis, a pathway often correlated with DDX3X-dependent cell proliferation and transformation [
14]. Based on these observations, and on the demonstration of a favorable pharmacokinetic profile and distribution in mouse models, we decided to test FHP01 activity also in a classic in vivo model for TNBC. Indeed, FHP01 demonstrated strong anti-tumor activity with no signs of specific organ toxicity and a good drug tolerance.
2. Materials and Methods
2.1. Cell Culture
Breast cancer cells were purchased from ATCC. All cells were cultured in the adequate medium at 37 °C in an atmosphere of 5% CO2/air. Specifically, MDA MB 231, T47D, MCF7, and 293T cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM), 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 units per mL penicillin-streptomycin; MDA MB 468 in RPMI 1640, 10% FBS, 2 mM L-glutamine, 100 units per mL penicillin-streptomycin; SKBR3 in RPMI 1640, 10% FBS, 250 mg glucose, 1 mM sodium pyruvate, 2 mM L-glutamine, 100 units per mL penicillin-streptomycin; MCF10A in DMEM/F12, 5% Horse serum, 20 ng/mL EGF, 0.5 μg/mL Hydrocortisone, 100 ng/mL Cholera Toxin, 10 μg /mL Insulin, 2 mM L-glutamine, 100 units per mL penicillin-streptomycin. Peripheral blood mononuclear cells (PBMCs) were cultured in RPMI 1640 supplemented with 10% of heat-inactivated fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, interleukin-2 20 U/mL, HEPES 12.5 mM.
2.2. Helicase Assay
293T cells (3 × 10
5) were seeded in 6-well plates and, after 24 h, transfected with 1 μg of HA-DDX3 (Addgene plasmid # 44975) [
15] by using the Lipofectamine reagent (Thermo Fischer Scientific, Waltham, MA, USA). After 36 h, cells were lysed in NP-40 Lysis Buffer (20 mM HEPES pH = 7.5, 10 mM EGTA pH = 8, 40 mM β-glycerolphosphate, 1% NP40, 2.5 mM MgCl
2, 2 mM Orthovanadate, 2 mM NaF, 1 mM DTT, and 1X protease inhibitors mixture) and 2 mg of proteins were immunoprecipitated with anti-HA antibodies (Covance, Princeton, NJ, USA) in NP40 lysis buffer. Immunoprecipitated samples were washed twice with lysis buffer and one time in helicase reaction buffer (50 mM Tris HCl pH = 8, 1 mM DTT, 5% glycerol, 10 mM MgCl
2). Beads were then resuspended in 100 μL in helicase reaction buffer.
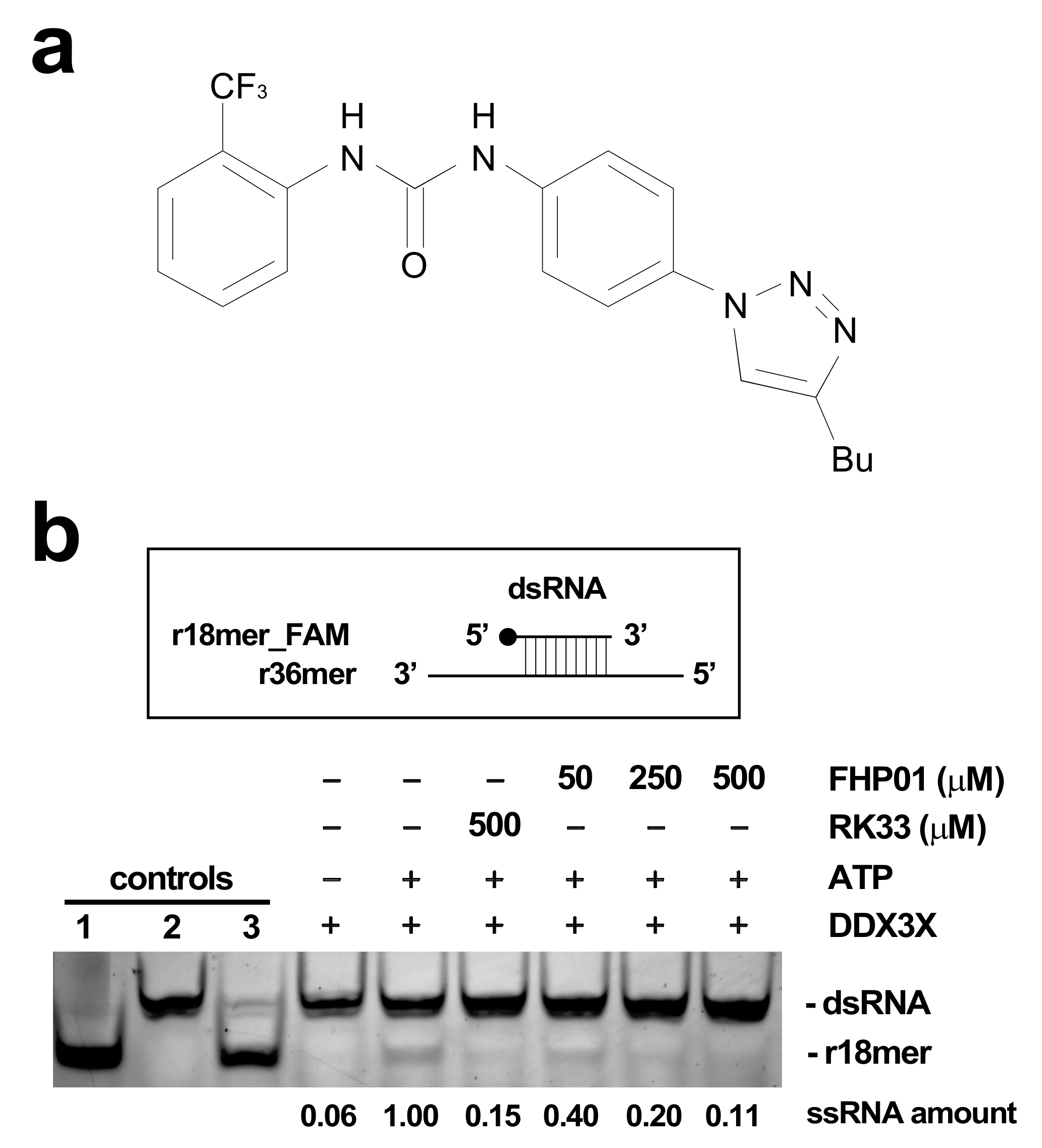
The helicase activity of DDX3 was tested by measuring the unwinding of an 18/36mer double stranded (ds) RNA oligonucleotide: r36mer 5′-ACCAGCUUUGUUCCUUGGGUUCUUGGGAGCAGCAGG-3′ and r18mer_FAM [6FAM]-5′-CCCAAGAACCCAAGGAAC-3′ [
16]. The 36mer oligonucleotides sequence is complementary to the 18mer. An RNA oligonucleotide homologous to the 18mer (without FAM modification) was used as competitor to avoid reannealing of the probed RNA after unwinding. Annealing reactions were performed in 50 mM Tris HCl pH = 8, 10 mM MgCl
2, 0.2 μM of r18mer, and 0.2 μM of r36mer. A sample with the final concentration of 20 nM of dsRNA plus 100 nM of the competitor were heated at 90 °C for 1 min to test efficiency of competition.
Ultimately, reactions were performed in a final volume 100 μL of helicase reaction buffer plus ATP 4 mM, 30 μL of beads used to immunoprecipitate HA-DDX3X, 20 nM of 18/36mer dsRNA, and 100 nM of competitor at 37 °C for 1 h and stopped by adding 10 μL of a stopping solution (EDTA 60 mM pH = 8, 40% glycerol, 0,6% SDS, 1% Orange G). Samples were next loaded on a 15% polyacrylamide TBE gel containing 1% SDS in 1× TBE buffer with 0.1% SDS and run at 4 °C for 1.5 h. The product of the unwinding reaction, r18mer_FAM and the dsRNA were visualized by an Image-Quant Las4000 (GE Healthcare, Chicago, IL, USA) with Cy3 detection filter and Green Epi light. Quantification of the ssRNA was performed by the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.3. Western Blots
Cells (1 × 105) were seeded in 6-well plates. After 24 h, cells were treated for indicated times with DMSO (vehicle) or FHP01 or XAV939 (Merck). After treatments, cells were washed with cold PBS and total protein extracts obtained by adding 80 µL of “RIPA Lysis buffer” (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5% Sodium deoxycholate, 0.1% SDS, 1% NP-40, 2 mM Orthovanadate, 2mM Sodium fluoride, 1mM Dithiothreitol and 1× protease inhibitors). After mechanicals detachment with cell scrapers, total lysates were collected in tubes, vortexed, and incubated for 15 min on ice. Tumors from mice were collected and disrupted by bead homogenization in Lysing Matrix D tubes using a FastPrep-24 5G homogenizer (MP Biomedicals, Solon, OH, USA) filled with “RIPA Lysis buffer” w:v. Next, samples were centrifuged for 10 min at 16,000× g, and supernatants were collected, representing total cell lysates.
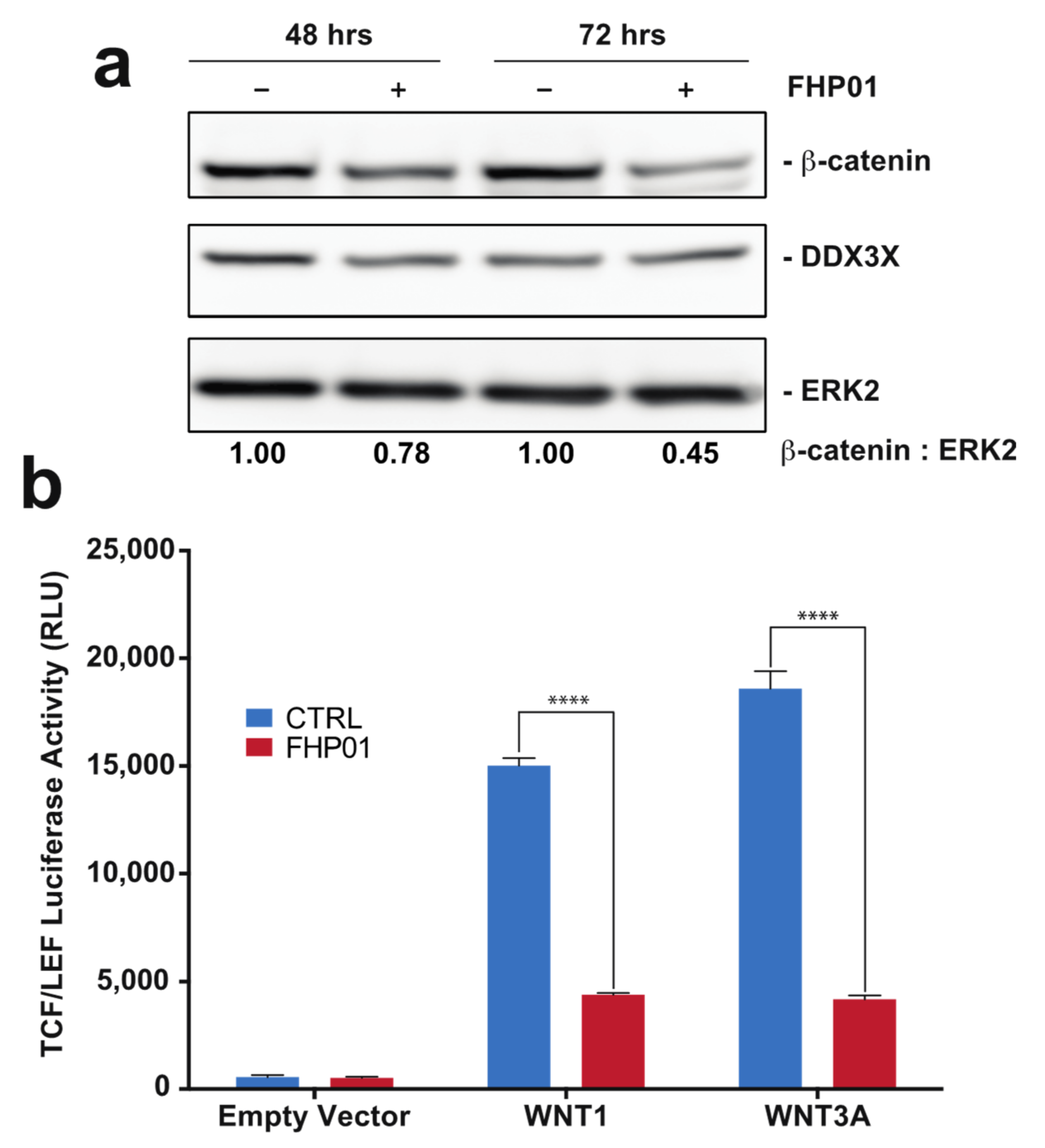
For Western blot analysis, 20 μg of proteins derived from total lysates was loaded on 8% polyacrylamide gels with 1× of Laemmli buffer and resolved by SDS-PAGE, transferred to Immobilon-P PVDF membrane (Millipore, IPVH00010), probed with anti-DDX3 (Cell Signaling, 2635), anti-β-catenin (c19220, BD Biosciences, Franklin Lakes, NJ, USA), anti-phospho β-catenin (9561, Cell Signaling, Danvers, MA, USA), anti-ERK2 (sc-154, Santa Cruz, Dallas, TX, USA), and revealed by enhanced chemiluminescence detection (ECL Plus; GE Healthcare, RPN2132). Densitometric analysis of western blots was performed with NIH Image J (National Institutes of Health).
2.4. Luciferase Assays
MDA MB 231 cells were seeded at 3 × 10
4 cells/well density in 12-well plates, in triplicate, and transfected with 250 ng of the M50 Super 8× TOPFlash reporter vector (Addgene Plasmid #12456) [
17] and 1 μg of expression vectors inducing the WNT pathway (Wnt1-V5, Addgene plasmid #43807, and Wnt3A-V5, Addgene plasmid #43810) [
18]. Eight hours after transfection, cells were treated with FHP01, XAV939 (Merck, Kenilworth, NJ, USA) or DMSO for indicated times prior to lysis in Passive Lysis Buffer (Promega, Madison, WI, USA). Luciferase activity in cellular lysates was assessed on a Glomax 20/20 luminometer (Promega) using the Luciferase Assay System (Promega). Results were normalized for GFP expression, upon transfection with GFP coding vector (1 μg). Representative results from 3 independent experiments are shown (
n = 3). Error bars represent the Standard Error of the Mean. All samples were read at least in triplicate.
2.5. Cell Viability Assays in Cancer Cells
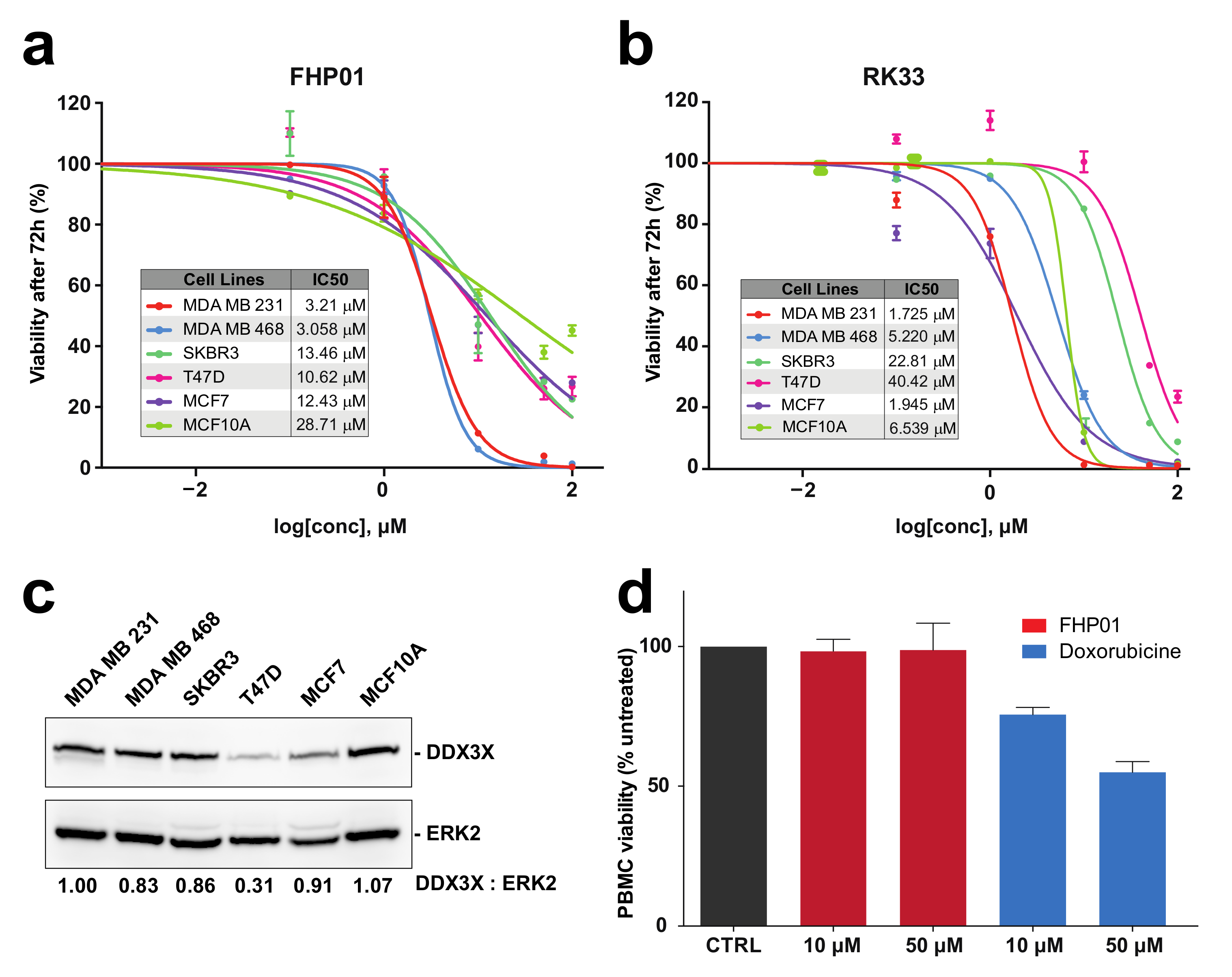
Cells were seeded at 3–5 × 104 cells/well density (depending on cell lines) in 12-well plates, in triplicate, and maintained at 37 °C in 5% CO2. After 24 h, cells were treated with different concentrations of indicated drugs in a range from 0 to 100 μM (0, 0.1, 1, 10, 50, 100 μM). After 72 h incubation, cells were washed in PBS and harvested. Cell number of each sample was determined with Z2 Coulter Counter (Beckman Coulter, Brea, CA, USA), in triplicate. Data about cell viability were plotted in GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA) to draw dose-response curve and to calculate IC50.
2.6. Cell Viability Assays in Peripheral Blood Mononuclear Cells (PBMCs)
Two days before the assay, phytohemagglutinin-A (PHA) 2 µg/mL was added to the PBMC culture. Cells were seeded at 13 × 104 cells/well in triplicate and maintained at 37 °C in 5% CO2. After 24 h, cells were treated with 10 μM and 50 μM of indicated drugs. After 72 h incubation, cells were washed in PBS and harvested. Cell number of each sample was determined with Z2 Coulter Counter (Beckman Coulter), in triplicate. Data about cell viability were plotted in GraphPad Prism 8.0 software 100% viability corresponding to vehicle (DMSO) treated PBMCs.
2.7. Pharmacokinetic Studies in Animal
These studies were performed at the GVKBioscience Pvt. LTD Contract Research and Development Organization (CRDO). Animals were housed in cages with clean bedding. Adult Swiss Albino male mice (weight = 20–35 g) were maintained and monitored for good health in accordance with GVKBioscience Pvt. LTD Test Facility SOPs and at the discretion of the laboratory animal veterinarian. Certified rodent diet was provided, and water was available ad libitum. A periodic analysis of the water was performed, and the results have been archived at the Test Facility. Environmental controls for the animal room were set to maintain a temperature of 22 to 25 °C, humidity of 40–70% relative humidity, and a 12-h light/12-h dark cycle. Animals have been dosed i.v. with 1 mg/kg of FHP01 i.p. with 10 mg/kg of compound or p.o. through gastric gavage needle with 50 mg/kg of compound. Next, mice were anesthetized using gaseous anesthesia and blood samples were collected at each time point, from 3 mice of respective group. After collection of blood samples at each time point, the blood samples were stored on ice, prior to centrifugation. Blood samples were centrifuged at 1540 rcf (5000 rpm), 4 °C for 10 min to separate plasma. Plasma was transferred to pre-labeled micro-centrifuge tubes and promptly frozen at −80 °C until analysis. Samples have been identified by test item, group, animal number, and collection time point. A fit-for-purpose bioanalytical method was developed for analyzing the plasma samples. One set of nine standards have been run before the sample batch and used for plotting the calibration curve. Quality control (QC) samples have been prepared at a minimum of three concentrations, i.e., LQC (not more than 5 times to that of lowest standard concentration), HQC (not less than 75% of the highest standard concentration), and MQC (between the low and high concentration). Minimum of 6 QC samples (three concentrations in duplicate). One Set of QC (LQC, MQC, and HQC) samples have been analyzed before and after the sample batch.
For the analyses of drug distribution to brain and liver, whole body perfusion was performed using chilled saline in the defined group of animals followed by decapitation for tissue collection. The chest of the mice was exposed, the abdominal aorta was clamped, and both jugular veins were cut. Intra-cardial perfusion was performed through an insertion in the left ventricle. Mice brains and livers have been collected and immediately washed, dried, weighed and freeze at −80 °C until homogenization. The protocol has been reviewed and approved by the Institutional Animal Ethics Committee (IAEC).
2.8. Cancer Model Animal Study
Animal experiments were performed in accordance with current Italian legislation and the animal protocol has been reviewed and approved by Italian Ministero della Salute (authorization n. 886/2020-PR). The study was designed to verify the effect of newly synthetized molecule with DDX3X inhibitor activity administered via i.p at a concentration of 45 mg/kg on the tumor growth of xenograft MDA MB 231 human breast cancer cells. In the study, n refers to the number of tumors expressed on both flanks of the animals. The sample size was calculated by g-power calculator placing a predictable large (2-fold increase) effect size, alpha as 0.05, and a confidence interval of 95% and we set our experimental unit number of 10 (corresponding to 5 animals). Total number of animals used in the study was 10. Six-week-old athymic nude mice were purchased from Charles River Laboratories. Five animals in each cage acclimatized in our animal facilities for 5 days in individually ventilated cages. Animals were fed with the lab diet ad libitum and water was accessible at all times and kept under standard specific pathogen-free conditions of 12 h light/dark cycle. Observational analysis was performed at the beginning of experimental procedure to ensure animal wellbeing (
Table S1).
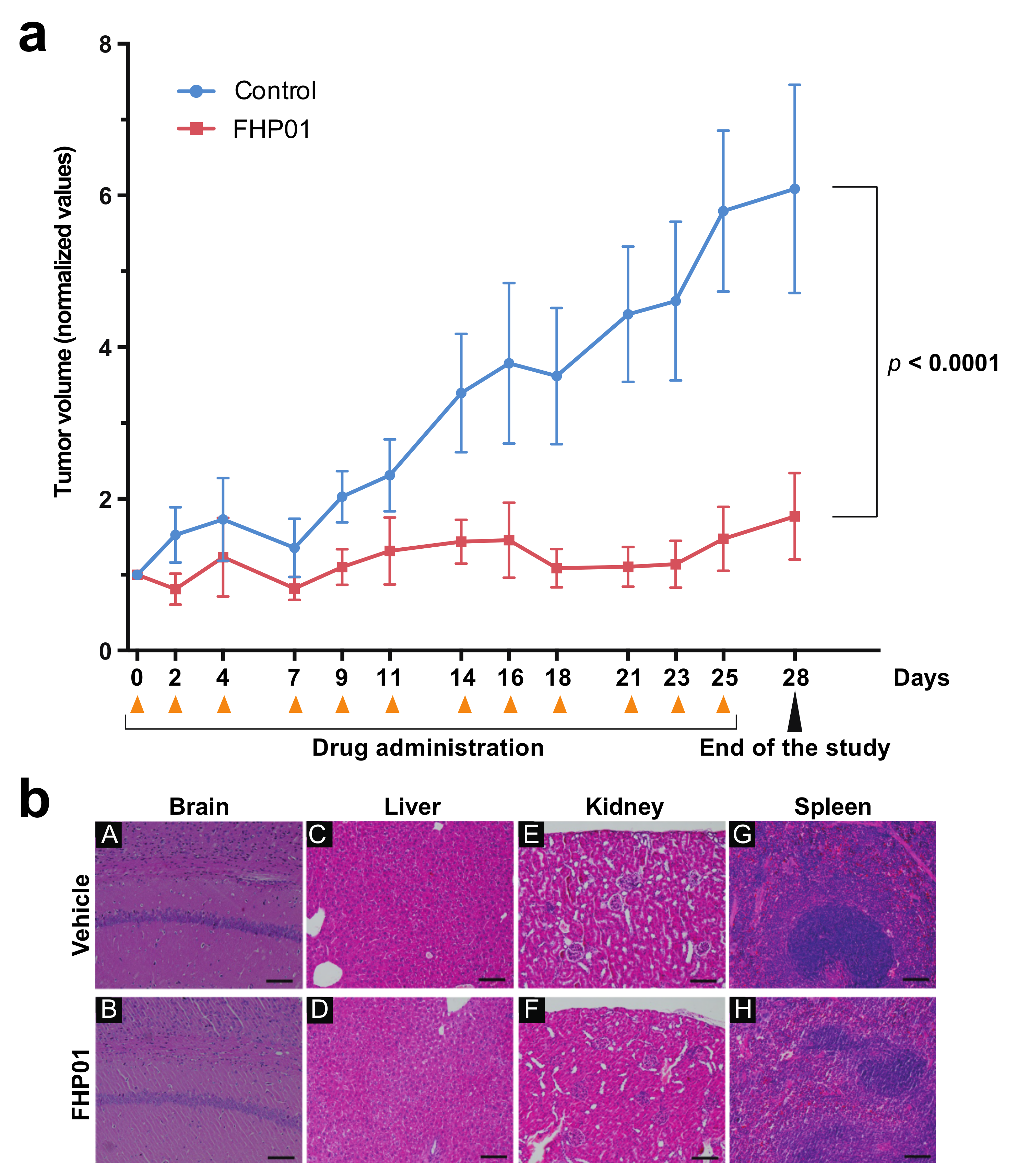
To establish xenograft tumors, MDA MB 231 cells were cultured in media as described above. On the day of experiment, cells (2.8 × 106) were suspended in 150 μL PBS 1:1 Matrigel and injected subcutaneously on both size of the flank region of nude mice. In order to determine tumor volume by manual caliper, the greatest longitudinal diameter (length) and the greatest transverse diameter (width) were determined. Tumor volumes based on caliper measurements were calculated by the modified ellipsoidal formula Tumor volume = 1/2(length × width2).
Group allocation and treatments: once tumors reached a mean size of 24.82 ± 2.56 mm3, animals were allocated in two groups by minimization strategy according to tumor size: the treatment group received 100 μL of a 45 mg/kg FHP01 suspended in a vehicle consisting of 10% DMSO, 5% Tween 80, 85% H2O via i.p. injection three times per week for 4 weeks; the control group included animals treated with 100 μL of vehicle (10% DMSO, 5% Tween 80, 85% H2O). One control animal reached HEP and was sacrificed and was not account for in the analysis. The FHP01 treatment group showed an outlier that was not accounted for. At the end of the experiment, animals were euthanized according to FELASA protocol and internal organs of interest were collected for postmortem histology.
For the final statistics, the number of tumors analyzed was 8 in the control group (one animal died for unknown reasons as certified by the AWB designed veterinary) and 9 for the treated group (the volume of one tumor was considered outlier). Results were reported as tumor volume increase, normalizing the value of each measurement to the baseline value of each sample and expressed as mean ± SEM. Two-way RM ANOVA was used to describe the significant effect of treatment versus time, considered significant when p < 0.05.
2.9. Histology Sampling
Animals were euthanized and internal organ samples collected. Spleen, kidney, lung, and brain from each accounted animal were formalin fixed and paraffin embedded. H&E staining of 4-µm-thick sections was performed to unveil sign of toxicity and loss of morphology.
2.10. Survival Analysis
Gene expression and clinical data were retrieved from GEO database (accession number [GSE31519],
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31519 accessed on 21 August 2020). We analyzed DDX3X probe specific expression vs. Event-Free Survival. After splitting patients according to DDX3X median expression level of 4 probes, we performed survival analysis using R software (Version R-4.0.1, R Foundation for Statistical Computing, Vienna, Austria) [
19]. Specifically, Kaplan-Meier survival curves were built, visualized, and compared (through Log-rank test) by using “survival” and “surviminer” CRAN packages.
2.11. Statistical Analysis
For each analysis, data are represented as the mean ± SEM. For comparison, statistical significance was tested using t-test for selected time point. Two-way RM ANOVA was applied to verify the effect of the two treatments in vivo. All p-values were based on two-sided statistical analyses, and p < 0.05 was considered statistically significant.
4. Discussion
Altogether, our results clearly indicate that the FHP01 compound exerts powerful in vivo anticancer activity as a single agent against a TNBC tumor model. In turn, this supports the possibility of using DDX3X helicase inhibitors against breast cancer. Nonetheless, while DDX3X has a clear pro-tumorigenic role in breast cancer, there are examples of context-dependent pro- or anti-proliferative effects for DDX3X in different or even in the same cancer types [
7,
8]. This aspect needs, therefore, to be considered in the hypothesis of using DDX3X inhibitors for cancer therapy in humans. In this context, DDX3X mutations have also been identified in some cancer types, i.e., medulloblastomas, head and neck squamous cell carcinomas (HNSCC), and hematological malignancies [
6]. Specifically, in medulloblastomas and natural killer/T cell lymphoma (NKTCL), DDX3X represents one of the most frequently mutated genes [
29,
30]. Unfortunately, the effects of these mutations on DDX3X functions are still debated [
29,
30,
31], making it difficult to define whether inhibition rather than stimulation of the DDX3X activity would be necessary to target these specific tumors.
Based on their potential anticancer and antiviral effects, multiple DDX3X inhibitors have been identified and studied over the last 10–15 years [
8,
32]. As an ATP-dependent RNA helicase, DDX3X offers two main mechanisms that can be exploited for inhibition: direct inhibition at the ATPase domain or, alternatively, occupation of the RNA-binding site. Drugs targeting the ATP-binding site block both the ATPase and helicase activity of DDX3X, therefore representing an efficient method to impair DDX3X functions. However, ATP-binding sites are present in several classes of enzymes with a consequent intrinsic potential for non-specific binding to these proteins, and increased probability of toxic effects. Conversely, drugs targeting RNA binding to the helicase domain will likely represent a more selective class of inhibitors with a narrower pharmacological profile: nonetheless, most current DDX3X inhibitors mostly target its ATP binding site [
8,
32]. Unfortunately, while these ATP binding site inhibitors demonstrate efficient cytotoxicity in vitro, their single agent efficacy in vivo is poor. The only effects observed are, in fact, often limited to a radio-sensitizing effect, as shown previously in breast cancer preclinical models, possibly because of selectivity or bioavailability issues [
8,
32,
33]. Conversely, all of them showed low toxicity in in vivo models, suggesting that inhibition of DDX3X activity per se is not toxic and that drugs with improved physicochemical and pharmacodynamic characteristics might represent a promising approach for breast cancer treatment. In fact, more selective DDX3X inhibitors that solely target its helicase activity have been recently identified and shown to exert strong antiviral efficacy and low toxicity [
11]. Consequently, we hypothesized that these types of drugs could represent good candidates to inhibit breast cancer cells’ proliferation with a novel targeted approach. Here, we demonstrated for the first time that, indeed, FHP01 showed a significant antiproliferative effect against several human breast cancer cells and this effect was particularly evident for TNBC cells. As TNBC is the subtype with the most unfavorable outcomes, the need for new targets is paramount [
2]. Importantly, the effect of in vivo FHP01 treatment, as a single agent, in TNBC mouse models revealed very high efficacy and low toxicity, overall demonstrating a strong potential for this class of molecules as a more effective therapeutic approach for this very aggressive type of cancer.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}