
Cancer Stem Cell for Tumor Therapy



Abstract
Simple Summary
Abstract
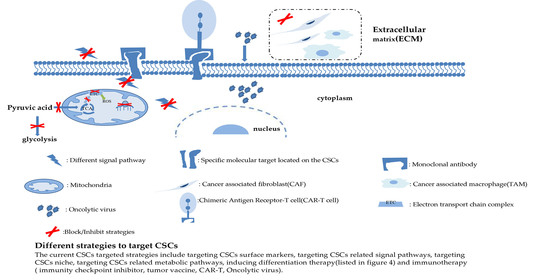
1. Introduction
2. Biological Characteristics
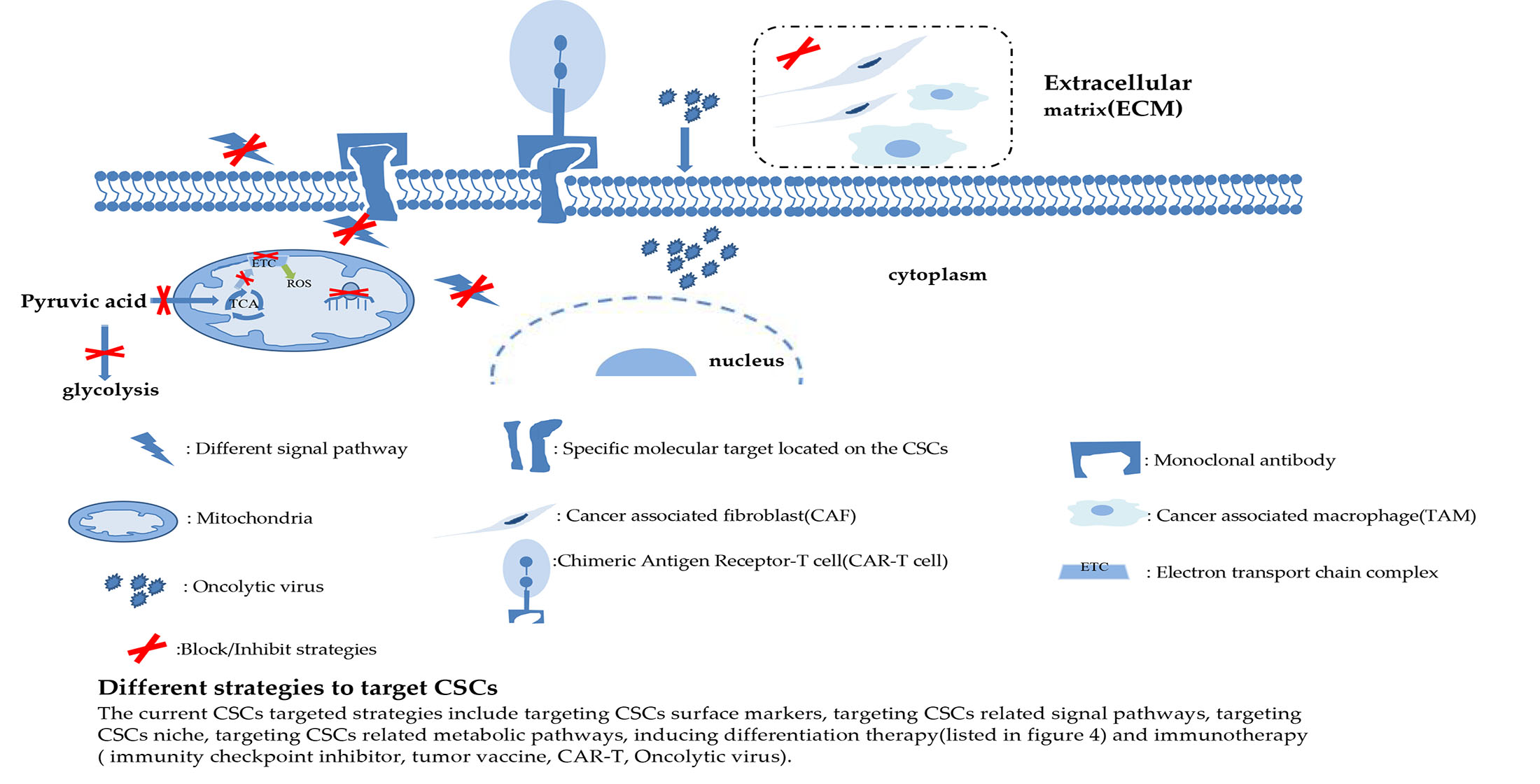
2.1. Mitotic Division Pattern of CSCs
2.2. Metabolic Pattern of CSCs
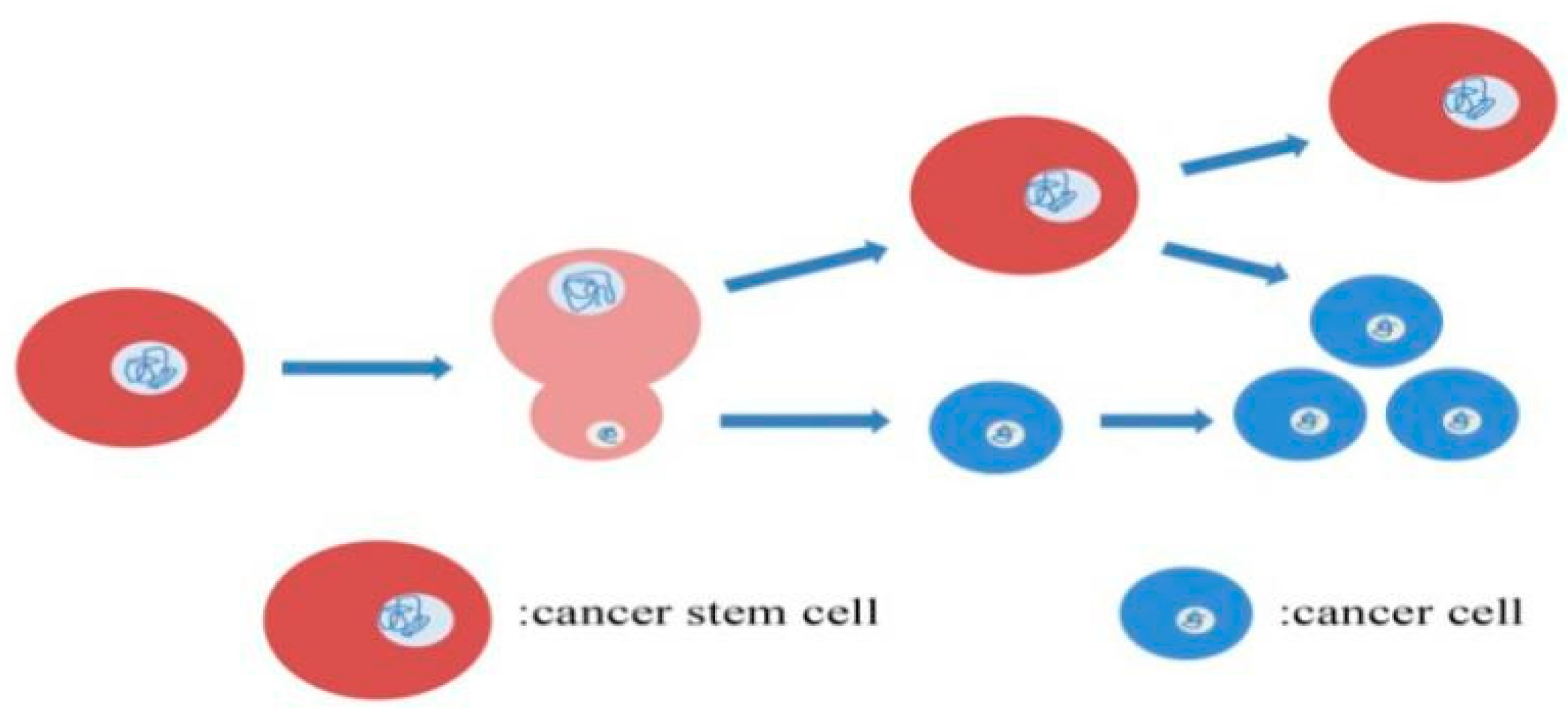
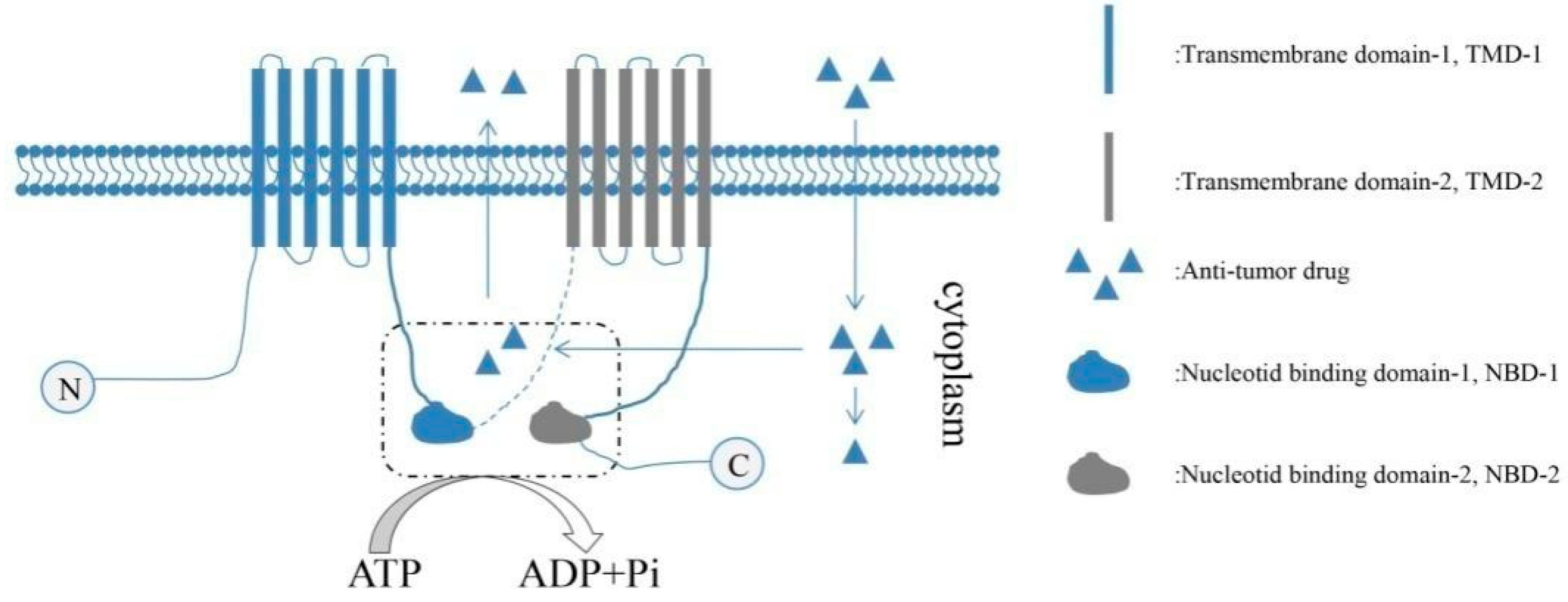
2.3. Therapeutic Resistance of CSCs
3. Targeted Strategies of CSCs
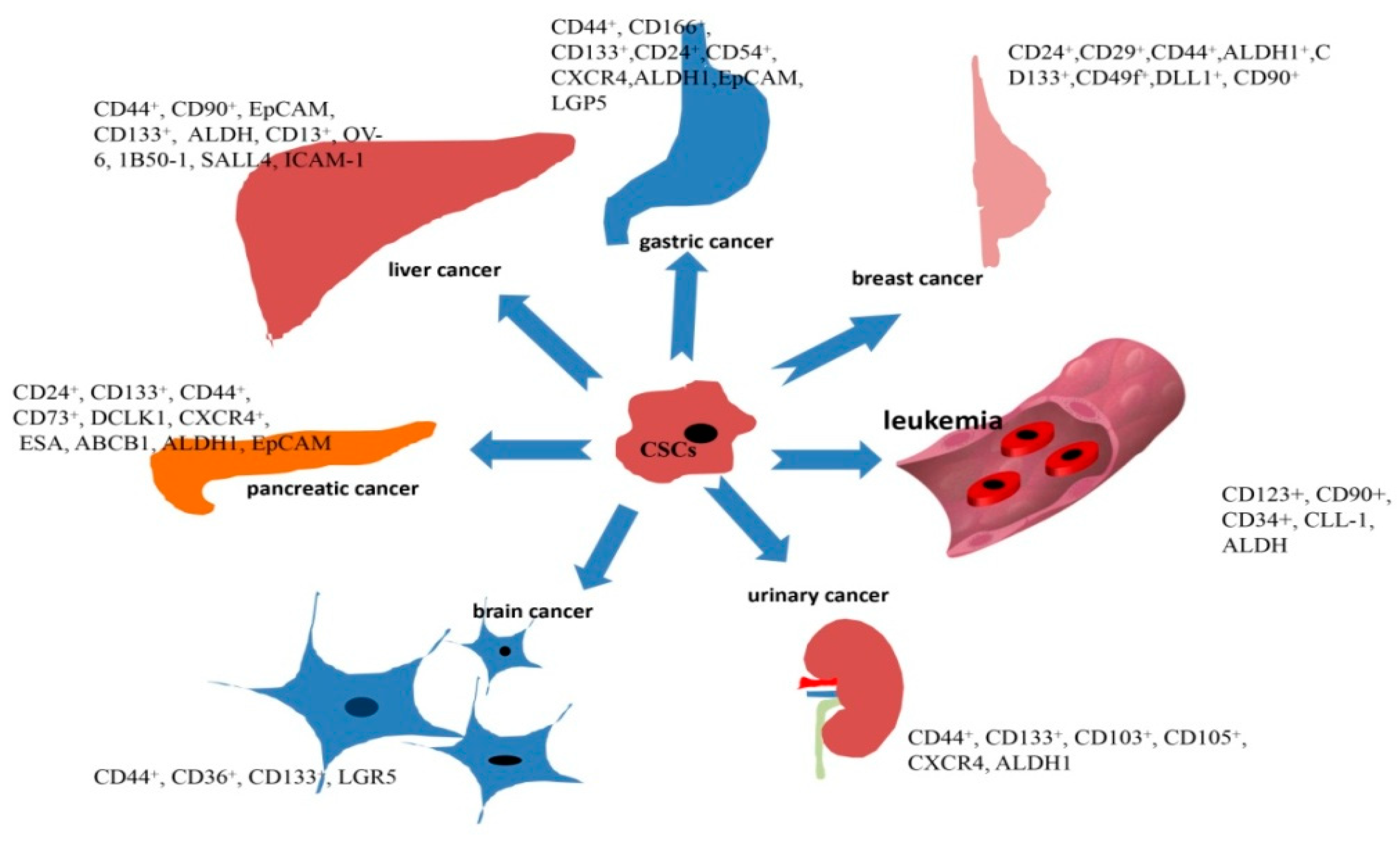
3.1. Targeting Surface Molecules of CSCs
3.2. Targeting Signal Pathways of CSCs
3.2.1. Notch Signal Pathway Inhibitor
γ-Secretase Inhibitors (GSIs)
Anti- Delta-like Ligand 4 (DLL4) Monoclonal Antibody
3.2.2. Hedgehog(Hh) Signal Pathway Inhibitor
3.2.3. Wnt Signal Pathway Inhibitor
3.2.4. Other Signal Pathway Inhibitors
3.3. Targeting Metabolic Pathways of CSCs
3.4. Targeting CSC Niches
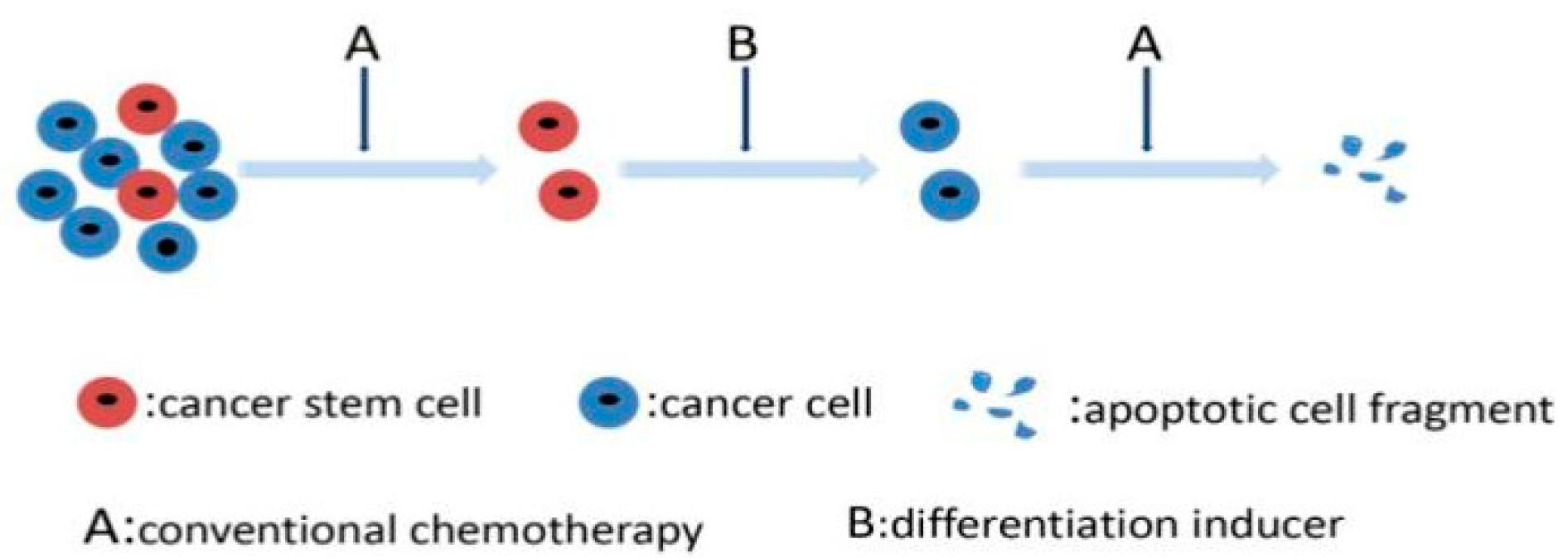
3.5. Targeting Differentiation Mechanisms of CSCs
3.6. Immunity Method of CSCs
3.6.1. Tumor Vaccine
3.6.2. Chimeric Antigen Receptor T Cell Therapy (CAR-T)
3.6.3. Oncolytic Virus
3.6.4. Targeting CSC-Immune Cell Crosstalk
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Heron, M.; Anderson, R.N. Changes in the Leading Cause of Death: Recent Patterns in Heart Disease and Cancer Mortality. NCHS Data Brief 2016, 254, 1–8. [Google Scholar]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Ma, S.; Cao, K.; Zhou, S.; Zhao, A.; Li, M.; Qian, F.; Zhu, C. Therapeutic approaches targeting cancer stem cells. J. Cancer Res. Ther. 2018, 14, 1469–1475. [Google Scholar] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Najafi, M.; Mortezaee, K.; Ahadi, R. Cancer stem cell (a)symmetry & plasticity: Tumorigenesis and therapy relevance. Life Sci. 2019, 231, 116520. [Google Scholar]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Knoblich, J.A. Mechanisms of asymmetric stem cell division. Cell 2008, 132, 583–597. [Google Scholar] [CrossRef]
- Bai, S.; Ingram, P.; Chen, Y.C.; Deng, N.; Pearson, A.; Niknafs, Y.S.; O’Hayer, P.; Wang, Y.; Zhang, Z.Y.; Boscolo, E.; et al. EGFL6 Regulates the Asymmetric Division, Maintenance, and Metastasis of ALDH+ Ovarian Cancer Cells. Cancer Res. 2016, 76, 6396–6409. [Google Scholar] [CrossRef] [PubMed]
- Sonbol, M.B.; Ahn, D.H.; Bekaii-Saab, T. Therapeutic Targeting Strategies of Cancer Stem Cells in Gastrointestinal Malignancies. Biomedicines 2019, 7, 17. [Google Scholar] [CrossRef]
- Smith, P.; Azzam, M.; Hinck, L. Extracellular Regulation of the Mitotic Spindle and Fate Determinants Driving Asymmetric Cell Division. Results Probl. Cell Differ. 2017, 61, 351–373. [Google Scholar] [PubMed]
- Lathia, J.D.; Hitomi, M.; Gallagher, J.; Gadani, S.P.; Adkins, J.; Vasanji, A.; Liu, L.; Eyler, C.E.; Heddleston, J.M.; Wu, Q.; et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011, 2, e200. [Google Scholar] [CrossRef]
- Kaseb, H.O.; Lewis, D.W.; Saunders, W.S.; Gollin, S.M. Cell division patterns and chromosomal segregation defects in oral cancer stem cells. Genes Chromosomes Cancer 2016, 55, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, M.; Chumakova, A.P.; Silver, D.J.; Knudsen, A.M.; Pontius, W.D.; Murphy, S.; Anand, N.; Kristensen, B.W.; Lathia, J.D. Asymmetric cell division promotes therapeutic resistance in glioblastoma stem cells. JCI Insight 2021, 6, e130510. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef]
- Gao, C.; Shen, Y.; Jin, F.; Miao, Y.; Qiu, X. Cancer Stem Cells in Small Cell Lung Cancer Cell Line H446: Higher Dependency on Oxidative Phosphorylation and Mitochondrial Substrate-Level Phosphorylation than Non-Stem Cancer Cells. PLoS ONE 2016, 11, e0154576. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, A.; Sahara, H. The Metabolic Heterogeneity and Flexibility of Cancer Stem Cells. Cancers 2020, 12, 2780. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar] [PubMed]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2019, 46, 100645. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Abad, E.; Graifer, D.; Lyakhovich, A. DNA damage response and resistance of cancer stem cells. Cancer Lett. 2020, 474, 106–117. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, T.; Dong, Q.; Nice, E.C.; Huang, C.; Wei, Y. Redox homeostasis: The linchpin in stem cell self-renewal and differentiation. Cell Death Dis. 2013, 4, e537. [Google Scholar] [CrossRef]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef]
- Kazi, J.U. Mechanisms of Anticancer Therapy Resistance: The Role of Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21, 9006. [Google Scholar] [CrossRef]
- Hou, H.; Sun, H.; Lu, P.; Ge, C.; Zhang, L.; Li, H.; Zhao, F.; Tian, H.; Zhang, L.; Chen, T.; et al. Tunicamycin potentiates cisplatin anticancer efficacy through the DPAGT1/Akt/ABCG2 pathway in mouse Xenograft models of human hepatocellular carcinoma. Mol. Cancer Ther. 2013, 12, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Mortezaee, K.; Majidpoor, J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019, 234, 116781. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Xu, X.; Lin, S.; Zhang, Y.; Liu, H.; Zhang, C.; Mo, R. A nanotherapeutic strategy to overcome chemotherapeutic resistance of cancer stem-like cells. Nat. Nanotechnol. 2021, 16, 104–113. [Google Scholar] [CrossRef]
- Nunes, T.; Hamdan, D.; Leboeuf, C.; El Bouchtaoui, M.; Gapihan, G.; Nguyen, T.T.; Meles, S.; Angeli, E.; Ratajczak, P.; Lu, H.; et al. Targeting Cancer Stem Cells to Overcome Chemoresistance. Int. J. Mol. Sci. 2018, 19, 4036. [Google Scholar] [CrossRef]
- Zhou, H.M.; Zhang, J.G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef]
- Majeti, R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene 2011, 30, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Buishand, F.O.; Arkesteijn, G.J.; Feenstra, L.R.; Oorsprong, C.W.; Mestemaker, M.; Starke, A.; Speel, E.J.; Kirpensteijn, J.; Mol, J.A. Identification of CD90 as Putative Cancer Stem Cell Marker and Therapeutic Target in Insulinomas. Stem Cells Dev. 2016, 25, 826–835. [Google Scholar] [CrossRef]
- Koyama, S.; Tsuchiya, H.; Amisaki, M.; Sakaguchi, H.; Honjo, S.; Fujiwara, Y.; Shiota, G. NEAT1 is Required for the Expression of the Liver Cancer Stem Cell Marker CD44. Int. J. Mol. Sci. 2020, 21, 1927. [Google Scholar] [CrossRef]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Brown, R.L.; Wei, Y.; Zhao, P.; Liu, S.; Liu, X.; Deng, Y.; Hu, X.; Zhang, J.; Gao, X.D.; et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. 2019, 33, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Dosch, J.; Simeone, D.M. Pancreatic cancer stem cells. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 2806–2812. [Google Scholar] [CrossRef] [PubMed]
- Rivas, S.; Antón, I.M.; Wandosell, F. WIP-YAP/TAZ as A New Pro-Oncogenic Pathway in Glioma. Cancers 2018, 10, 191. [Google Scholar] [CrossRef]
- Li, X.; Ma, X.; Chen, L.; Gu, L.; Zhang, Y.; Zhang, F.; Ouyang, Y.; Gao, Y.; Huang, Q.; Zhang, X. Prognostic value of CD44 expression in renal cell carcinoma: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 13157. [Google Scholar] [CrossRef][Green Version]
- Wang, C.; Xie, J.; Guo, J.; Manning, H.C.; Gore, J.C.; Guo, N. Evaluation of CD44 and CD133 as cancer stem cell markers for colorectal cancer. Oncol. Rep. 2012, 28, 1301–1308. [Google Scholar] [CrossRef]
- Geng, S.; Guo, Y.; Wang, Q.; Li, L.; Wang, J. Cancer stem-like cells enriched with CD29 and CD44 markers exhibit molecular characteristics with epithelial-mesenchymal transition in squamous cell carcinoma. Arch. Dermatol. Res. 2013, 305, 35–47. [Google Scholar] [CrossRef]
- Sharma, R.B.; Wang, Q.; Khillan, J.S. Amplification of tumor inducing putative cancer stem cells (CSCs) by vitamin A/retinol from mammary tumors. Biochem. Biophys. Res. Commun. 2013, 436, 625–631. [Google Scholar] [CrossRef]
- Fang, D.D.; Kim, Y.J.; Lee, C.N.; Aggarwal, S.; McKinnon, K.; Mesmer, D.; Norton, J.; Birse, C.E.; He, T.; Ruben, S.M.; et al. Expansion of CD133(+) colon cancer cultures retaining stem cell properties to enable cancer stem cell target discovery. Br. J. Cancer 2010, 102, 1265–1275. [Google Scholar] [CrossRef]
- Li, C.; Liu, S.; Yan, R.; Han, N.; Wong, K.K.; Li, L. CD54-NOTCH1 axis controls tumor initiation and cancer stem cell functions in human prostate cancer. Theranostics 2017, 7, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Knight, D.A.; Smyth, M.J.; Stewart, T.J. Sensitivity of a novel model of mammary cancer stem cell-like cells to TNF-related death pathways. Cancer Immunol. Immunother. 2012, 61, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- Sugita, M.; Guzman, M.L. CD123 as a Therapeutic Target Against Malignant Stem Cells. Hematol./Oncol. Clin. N. Am. 2020, 34, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Konno, M.; Hamabe, A.; Hasegawa, S.; Kano, Y.; Ohta, K.; Fukusumi, T.; Sakai, D.; Kudo, T.; Haraguchi, N.; et al. Aldehyde dehydrogenase high gastric cancer stem cells are resistant to chemotherapy. Int. J. Oncol. 2013, 42, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Long, C.; Tran, K.A.M.; Lee, J. A Synthetic Binder of Breast Cancer Stem Cells. Chemistry 2018, 24, 3694–3698. [Google Scholar] [CrossRef]
- Ishiwata, T.; Matsuda, Y.; Yoshimura, H.; Sasaki, N.; Ishiwata, S.; Ishikawa, N.; Takubo, K.; Arai, T.; Aida, J. Pancreatic cancer stem cells: Features and detection methods. Pathol. Oncol. Res. 2018, 24, 797–805. [Google Scholar] [CrossRef]
- Wang, L.; Park, P.; La Marca, F.; Than, K.D.; Lin, C.Y. BMP-2 inhibits tumor-initiating ability in human renal cancer stem cells and induces bone formation. J. Cancer Res. Clin. Oncol. 2015, 141, 1013–1024. [Google Scholar] [CrossRef]
- Giraud, J.; Failla, L.M.; Pascussi, J.M.; Lagerqvist, E.L.; Ollier, J.; Finetti, P.; Bertucci, F.; Ya, C.; Gasmi, I.; Bourgaux, J.F.; et al. Autocrine Secretion of Progastrin Promotes the Survival and Self-Renewal of Colon Cancer Stem-like Cells. Cancer Res. 2016, 76, 3618–3628. [Google Scholar] [CrossRef]
- Prince, M.E.P.; Zhou, L.; Moyer, J.S.; Tao, H.; Lu, L.; Owen, J.; Etigen, M.; Zheng, F.; Chang, A.E.; Xia, J.; et al. Evaluation of the immunogenicity of ALDH(high) human head and neck squamous cell carcinoma cancer stem cells in vitro. Oral Oncol. 2016, 59, 30–42. [Google Scholar] [CrossRef]
- Katsuta, E.; Tanaka, S.; Mogushi, K.; Shimada, S.; Akiyama, Y.; Aihara, A.; Matsumura, S.; Mitsunori, Y.; Ban, D.; Ochiai, T.; et al. CD73 as a therapeutic target for pancreatic neuroendocrine tumor stem cells. Int. J. Oncol. 2016, 48, 657–669. [Google Scholar] [CrossRef]
- Khan, A.I.; Kerfoot, S.M.; Heit, B.; Liu, L.; Andonegui, G.; Ruffell, B.; Johnson, P.; Kubes, P. Role of CD44 and hyaluronan in neutrophil recruitment. J. Immunol. 2004, 173, 7594–7601. [Google Scholar] [CrossRef] [PubMed]
- Flynn, K.M.; Michaud, M.; Canosa, S.; Madri, J.A. CD44 regulates vascular endothelial barrier integrity via a PECAM-1 dependent mechanism. Angiogenesis 2013, 16, 689–705. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.N.; Nölle, B.; Duncker, G. Expression of adhesion molecule CD44 on human corneas. Br. J. Ophthalmol. 1997, 81, 80–84. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Girgrah, N.; Letarte, M.; Becker, L.E.; Cruz, T.F.; Theriault, E.; Moscarello, M.A. Localization of the CD44 glycoprotein to fibrous astrocytes in normal white matter and to reactive astrocytes in active lesions in multiple sclerosis. J. Neuropathol. Exp. Neurol. 1991, 50, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Li, S.; Zou, B.; Liu, H.; Wang, S. Expressions and clinical significance of HER4 and CD44 in sinonasal mucosal malignant melanoma. Melanoma Res. 2018, 28, 105–110. [Google Scholar] [CrossRef]
- Reid, S.D.; Pockley, A.G. Cytokine regulation of CD44 expression on rat intestinal epithelial cells. Immunol. Investig. 2000, 29, 271–286. [Google Scholar] [CrossRef]
- Malan, D.; Wenzel, D.; Schmidt, A.; Geisen, C.; Raible, A.; Bölck, B.; Fleischmann, B.K.; Bloch, W. Endothelial beta1 integrins regulate sprouting and network formation during vascular development. Development 2010, 137, 993–1002. [Google Scholar] [CrossRef]
- Togarrati, P.P.; Dinglasan, N.; Desai, S.; Ryan, W.R.; Muench, M.O. CD29 is highly expressed on epithelial, myoepithelial, and mesenchymal stromal cells of human salivary glands. Oral Dis. 2018, 24, 561–572. [Google Scholar] [CrossRef]
- Kurata, R.; Futaki, S.; Nakano, I.; Tanemura, A.; Murota, H.; Katayama, I.; Sekiguchi, K. Isolation and characterization of sweat gland myoepithelial cells from human skin. Cell Struct. Funct. 2014, 39, 101–112. [Google Scholar] [CrossRef][Green Version]
- Zhang, M.; Che, Y.; Zhao, S.; Xia, X.; Liu, H.; Liu, J.; Wang, Y.; Han, W.; Yang, Y.; Zhou, C.; et al. TGF-β1 promoted the infection of bovine mammary epithelial cells by Staphylococcus aureus through increasing expression of cells’ fibronectin and integrin β1. Vet. Microbiol. 2019, 237, 108420. [Google Scholar] [CrossRef]
- Maio, M.; Del Vecchio, L. Expression and functional role of CD54/Intercellular Adhesion Molecule-1 (ICAM-1) on human blood cells. Leuk. Lymphoma 1992, 8, 23–33. [Google Scholar] [CrossRef]
- Takashi, S.; Okubo, Y.; Horie, S. Contribution of CD54 to human eosinophil and neutrophil superoxide production. J. Appl. Physiol. 2001, 91, 613–622. [Google Scholar] [CrossRef]
- Morikawa, Y.; Tohya, K.; Hara, T.; Kitamura, T.; Miyajima, A. Expression of IL-3 receptor in testis. Biochem. Biophys. Res. Commun. 1996, 226, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Chritton, S.L.; Sheng, M. CYRL, a novel cytokine receptor-like protein expressed in testis, lung, and spleen. Biochem. Biophys. Res. Commun. 2000, 267, 697–702. [Google Scholar] [CrossRef]
- Han, X.; Jorgensen, J.L.; Brahmandam, A.; Schlette, E.; Huh, Y.O.; Shi, Y.; Awagu, S.; Chen, W. Immunophenotypic study of basophils by multiparameter flow cytometry. Arch. Pathol. Lab. Med. 2008, 132, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, D.; Boström, A.K.; Nilsson, K.; Hansson, J.; Sjölund, J.; Möller, C.; Jirström, K.; Nilsson, E.; Landberg, G.; Axelson, H.; et al. Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am. J. Pathol. 2011, 178, 828–837. [Google Scholar] [CrossRef]
- Eichin, D.; Pessia, A.; Takeda, A.; Laakkonen, J.; Bellmann, L.; Kankainen, M.; Imhof, B.A.; Stoitzner, P.; Tang, J.; Salmi, M.; et al. CD73 contributes to anti-inflammatory properties of afferent lymphatic endothelial cells in humans and mice. Eur. J. Immunol. 2021, 51, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, E.; Palomo, I. Extracellular ATP metabolism on vascular endothelial cells: A pathway with pro-thrombotic and anti-thrombotic molecules. Vasc. Pharmacol. 2015, 75, 1–6. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Ordóñez-Morán, P.; Dafflon, C.; Imajo, M.; Nishida, E.; Huelsken, J. HOXA5 Counteracts Stem Cell Traits by Inhibiting Wnt Signaling in Colorectal Cancer. Cancer Cell 2015, 28, 815–829. [Google Scholar] [CrossRef]
- Fendler, A.; Bauer, D.; Busch, J.; Jung, K.; Wulf-Goldenberg, A.; Kunz, S.; Song, K.; Myszczyszyn, A.; Elezkurtaj, S.; Erguen, B.; et al. Inhibiting WNT and NOTCH in renal cancer stem cells and the implications for human patients. Nat. Commun. 2020, 11, 929. [Google Scholar] [CrossRef]
- Zhou, M.; Hou, Y.; Yang, G.; Zhang, H.; Tu, G.; Du, Y.E.; Wen, S.; Xu, L.; Tang, X.; Tang, S.; et al. LncRNA-Hh Strengthen Cancer Stem Cells Generation in Twist-Positive Breast Cancer via Activation of Hedgehog Signaling Pathway. Stem Cells 2016, 34, 55–66. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e5. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, C.J.; Choi, J.H.; Kim, J.H.; Kim, J.W.; Kim, J.Y.; Nam, J.S. The JAK2/STAT3/CCND2 Axis promotes colorectal Cancer stem cell persistence and radioresistance. J. Exp. Clin. Cancer Res. CR 2019, 38, 399. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef]
- Moon, C.M.; Kwon, J.H.; Kim, J.S.; Oh, S.H.; Jin Lee, K.; Park, J.J.; Pil Hong, S.; Cheon, J.H.; Kim, T.I.; Kim, W.H. Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer. Int. J. Cancer 2014, 134, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef] [PubMed]
- Leon, G.; MacDonagh, L.; Finn, S.P.; Cuffe, S.; Barr, M.P. Cancer stem cells in drug resistant lung cancer: Targeting cell surface markers and signaling pathways. Pharmacol. Ther. 2016, 158, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Q.; Li, D.; Ching, K.; Zhang, C.; Zheng, X.; Ozeck, M.; Shi, S.; Li, X.; Wang, H.; et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a γ-secretase inhibitor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1487–1496. [Google Scholar] [CrossRef]
- Stoeck, A.; Lejnine, S.; Truong, A.; Pan, L.; Wang, H.; Zang, C.; Yuan, J.; Ware, C.; MacLean, J.; Garrett-Engele, P.W.; et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014, 4, 1154–1167. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Nakanishi, M.; Rosenberg, D.W. Suppression of colon carcinogenesis by targeting Notch signaling. Carcinogenesis 2013, 34, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Moon, J.; Redman, B.G.; Chidiac, T.; Flaherty, L.E.; Zha, Y.; Othus, M.; Ribas, A.; Sondak, V.K.; Gajewski, T.F.; et al. Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer 2015, 121, 432–440. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Yeatman, T.; Weber, J.; Coppola, D.; Schell, M.J.; Han, G.; Almhanna, K.; Kim, R.; Valone, T.; Jump, H.; et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur. J. Cancer 2012, 48, 997–1003. [Google Scholar] [CrossRef]
- Diaz-Padilla, I.; Wilson, M.K.; Clarke, B.A.; Hirte, H.W.; Welch, S.A.; Mackay, H.J.; Biagi, J.J.; Reedijk, M.; Weberpals, J.I.; Fleming, G.F.; et al. A phase II study of single-agent RO4929097, a gamma-secretase inhibitor of Notch signaling, in patients with recurrent platinum-resistant epithelial ovarian cancer: A study of the Princess Margaret, Chicago and California phase II consortia. Gynecol. Oncol. 2015, 137, 216–222. [Google Scholar] [CrossRef]
- Diaz-Padilla, I.; Hirte, H.; Oza, A.M.; Clarke, B.A.; Cohen, B.; Reedjik, M.; Zhang, T.; Kamel-Reid, S.; Ivy, S.P.; Hotte, S.J.; et al. A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Pandya, K.; Meeke, K.; Clementz, A.G.; Rogowski, A.; Roberts, J.; Miele, L.; Albain, K.S.; Osipo, C. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br. J. Cancer 2011, 105, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Aleksic, T.; Feller, S.M. Gamma-secretase inhibition combined with platinum compounds enhances cell death in a large subset of colorectal cancer cells. Cell Commun. Signal. 2008, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G.; Noguera-Troise, I.; Yancopoulos, G.D. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat. Rev. Cancer 2007, 7, 327–331. [Google Scholar] [CrossRef]
- Kuhnert, F.; Chen, G.; Coetzee, S.; Thambi, N.; Hickey, C.; Shan, J.; Kovalenko, P.; Noguera-Troise, I.; Smith, E.; Fairhurst, J.; et al. Dll4 Blockade in Stromal Cells Mediates Antitumor Effects in Preclinical Models of Ovarian Cancer. Cancer Res. 2015, 75, 4086–4096. [Google Scholar] [CrossRef]
- Chiorean, E.G.; LoRusso, P.; Strother, R.M.; Diamond, J.R.; Younger, A.; Messersmith, W.A.; Adriaens, L.; Liu, L.; Kao, R.J.; DiCioccio, A.T.; et al. A Phase I First-in-Human Study of Enoticumab (REGN421), a Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Hoey, T.; Yen, W.C.; Axelrod, F.; Basi, J.; Donigian, L.; Dylla, S.; Fitch-Bruhns, M.; Lazetic, S.; Park, I.K.; Sato, A.; et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 2009, 5, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, C.; Xin, P.; Zheng, Y.; Peng, Q.; Xu, Y.; Luo, Y.; Wu, Y.; Zhu, X. Sonidegib, a Smoothened Inhibitor, Promotes Apoptosis and Suppresses Proliferation of Natural Killer/T-Cell Lymphoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 8579–8586. [Google Scholar] [CrossRef]
- Martinelli, G.; Oehler, V.G.; Papayannidis, C.; Courtney, R.; Shaik, M.N.; Zhang, X.; O’Connell, A.; McLachlan, K.R.; Zheng, X.; Radich, J.; et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: A phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015, 2, e339–e346. [Google Scholar] [CrossRef]
- Shaik, N.; Hee, B.; Liang, Y.; LaBadie, R.R. Absolute Oral Bioavailability of Glasdegib (PF-04449913), a Smoothened Inhibitor, in Randomized Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2019, 8, 895–902. [Google Scholar] [CrossRef] [PubMed]
- AlMuraikhi, N.; Almasoud, N.; Binhamdan, S.; Younis, G.; Ali, D.; Manikandan, M.; Vishnubalaji, R.; Atteya, M.; Siyal, A.; Alfayez, M.; et al. Hedgehog Signaling Inhibition by Smoothened Antagonist BMS-833923 Reduces Osteoblast Differentiation and Ectopic Bone Formation of Human Skeletal (Mesenchymal) Stem Cells. Stem Cells Int. 2019, 2019, 3435901. [Google Scholar] [CrossRef]
- Wu, C.; Hu, S.; Cheng, J.; Wang, G.; Tao, K. Smoothened antagonist GDC-0449 (Vismodegib) inhibits proliferation and triggers apoptosis in colon cancer cell lines. Exp. Ther. Med. 2017, 13, 2529–2536. [Google Scholar] [CrossRef]
- Riedlinger, D.; Bahra, M.; Boas-Knoop, S.; Lippert, S.; Bradtmöller, M.; Guse, K.; Seehofer, D.; Bova, R.; Sauer, I.M.; Neuhaus, P.; et al. Hedgehog pathway as a potential treatment target in human cholangiocarcinoma. J. Hepato-Biliary-Pancreat. Sci. 2014, 21, 607–615. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef]
- Hou, X.; Chen, X.; Zhang, P.; Fan, Y.; Ma, A.; Pang, T.; Song, Z.; Jin, Y.; Hao, W.; Liu, F.; et al. Inhibition of hedgehog signaling by GANT58 induces apoptosis and shows synergistic antitumor activity with AKT inhibitor in acute T cell leukemia cells. Biochimie 2014, 101, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hao, C.; Zhang, R.; Pei, X.; Li, J.; Wang, L. A Gli inhibitor GANT61 suppresses cell proliferation, promotes cell apoptosis and induces G1/G0 cycle retardation with a dose- and time-dependent manner through inhibiting Notch pathway in multiple myeloma. Cell Cycle 2020, 19, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, J.; Zhao, S.; Yao, K.; Sun, Y.; Li, Y.; Chen, L.; Li, R.; Zhai, X.; Zhang, J.; et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J. Exp. Clin. Cancer Res. CR 2016, 35, 184. [Google Scholar] [CrossRef]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- El-Sahli, S.; Xie, Y.; Wang, L.; Liu, S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers 2019, 11, 904. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef]
- Shao, Y.C.; Wei, Y.; Liu, J.F.; Xu, X.Y. The role of Dickkopf family in cancers: From Bench to Bedside. Am. J. Cancer Res. 2017, 7, 1754–1768. [Google Scholar]
- Liang, C.J.; Wang, Z.W.; Chang, Y.W.; Lee, K.C.; Lin, W.H.; Lee, J.L. SFRPs Are Biphasic Modulators of Wnt-Signaling-Elicited Cancer Stem Cell Properties beyond Extracellular Control. Cell Rep. 2019, 28, 1511–1525.e5. [Google Scholar] [CrossRef]
- Xavier, C.P.; Melikova, M.; Chuman, Y.; Üren, A.; Baljinnyam, B.; Rubin, J.S. Secreted Frizzled-related protein potentiation versus inhibition of Wnt3a/β-catenin signaling. Cell. Signal. 2014, 26, 94–101. [Google Scholar] [CrossRef]
- Chen, C.C.; Chen, H.Y.; Su, K.Y.; Hong, Q.S.; Yan, B.S.; Chen, C.H.; Pan, S.H.; Chang, Y.L.; Wang, C.J.; Hung, P.F.; et al. Shisa3 is associated with prolonged survival through promoting β-catenin degradation in lung cancer. Am. J. Respir. Crit. Care Med. 2014, 190, 433–444. [Google Scholar] [CrossRef]
- Maffei, R.; Fiorcari, S.; Martinelli, S.; Benatti, S.; Bulgarelli, J.; Rizzotto, L.; Debbia, G.; Santachiara, R.; Rigolin, G.M.; Forconi, F.; et al. Increased SHISA3 expression characterizes chronic lymphocytic leukemia patients sensitive to lenalidomide. Leuk. Lymphoma 2018, 59, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.K.; Hipolito, C.J.; Ten Dijke, P. A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Banerjee, S.; Mondal, A.; Chakraborty, U.; Pumarol, J.; Croley, C.R.; Bishayee, A. Targeting the JAK/STAT Signaling Pathway Using Phytocompounds for Cancer Prevention and Therapy. Cells 2020, 9, 1451. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Jiao, X.X.; Lin, S.Y.; Lian, S.X.; Qiu, Y.R.; Li, Z.H.; Chen, Z.H.; Lu, W.Q.; Zhang, Y.; Deng, L.; Jiang, Y.; et al. The inhibition of the breast cancer by PPARγ agonist pioglitazone through JAK2/STAT3 pathway. Neoplasma 2020, 67, 834–842. [Google Scholar] [CrossRef]
- Cai, X.; Yao, Z.; Li, L.; Huang, J. Role of DKK4 in Tumorigenesis and Tumor Progression. Int. J. Biol. Sci. 2018, 14, 616–621. [Google Scholar] [CrossRef]
- Courtois, S.; Durán, R.V.; Giraud, J.; Sifré, E.; Izotte, J.; Mégraud, F.; Lehours, P.; Varon, C.; Bessède, E. Metformin targets gastric cancer stem cells. Eur. J. Cancer 2017, 84, 193–201. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, K.J.; Seo, Y.; Kwon, J.H.; Yoon, J.P.; Kang, J.Y.; Lee, H.J.; Park, S.J.; Hong, S.P.; Cheon, J.H.; et al. Effects of metformin on colorectal cancer stem cells depend on alterations in glutamine metabolism. Sci. Rep. 2018, 8, 409. [Google Scholar] [CrossRef]
- Shank, J.J.; Yang, K.; Ghannam, J.; Cabrera, L.; Johnston, C.J.; Reynolds, R.K.; Buckanovich, R.J. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol. Oncol. 2012, 127, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.J.; Klotz, L.H.; Venkateswaran, V. Metformin and prostate cancer stem cells: A novel therapeutic target. Prostate Cancer Prostatic Dis. 2015, 18, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, L.; Xiong, Y.; Yu, S.; Li, H.; Fan, J.; Li, F.; Su, Z.; Song, J.; Sun, Q.; et al. Salinomycin exerts anti-colorectal cancer activity by targeting the β-catenin/T-cell factor complex. Br. J. Pharmacol. 2019, 176, 3390–3406. [Google Scholar] [CrossRef]
- Managò, A.; Leanza, L.; Carraretto, L.; Sassi, N.; Grancara, S.; Quintana-Cabrera, R.; Trimarco, V.; Toninello, A.; Scorrano, L.; Trentin, L.; et al. Early effects of the antineoplastic agent salinomycin on mitochondrial function. Cell Death Dis. 2015, 6, e1930. [Google Scholar] [CrossRef]
- Mitani, M.; Yamanishi, T.; Miyazaki, Y.; Otake, N. Salinomycin effects on mitochondrial ion translocation and respiration. Antimicrob. Agents Chemother. 1976, 9, 655–660. [Google Scholar] [CrossRef]
- Guo, F.; Yang, Z.; Kulbe, H.; Albers, A.E.; Sehouli, J.; Kaufmann, A.M. Inhibitory effect on ovarian cancer ALDH+ stem-like cells by Disulfiram and Copper treatment through ALDH and ROS modulation. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 118, 109371. [Google Scholar] [CrossRef]
- Eskandari, A.; Suntharalingam, K. A reactive oxygen species-generating, cancer stem cell-potent manganese(ii) complex and its encapsulation into polymeric nanoparticles. Chem. Sci. 2019, 10, 7792–7800. [Google Scholar] [CrossRef]
- Han, S.; Wei, R.; Zhang, X.; Jiang, N.; Fan, M.; Huang, J.H.; Xie, B.; Zhang, L.; Miao, W.; Butler, A.C.; et al. CPT1A/2-Mediated FAO Enhancement-A Metabolic Target in Radioresistant Breast Cancer. Front. Oncol. 2019, 9, 1201. [Google Scholar] [CrossRef]
- Cheng, S.; Wang, G.; Wang, Y.; Cai, L.; Qian, K.; Ju, L.; Liu, X.; Xiao, Y.; Wang, X. Fatty acid oxidation inhibitor etomoxir suppresses tumor progression and induces cell cycle arrest via PPARγ-mediated pathway in bladder cancer. Clin. Sci. 2019, 133, 1745–1758. [Google Scholar] [CrossRef]
- Tan, Z.; Xiao, L.; Tang, M.; Bai, F.; Li, J.; Li, L.; Shi, F.; Li, N.; Li, Y.; Du, Q.; et al. Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics 2018, 8, 2329–2347. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Regulation of Cancer Stem Cell Metabolism by Secreted Frizzled-Related Protein 4 (sFRP4). Cancers 2018, 10, 40. [Google Scholar] [CrossRef]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef]
- Zhong, D.; Xiong, L.; Liu, T.; Liu, X.; Liu, X.; Chen, J.; Sun, S.Y.; Khuri, F.R.; Zong, Y.; Zhou, Q.; et al. The glycolytic inhibitor 2-deoxyglucose activates multiple prosurvival pathways through IGF1R. J. Biol. Chem. 2009, 284, 23225–23233. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.R.; Wang, S.; Yan, S.C.; Zhang, Y.; Nelson, P.J.; Jia, H.L.; Qin, L.X.; Dong, Q.Z. Therapeutic Strategies Targeting Cancer Stem Cells and Their Microenvironment. Front. Oncol. 2019, 9, 1104. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J.; Ahadi, R. Hypoxia in solid tumors: A key promoter of cancer stem cell (CSC) resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31. [Google Scholar] [CrossRef]
- Bhuria, V.; Xing, J.; Scholta, T.; Bui, K.C.; Nguyen, M.L.T.; Malek, N.P.; Bozko, P.; Plentz, R.R. Hypoxia induced Sonic Hedgehog signaling regulates cancer stemness, epithelial-to-mesenchymal transition and invasion in cholangiocarcinoma. Exp. Cell Res. 2019, 385, 111671. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Izumikawa, T.; Higashide, M.; Mochizuki, N.; Chokchaitaweesuk, C.; Khansai, M.; Nakajima, K.; Kakizaki, I.; Kongtawelert, P.; et al. Hyaluronan Production Regulates Metabolic and Cancer Stem-like Properties of Breast Cancer Cells via Hexosamine Biosynthetic Pathway-coupled HIF-1 Signaling. J. Biol. Chem. 2016, 291, 24105–24120. [Google Scholar] [CrossRef]
- Bae, K.M.; Dai, Y.; Vieweg, J.; Siemann, D.W. Hypoxia regulates SOX2 expression to promote prostate cancer cell invasion and sphere formation. Am. J. Cancer Res. 2016, 6, 1078–1088. [Google Scholar]
- Tang, Y.A.; Chen, Y.F.; Bao, Y.; Mahara, S.; Yatim, S.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef]
- Bai, J.; Chen, W.B.; Zhang, X.Y.; Kang, X.N.; Jin, L.J.; Zhang, H.; Wang, Z.Y. HIF-2α regulates CD44 to promote cancer stem cell activation in triple-negative breast cancer via PI3K/AKT/mTOR signaling. World J. Stem Cells 2020, 12, 87–99. [Google Scholar] [CrossRef]
- Huang, T.X.; Guan, X.Y.; Fu, L. Therapeutic targeting of the crosstalk between cancer-associated fibroblasts and cancer stem cells. Am. J. Cancer Res. 2019, 9, 1889–1904. [Google Scholar]
- Li, Y.; Wang, R.; Xiong, S.; Wang, X.; Zhao, Z.; Bai, S.; Wang, Y.; Zhao, Y.; Cheng, B. Cancer-associated fibroblasts promote the stemness of CD24(+) liver cells via paracrine signaling. J. Mol. Med. 2019, 97, 243–255. [Google Scholar] [CrossRef] [PubMed]
- López-Gil, J.C.; Martin-Hijano, L.; Hermann, P.C.; Sainz, B., Jr. The CXCL12 Crossroads in Cancer Stem Cells and Their Niche. Cancers 2021, 13, 469. [Google Scholar] [CrossRef]
- Cui, C.P.; Wong, C.C.; Kai, A.K.; Ho, D.W.; Lau, E.Y.; Tsui, Y.M.; Chan, L.K.; Cheung, T.T.; Chok, K.S.; Chan, A.C.Y.; et al. SENP1 promotes hypoxia-induced cancer stemness by HIF-1α deSUMOylation and SENP1/HIF-1α positive feedback loop. Gut 2017, 66, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, C.; Qian, P.; Lu, X.; Sun, B.; Zhang, X.; Wang, L.; Gao, X.; Li, H.; Chen, Z.; et al. Gd-metallofullerenol nanomaterial as non-toxic breast cancer stem cell-specific inhibitor. Nat. Commun. 2015, 6, 5988. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Beck, B.; Driessens, G.; Goossens, S.; Youssef, K.K.; Kuchnio, A.; Caauwe, A.; Sotiropoulou, P.A.; Loges, S.; Lapouge, G.; Candi, A.; et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 2011, 478, 399–403. [Google Scholar] [CrossRef]
- Su, J.; Wu, S.; Wu, H.; Li, L.; Guo, T. CD44 is functionally crucial for driving lung cancer stem cells metastasis through Wnt/β-catenin-FoxM1-Twist signaling. Mol. Carcinog. 2016, 55, 1962–1973. [Google Scholar] [CrossRef]
- Chen, R.; Masuo, K.; Yogo, A.; Yokoyama, S.; Sugiyama, A.; Seno, H.; Yoshizawa, A.; Takaishi, S. SNAIL regulates gastric carcinogenesis through CCN3 and NEFL. Carcinogenesis 2021, 42, 190–201. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, B.; Deng, Q.; Sheng, D.; Xu, J.; He, X.; Zhang, L.; Liu, S. UCP1 regulates ALDH-positive breast cancer stem cells through releasing the suppression of Snail on FBP1. Cell Biol. Toxicol. 2021, 37, 277–291. [Google Scholar] [CrossRef]
- Vijay, G.V.; Zhao, N.; Den Hollander, P.; Toneff, M.J.; Joseph, R.; Pietila, M.; Taube, J.H.; Sarkar, T.R.; Ramirez-Pena, E.; Werden, S.J.; et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. BCR 2019, 21, 37. [Google Scholar] [CrossRef] [PubMed]
- Piasecka, D.; Braun, M.; Kordek, R.; Sadej, R.; Romanska, H. MicroRNAs in regulation of triple-negative breast cancer progression. J. Cancer Res. Clin. Oncol. 2018, 144, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ji, J.; Xu, Y.; Liu, Y.; Shi, L.; Liu, Y.; Lu, X.; Zhao, Y.; Luo, F.; Wang, B.; et al. MicroRNA-191, by promoting the EMT and increasing CSC-like properties, is involved in neoplastic and metastatic properties of transformed human bronchial epithelial cells. Mol. Carcinog. 2015, 54 (Suppl. S1), E148–E161. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Hu, W.; Xu, J.; Kaufmann, A.M.; Albers, A.E. MicroRNA-34a regulates epithelial-mesenchymal transition and cancer stem cell phenotype of head and neck squamous cell carcinoma in vitro. Int. J. Oncol. 2015, 47, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.B.; Yan, C.; Mu, L.; Mi, Y.L.; Zhao, H.; Hu, H.; Li, X.L.; Tao, D.D.; Wu, Y.Q.; Gong, J.P.; et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene 2019, 38, 1951–1965. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.C.; Kats, L.M. Non-genetic heterogeneity, altered cell fate and differentiation therapy. EMBO Mol. Med. 2021, 13, e12670. [Google Scholar] [CrossRef] [PubMed]
- Azzi, S.; Bruno, S.; Giron-Michel, J.; Clay, D.; Devocelle, A.; Croce, M.; Ferrini, S.; Chouaib, S.; Vazquez, A.; Charpentier, B.; et al. Differentiation therapy: Targeting human renal cancer stem cells with interleukin 15. J. Natl. Cancer Inst. 2011, 103, 1884–1898. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Fadera, S.; Liu, H.L. Strategy of differentiation therapy: Effect of dual-frequency ultrasound on the induction of liver cancer stem-like cells on a HA-based multilayer film system. J. Mater. Chem. B 2019, 7, 5401–5411. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Amante, J.J.; Goel, H.L.; Zhang, X.; Walker, M.R.; Luther, D.C.; Mercurio, A.M.; Rotello, V.M. Differentiation of Cancer Stem Cells through Nanoparticle Surface Engineering. ACS Nano 2020, 14, 15276–15285. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Hu, G.; Cai, X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit. Rev. Food Sci. Nutr. 2019, 59, S71–S80. [Google Scholar] [CrossRef] [PubMed]
- Mansour, F.A.; Al-Mazrou, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 is overexpressed on breast cancer stem cells through notch3/mTOR axis. Oncoimmunology 2020, 9, 1729299. [Google Scholar] [CrossRef]
- Zhang, B.; Dang, J.; Ba, D.; Wang, C.; Han, J.; Zheng, F. Potential function of CTLA-4 in the tumourigenic capacity of melanoma stem cells. Oncol. Lett. 2018, 16, 6163–6170. [Google Scholar] [CrossRef]
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018, 18, 469. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Hu, A.; Zhu, J.; Yu, J.; Talebian, F.; Bai, X.F. CD200-CD200R Pathway in the Regulation of Tumor Immune Microenvironment and Immunotherapy. Adv. Exp. Med. Biol. 2020, 1223, 155–165. [Google Scholar] [PubMed]
- Tsuchiya, H.; Shiota, G. Immune evasion by cancer stem cells. Regen. Ther. 2021, 17, 20–33. [Google Scholar] [CrossRef]
- Lu, L.; Tao, H.; Chang, A.E.; Hu, Y.; Shu, G.; Chen, Q.; Egenti, M.; Owen, J.; Moyer, J.S.; Prince, M.E.; et al. Cancer stem cell vaccine inhibits metastases of primary tumors and induces humoral immune responses against cancer stem cells. Oncoimmunology 2015, 4, e990767. [Google Scholar] [CrossRef]
- Guo, M.; Luo, B.; Pan, M.; Li, M.; Zhao, F.; Dou, J. MUC1 plays an essential role in tumor immunity of colorectal cancer stem cell vaccine. Int. Immunopharmacol. 2020, 85, 106631. [Google Scholar] [CrossRef]
- El-Ashmawy, N.E.; Salem, M.L.; Khedr, E.G.; El-Zamarany, E.A.; Ibrahim, A.O. Dual-targeted therapeutic strategy combining CSC-DC-based vaccine and cisplatin overcomes chemo-resistance in experimental mice model. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2020, 22, 1155–1165. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Badieyan, Z.S.; Hoseini, S.S. Adverse Effects Associated with Clinical Applications of CAR Engineered T Cells. Arch. Immunol. Ther. Exp. 2018, 66, 283–288. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, K.; Cheng, H.; Cao, J.; Shi, M.; Qiao, J.; Yan, Z.; Jing, G.; Pan, B.; Sang, W.; et al. Coagulation Disorders after Chimeric Antigen Receptor T Cell Therapy: Analysis of 100 Patients with Relapsed and Refractory Hematologic Malignancies. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2020, 26, 865–875. [Google Scholar] [CrossRef]
- Raja, J.; Ludwig, J.M.; Gettinger, S.N.; Schalper, K.A.; Kim, H.S. Oncolytic virus immunotherapy: Future prospects for oncology. J. Immunother. Cancer 2018, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, S.; Chen, N.G.; Warner, S.G. Oncolytic Virotherapy versus Cancer Stem Cells: A Review of Approaches and Mechanisms. Cancers 2018, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hsu, W.H.; Han, J.; Xia, Y.; DePinho, R.A. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021, 34, 108597. [Google Scholar] [CrossRef] [PubMed]
- Gomez, K.E.; Wu, F.; Keysar, S.B.; Morton, J.J.; Miller, B.; Chimed, T.S.; Le, P.N.; Nieto, C.; Chowdhury, F.N.; Tyagi, A.; et al. Cancer Cell CD44 Mediates Macrophage/Monocyte-Driven Regulation of Head and Neck Cancer Stem Cells. Cancer Res. 2020, 80, 4185–4198. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef]
- Komura, N.; Mabuchi, S.; Shimura, K.; Yokoi, E.; Kozasa, K.; Kuroda, H.; Takahashi, R.; Sasano, T.; Kawano, M.; Matsumoto, Y.; et al. The role of myeloid-derived suppressor cells in increasing cancer stem-like cells and promoting PD-L1 expression in epithelial ovarian cancer. Cancer Immunol. Immunother. 2020, 69, 2477–2499. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Wu, W.; Gao, H.; Liu, N.; Zhan, G.; Li, L.; Han, L.; Guo, X. Myeloid-derived suppressor cells promote epithelial ovarian cancer cell stemness by inducing the CSF2/p-STAT3 signalling pathway. FEBS J. 2020, 287, 5218–5235. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agents | Target | NCT Number | Phase | Current State | Condition |
|---|---|---|---|---|---|
| BIWA1 | CD44v6 | NCT02204046 | I | Completed | Breast cancer |
| NCT02204059 | I | Completed | Lung cancer | ||
| RO5429083 | CD44 | NCT01358903 | I | Completed | Malignant solid tumors |
| NCT01641250 | I | Completed | Leukemia | ||
| 17-1A | EpCAM | NCT02915445 | Recruiting | Nasopharyngeal/breast | |
| cancer | |||||
| NCT00309517 | Terminated | Rectal carcinoma | |||
| Catumaxomab | EpCAM | NCT01815528 | II | Completed | Ovarian cancer |
| NCT01246440 | II | Completed | Ovarian cancer | ||
| NCT00326885 | II | Completed | Malignant ascites | ||
| NCT00464893 | II | Completed | Gastric cancer | ||
| Adecatumumab | EpCAM | NCT00866944 | II | Completed | Colorectal liver |
| Metastases | |||||
| ING-1 | EpCAM | NCT00051675 | I | Completed | Adenocarcinomas |
| Edrecolomab | EpCAM | NCT00002968 | III | Completed | Colon cancer |
| CSL-360 | CD123 | NCT00401739 | I | Completed | Acute Leukemia |
| Talacotuzumab | CD123 | NCT02472145 | II/III | Completed | Acute Leukemia |
| anti-ICAM-1 Mab | CD54 | NCT01025206 | I | Completed | Multiple myeloma |
| Surface Marker | CSCs | Normal Cells/Organs |
|---|---|---|
| CD44 | Liver [42], stomach [43], breast [44], pancreas [45], glioma [46], kidney [47], colon, rectum [48] | Lymphocytes [62], vascular endothelial cells [63] corneal cells [64], astrocytes [65], mucosal cells [66] intestinal epithelial cells [67] |
| CD29 | squamous cell carcinoma [49], breast [50], colon [51] | vascular endothelial cells [68], salivary glands [69] myoepithelial cells [70], epithelial cells [71] |
| CD54 | prostate [52], breast [53] | hematopoietic cells [72,73] |
| CD123 | leukemic stem cells [54] | testis [74], lung, brain [75], basophil [76] |
| ALDH | stomach [55], breast [56], pancreas [57], kidney [58], colon [59] squamous cancer [60] | breast stem cells [77], renal tubular stem cells [78] |
| CD73 | pancreas [61] | lymphatic endothelial cells [79], vascular endothelial cells [80] |
| Drug Name | Target | NCT Number | Phase | Current State | Condition |
|---|---|---|---|---|---|
| RO4929097 | Notch | NCT01120275 | II | Terminated | Melanoma |
| NCT01238133 | I | Terminated | Breast cancer | ||
| NCT01175343 | II | Completed | Ovarian cancer | ||
| NCT01154452 | I/II | Completed | Sarcoma | ||
| NCT01198535 | I | Terminated | Colorectal cancer | ||
| NCT01232829 | II | Completed | Pancreatic cancer | ||
| NCT01122901 | II | Terminated | Glioblastoma | ||
| NCT01131234 | I | Completed | Solid tumors | ||
| LY900009 | Notch | NCT01158404 | I | Completed | Advanced tumors |
| MK-0752 | Notch | NCT00645333 | I/II | Completed | Breast cancer |
| NCT00106145 | I | Completed | Breast cancer | ||
| NCT00572182 | I | Terminated | CNS cancer | ||
| OMP-54F28 | Wnt | NCT02069145 | I | Completed | Hepatocellular cancer |
| NCT02092363 | I | Completed | Ovarian cancer | ||
| NCT02050178 | I | Completed | Pancreatic cancer | ||
| NCT01608867 | I | Completed | Solid tumors | ||
| LGK974 | Wnt | NCT01351103 | I | Recruiting | Malignancies |
| WNT974 | Wnt | NCT02278133 | II | Completed | colorectal cancer |
| DKN01 | Wnt | NCT04681248 | Available | solid tumors | |
| NCT01457417 | I | Completed | Lung cancer | ||
| NCT03645980 | I/II | Recruiting | liver cancer | ||
| BMS-833923 | Hh | NCT00670189 | I | Completed | Advanced cancer |
| NCT01413906 | I | Completed | Solid tumors | ||
| NCT01357655 | II | Completed | Chronic myeloid leukemia | ||
| GDC-0449 | Hh | NCT01088815 | II | Completed | Pancreatic cancer |
| NCT00607724 | I | Completed | solid tumors | ||
| AVID200 | TGF-β | NCT03834662 | I | Active | Malignancies |
| LY2157299 | TGF-β | NCT02452008 | II | Recruiting | prostatic cancer |
| NCT02240433 | I | Completed | Hepatocellular cancer | ||
| NCT02688712 | II | Recruiting | Rectal cancer | ||
| GC1008 | TGF-β | NCT01112293 | II | Completed | Mesothelioma |
| NCT01401062 | II | Completed | breast cancer | ||
| Itacinib | JAK-STAT | NCT04358185 | II | Recruiting | hepatocellular cancer |
| NCT03989466 | I | Recruiting | T-Cell prolymphocytic | ||
| Leukemia | |||||
| Ruxolitinib | JAK-STAT | NCT04906746 | I | Not recruiting | Lung cancer |
| NCT01895842 | I | Completed | Leukemia | ||
| NCT03514069 | I | Recruiting | Glioma | ||
| NCT01594216 | III | Completed | breast cancer | ||
| SB1518 | JAK-STAT | NCT00719836 | I/II | Completed | Myeloid malignancies |
| NCT04635059 | Recruiting | Prostate cancer | |||
| NCT02323607 | I | Completed | Acute myeloid leukemia | ||
| Alpelisib | PI3K/AKT/mTOR | NCT04526470 | II | Not recruiting | Gastric cancer |
| NCT04544189 | II | Recruiting | Breast cancer | ||
| NCT01300962 | I | Completed | Breast cancer | ||
| Temsirolimus | PI3K/AKT/mTOR | NCT01072890 | I | Completed | Solid tumors |
| NCT01050985 | I | Completed | Advanced malignancies | ||
| Copanlisib | PI3K/AKT/mTOR | NCT03498430 | I | Completed | Non-Hodgkin’s lymphoma |
| NCT04750941 | II | Recruiting | Endometrial cancer | ||
| Capivasertib | PI3K/AKT/mTOR | NCT04742036 | I | Recruiting | Solid Tumors |
| NCT04087174 | I | Completed | Prostate cancer | ||
| NCT04862663 | I/III | Recruiting | Breast cancer | ||
| Pioglitazone | PPAR | NCT02133625 | I | Completed | Solid tumors |
| Agents | Target | NCT Number | Phase | Current State | Condition |
|---|---|---|---|---|---|
| NCT00897884 | Completed | Breast cancer | |||
| NCT01266486 | II | Completed | Breast cancer | ||
| Metformin | OXPHS | NCT01243385 | II | Completed | Prostate cancer |
| NCT02437656 | II | Completed | Rectal cancer | ||
| NCT03359681 | Recruiting | Colon cancer | |||
| Disulfiram | OXPHS | NCT01118741 | Completed | Prostate cancer | |
| NCT02678975 | Completed | Glioblastoma | |||
| NCT03584009 | II | Completed | Breast cancer | ||
| NCT03000257 | I | Not yet recruiting | Solid tumors | ||
| Venetoclax | BCL-2 | NCT03082209 | I | Recruiting | Solid tumors, |
| Hematologic malignancies | |||||
| NCT04161885 | III | Recruiting | Acute myeloid leukemia | ||
| NCT02265731 | I/I | Completed | Hematological malignancie | ||
| Ketoconazole | HK-II | NCT03763396 | Not yet recruiting | Glioma | |
| NCT01036594 | Completed | Prostate cancer | |||
| 2-DG | Glut | NCT00096707 | I | Completed | Solid tumors |
| Agents | NCT Number | Phase | Current State | Condition |
|---|---|---|---|---|
| CSCs niche inhibitor | ||||
| LY2510924 | NCT02652871 | I | Completed | Acute myeloid leukemia |
| NCT01439568 | II | Completed | Lung cancer | |
| BKT140 | NCT01010880 | I/II | Completed | Multiple myeloma |
| AMD3100 | NCT00512252 | I/II | Completed | Acute myeloid leukemia |
| BL-8040 | NCT01838395 | II | Completed | Acute myeloid leukemia |
| Bevacizumab | NCT01190345 | II | Completed | Breast cancer |
| NCT01137968 | II | Completed | Lung cancer | |
| NCT03632798 | III | Recruiting | Ovarian cancer | |
| Topotecan | NCT00320983 | I | Completed | Cervical cancer |
| NCT00477282 | III | Completed | Epithelial ovarian cancer | |
| NCT01630018 | II | Completed | Ovarian cancer | |
| Digoxin | NCT01162135 | II | Completed | Prostate cancer |
| NCT00650910 | I | Completed | Breast cancer | |
| NCT02106845 | I | Completed | Solid tumors | |
| PT2385 | NCT03216499 | II | Completed | Recurrent glioblastoma |
| NCT04989959 | I | Recruiting | Renal cell cancer | |
| EZN-2986 | NCT01120288 | I | Completed | Solid tumors |
| NCT00466583 | I | Completed | Solid tumors/lymphoma | |
| Differentiation inducer | ||||
| retinoic acid | NCT01276730 | II | Completed | Cervical cancer |
| NCT00002586 | II | Completed | Lung cancer | |
| NCT01048645 | II | Completed | Lung cancer | |
| NCT00004149 | II | Completed | Prostate cancer | |
| arsenic trioxide | NCT00128596 | II | Completed | Metastatic liver cancer |
| NCT00005069 | II | Completed | Metastatic kidney cancer | |
| dimethylsulfoxide | NCT04439318 | II | Not yet recruiting | Multiple myeloma |
| Agents | Target | NCT Number | Phase | Current State | Condition |
|---|---|---|---|---|---|
| CD133 | NCT02541370 | I/II | Completed | Advanced malignancies | |
| CAR-T | CD44v6 | NCT04097301 | II | Recruiting | AML, MM |
| NCT04427449 | I/II | Recruiting | Tumors (CD44v6+) | ||
| EpCAM | NCT03563326 | I | Recruiting | Gastric cancer | |
| NCT02915445 | I | Recruiting | Nasopharyngeal cancer, | ||
| Breast cancer | |||||
| NCT04151186 | Not yet recruiting | Recurrent/Refractory | |||
| Solid tumors | |||||
| NCT02729493 | II | Unknown | Liver cancer | ||
| CD19 | NCT04532281 | I | Recruiting | AML, Non-Hodgkin’s | |
| lymphoma | |||||
| NCT02975687 | I | Completed | AML | ||
| NCT04833504 | Completed | B Cell lymphoma | |||
| NCT03811457 | Completed | Leukemia, lymphoma | |||
| CD123 | NCT04272125 | I/II | Recruiting | AML | |
| NCT04265963 | I/II | Recruiting | AML | ||
| CD34 | NCT03473457 | Recruiting | Relapsed/Refractory AML | ||
| CXCR4 | NCT04727008 | I | Not yet recruiting | Refractory/Relapsed MM | |
| DC vaccine | ALDH | NCT02176746 | I/II | Completed | Colorectal cancer |
| NCT02178670 | I/II | Completed | Ovarian cancer | ||
| NCT02063893 | Completed | Breast cancer | |||
| NCT02084823 | I/II | Completed | Lung cancer | ||
| NCT02115958 | I/II | Completed | nasopharyngeal cancer | ||
| NCT02074046 | I/II | Completed | pancreatic cancer | ||
| NCT02089919 | I/II | Completed | liver cancer | ||
| Unknown | NCT00846456 | I/II | Completed | Glioblastoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, B.; Yan, X.; Li, Y. Cancer Stem Cell for Tumor Therapy. Cancers 2021, 13, 4814. https://doi.org/10.3390/cancers13194814
Huang B, Yan X, Li Y. Cancer Stem Cell for Tumor Therapy. Cancers. 2021; 13(19):4814. https://doi.org/10.3390/cancers13194814
Chicago/Turabian StyleHuang, Binjie, Xin Yan, and Yumin Li. 2021. "Cancer Stem Cell for Tumor Therapy" Cancers 13, no. 19: 4814. https://doi.org/10.3390/cancers13194814
APA StyleHuang, B., Yan, X., & Li, Y. (2021). Cancer Stem Cell for Tumor Therapy. Cancers, 13(19), 4814. https://doi.org/10.3390/cancers13194814