Simple Summary
Management of gastrointestinal (GI) cancers t is a worldwide challenge and some new tools are needed to guide it. Circulating tumor DNA (ctDNA) is a fraction of tumor DNA that can be detected by a liquid biopsy through a simple blood sample. In this work, we tried to summarize in a clinical review to what extend the analysis of ctDNA can improve therapeutic support in digestive oncology and how this circulating biomarker appears as a very promising improvement in addition to classic diagnostic, prognostic and theranostic methods. Although the level of evaluation of this tool is still different between the different GI cancers locations, it is in dynamic evolution in all of them.
Abstract
Gastrointestinal (GI) cancers are major health burdens worldwide and biomarkers are needed to improve the management of these diseases along their evolution. Circulating tumor DNA (ctDNA) is a promising non-invasive blood and other bodily-fluid-based biomarker in cancer management that can help clinicians in various cases for the detection, diagnosis, prognosis, monitoring and personalization of treatment in digestive oncology. In addition to the well-studied prognostic role of ctDNA, the main real-world applications appear to be the assessment of minimal residual disease to further guide adjuvant therapy and predict relapse, but also the monitoring of clonal evolution to tailor treatments in metastatic setting. Other challenges such as predicting response to treatment including immune checkpoint inhibitors could also be among the potential applications of ctDNA. Although the level of advancement of ctDNA development in the different tumor localizations is still inhomogeneous, it might be now reliable enough to be soon used in clinical routine for colorectal cancers and shows promising results in other GI cancers.
1. Introduction
Gastrointestinal (GI) cancers appear as major health burdens worldwide with high incidences and mortality rates. For these cancers, stage at diagnosis remains the most important prognostic factor for clinical outcome. However, the emergence of simple and reproducible biomarkers is needed for the management of these diseases along their evolution. Circulating cell-free DNA (cfDNA) can be detected in plasma, urine, and other bodily fluids for everyone, and is increased in inflammatory diseases, infections and cancers [1,2]. For patients with cancer, a fraction of this cfDNA, called circulating tumor DNA (ctDNA), contains tumor-specific molecular alterations [3,4]. Detection of ctDNA is challenging: First, for the majority of patients, quantities remain very low. Moreover, ctDNA is diluted within total cfDNA and its identification can be difficult. New approaches aretherefore in development to overcome this sensitivity challenge. Depending on cancers subtypes, specific molecular alterations can attest for the presence of ctDNA, which is a promising non-invasive biomarker in the era of personalized medicine. In this review, we tried to resume the molecular aspects of ctDNA and in what extent this biomarker can help clinicians in the detection, screening, diagnosis, prognosis, monitoring and personalization of treatment in patients with gastrointestinal cancers.
2. Molecular Aspects
The first challenge in detection of ctDNA was due to the low quantities of DNA extracted from plasma. While amounts of cfDNA are higher in plasma from patients with cancer, ranges of elevation remain around an average of 3–4 times the quantity of a healthy individual [5]. However, the most challenging technical aspect is the detection of the low ratio of ctDNA representing 1 to 10% of cfDNA for advanced stages and until <0.1% particularly in early stages [5] (Figure 1).
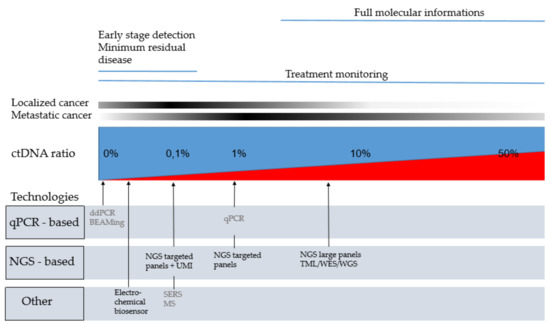
Figure 1.
Technologies that can be use regarding ctDNA ratio and the clinical application. In grey: technologies with limited targets suggesting prior knowledge of the mutation; In black: technologies with broad genes coverage; In blue: non tumoral cell free DNA. In red: circulating tumor DNA; BEAMing: beads, emulsion, amplification, magnetics; ctDNA: circulating tumor DNA; ddPCR: digital droplet PCR; MS: Mass spectrometry; NGS: new generation sequencing; qPCR: quantitative PCR; SERS: Surface-Enhanced Raman Scattering; TLM: tumor mutation load; UMI: unique molecular identifiers; WES: whole exome sequencing; WGS: whole exome sequencing.
The 1% allelic ratio threshold is commonly considered as the limit of detection for conventional quantitative polymerase chain reaction (PCR) technologies [6]. Recent advances in sequencing technologies allowed to detect these rare mutations within a background of wild type sequences, and to screen multiple genomic regions in a single run. The highly sensitive techniques that are required for liquid biopsy analyses can be broadly classified as digital PCR-based approaches and massive multiplexing next-generation sequencing (NGS)-based approaches [7].
Digital PCR (dPCR) techniques can identify specific known tumor mutations with high accuracy. Indeed, partitioning DNA molecules through the generation of millions of droplets and the possible additivity of reactions reduce template competition and allows to reach a theoretical detection threshold of mutations until 1/100,000 [8]. Therefore, digital droplet PCR (ddPCR) might appear as the most sensitive, suitable and fast technology [7,9,10,11]. Several ddPCR platforms are now being commercialized but still contain a restricted number of probes in each run. Therefore, their use in clinical routine remains limited to the detection of specific pre-known mutations [10].
NGS provides a comprehensive profile of molecular alterations occurring along tumor evolution, without necessity of their prior knowledge. First common NGS panels permitted to reach a detection threshold around 1–2%, stressing the need to improve their sensitivity for relevant liquid biopsies applications. Indeed, sequencing abilities are limited by several issues: depth of sequencing, background error noise and the methodological pipeline used to detect mutations.
Then, bioinformatics methods were developed to improve pipelines initially designed for tissue analysis. For example, Base Position Error Rate (BPER) is an algorithm based on the fact that 1–2% of error rate reported by NGS manufacturers refers to a mean of all type of error rates, which are highly variable along the genome because of sequencing environment. In this technique, a workflow recalculates an error rate for each position to be able to identify mutations at 0.3% ratio for single nucleotide variants and at 0.1% for indel >2 [12]. This algorithm provides a cost-effective solution to identify mutations in easily sequenced regions but however does not reach dPCR sensitivity.
Therefore, NGS sequencing technologies were upgraded to better discriminate a real variant from background noise. They can be divided in 3 categories: (1) capture of ctDNA with specific probes, (2) PCR amplification of selected regions, with short amplicons size (<150 bp), (3) anchored multiplex PCR, which theoretically enriches for highly fragmented ctDNA over high molecular weight genomic DNA. These technologies are frequently coupled with unique molecular identifiers (UMI), an initial random barcoding with short (8–16 bp) DNA sequences tagging each individual DNA fragment before amplification [13]. However, the required sequencing depth and the cost are more elevated. Elazezy et al. developed and compared various methods although they are not easily manageable by clinical laboratories yet [14]. Numerous commercial kits for ctDNA libraries are therefore available, with a range of targeted genes from 20 to >500 and using the UMI technology. Their heterogeneity of design and performances complicates the choice for clinical laboratories. Therefore, their evaluation and comparison in real life are strongly mandatory [15,16].
Another currently explored filed of interest is exosomes’ concentration and sequencing as these vesicles are actively excreted by tumor cells. However, in a recent cohort of 33 CRC, Thakur et al. did not report any improvement in the sensitivity for ctDNA detection [17].
Beyond the detection of point mutations, other technologies are now available through the analysis of liquid biopsy.
Shen et al. confirmed robust performance in universal cancer detection and classification across an extensive collection of plasma samples from several tumor types based on cell-free methylation patterns [18]. Recent validation results of the Circulation cell free Genome atlas study based on methylation and machine learning showed a specificity of 99.5% and a sensitivity of 50% for cancer detection [19].
More recently, Mathios et al. evaluated the cell free fragmentomes instead of ctDNA and reported good performances to detect lung cancer even at early stages (91% of stages I/II, and 96% of stages III/IV), thus opening a new paradigm for liquid biopsy [20].
Finally, other approaches are currently emerging to overcome sequencing technologies biases, such as plasmonic nanoparticles used in Surface-Enhanced Raman Scattering (SERS), mass spectrometry based assays and electrochemical biosensor technologies [7,21,22,23].
3. Early Cancer Detection through Circulating Tumor DNA and Molecular Profile Determination
The GI cancer diagnosis is currently based on a histological assessment and therefore requires tissue sample collected by surgical resection, endoscopic ultrasound, or biopsy of primitive tumor or accessible metastasis. Several studies assessed the interest of ctDNA as screening tool for early tumor stage. However, further studies are still required to prove the clinical utility of ctDNA in early diagnosis as stipulated by American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) in a recent report [24]. Some findings demonstrated that asymptomatic cancers could be detected years before conventional diagnosis through non-invasive blood tests. In a recent longitudinal study, analysis of ctDNA methylation was performed on plasma samples from 605 asymptomatic individuals. Among them, 191 later developed stomach, esophageal, colorectal, lung or liver cancer within four years of blood draw. This method was able to detect cancer in 95% of asymptomatic individuals who were later diagnosed [25]. However, future longitudinal studies are required to confirm these results. The main risk of this early screening would be over-diagnosis through false-positive results or through the detection of circulating genomic variants from cells that have taken the first step toward transformation but were never meant to become clinically important [24].
Tissue biopsy is usually only performed at diagnosis and can sometimes be hard to obtain. For these reasons, several studies have also evaluated in different GI cancers whether plasma molecular alterations can be detected with ctDNA and are correlated with tissue biopsies.
3.1. Colorectal Cancer
Tumor tissue is routinely used to search for KRAS or NRAS gene mutations that occur in around 55% of metastatic CRC (mCRC) and predict a lack of response to the EGFR-targeted monoclonal antibodies, such as cetuximab and panitumumab [12,26]. In the same context, BRAF mutation is another alteration known as a poor prognostic factor that can be targeted by a doublet-therapy combining an anti-BRAF kinase inhibitor (encorafenib) and anti-EGFR monoclonal antibody (cetuximab) [27,28,29].
In the context of mCRC, the quantitative PCR (Intplex qPCR) on ctDNA was described by Thierry et al. as a valuable detection method with a high rate of specificity and sensitivity, especially for BRAF V600E and KRAS mutations, in a prospective study on 106 patients with mCRC [30]. The digital droplet PCR (ddPCR) has also been validated by other group for detection of KRAS mutations in mCRC [31]. More recently, in a large prospective multicenter study. Another method consists in using the NGS-BEPER-method (22 genes), and two specific methylated biomarkers (WIF1 and NPY) as a second-step test for NGS-negative specimens. Bachet et al. used this technique to evaluate the concordance of RAS mutations between plasma and tissue among 406 chemotherapy-naive patients with mCRC with detectable ctDNA (n = 329/412). By comparing the results of RAS status in ctDNA and in matched tumor tissus, they founded an accuracy of 83% with NGS alone versus 93% with NGS plus methylated biomarkers [32]. Supplementary studies also suggested a good concordance rate between mutations observed in tumor biopsy and those identified on ctDNA [30,31,32,33,34,35,36].
3.2. Pancreatic Cancer
The KRAS gene mutations occur in more than 90% of pancreatic cancer (PC), and appears therefore as the best candidate to assess the presence of ctDNA in this tumor [37,38,39,40]. However, the ctDNA detection rate in metastatic PC varies widely from 40% to 80% and could therefore explain some discordance between tumor and plasma mutation assessment [41,42,43,44]. It could explain the results of the recent meta-analysis of Luchini et al. including 14 studies involving 369 patients, that reported a concordance rate of only 32% between ctDNA and tissue based on large NGS multi-gene mutation panels [45]. The overall pooled sensitivity and specificity of the mutational analysis on liquid biopsy compared to tumor tissue were 70% and 86% respectively. However, when focusing on studies analyzing KRAS mutations only, the sensitivity slightly decreased but the specificity increased and were 65% and 91%, respectively [45].
Indeed, apart from KRAS mutations for PC screening, adding NGS-based panel for other mutations such as SMAD4, CDKN2A, ROS1, BRAF and TP53 could lead to higher levels of ctDNA detection [46,47,48]. More recently, methylation of promoter of ADAMST1 and BNC1 genes were also described as potential tool to assess the presence of ctDNA in PC [49].
However, the use of highly sensitive detection methods of ctDNA might lead to false diagnosis of PC. Indeed, KRAS mutations can be detected in plasma in some non-cancerous diseases such as chronic pancreatitis. In a pilot study from Rashid et al., 21.8% of patients with chronic pancreatitis were tested positive for KRAS mutations in plasma [50]. Among these 64 patients, none developed a PC, with a mean follow-up duration (by clinic and by positron emission tomography or endoscopic ultrasound) of 2.5 years [50].
Quantitative ctDNA assessment, or combining biomarkers and methylation detection may improve the specificity of ctDNA detection and therefore help to discriminate benign from malignant pancreatic diseases, even at early tumor stages [51,52,53,54].
3.3. Esophageal and Gastric Cancer
In gastric cancer (GC), despite a low-frequency of genomic alterations [55,56], routine tissue-based NGS showed that at least 37% of patients harbor somatic mutations (TP53, KRAS) or gene amplification, such as HER2, MET, EGFR, and FGFR2 [57,58,59,60]. Some retrospective studies evaluated the feasibility of ctDNA detection by NGS among GC patients. In a recent study including 55 patients with GC tested by NGS, Kato et al. showed that 31 had concordant mutations between tumor tissue and ctDNA with levels ranged from 61.3% (for TP53 mutation) to 87.1% (for KRAS mutation) [61]. In their meta-analysis, Gao et al., reported that ctDNA detection might be a specific, but still a low sensitive test in GC patients [62]. More recently, the analysis of a large cohort of 1630 patients with GC revealed that ctDNA-NGS genomic landscape was similar but not identical to tissue-NGS [63]. This could reflect the molecular heterogeneity, with some targetable molecular alterations identified at higher frequency via ctDNA-NGS compared with previous matched primary tissue-NGS samples [63].
Despite increasing use of genomic alterations to detect ctDNA in GC, the most investigated technique to prove the presence of ctDNA is detection of hypermethylation of gene promoters which might result in an inappropriate silencing of tumor suppressor genes [62,64]. The promoter methylation of APC and RASSF1A in cfDNA was described as frequent epigenetic events in patients with early operable GC [65]. Aberrant methylation of other genes such as PCDH10, SOX17, TIMP3, MINT2 and WAF1 also showed promising results in GC [64].
In esophageal squamous cell carcinoma (ESCC), preliminary studies suggested the feasibility of ctDNA detection [66]. Luo et al. used exome or targeted sequencing to detect somatic mutations in 11 patients with ESCC and compared ctDNA from pre- and post-surgery plasma [66]. They compared plasma somatic mutations that were also identified in matched tumors and founded that mutant allelic franction (MAF) decreased after surgery [66].
3.4. Hepatocellular Carcinoma
The analysis of the mutational landscape of hepatocellular carcinoma (HCC) over 3000 samples in the Catalog of Somatic Mutation in Cancer showed that the most frequent tumor mutations were TP53 (27%), TERT (25%) and CTNNB1 (18%) [67,68,69]. Using targeted methods to detect these three genes mutations in plasma, ctDNA presence was proven from 20% to 55% of patients with HCC across different studies [68,69,70,71,72]. In one prospective study including 27 patients with proven ctDNA, only 22% of them (6/27) also had matched mutants in tumor tissues, underlying the heterogeneity of HCC [68]. Therefore, single specific molecular alterations do not seem to be sensitive or specific enough to be used as a diagnostic tool in HCC. Moreover, some molecular alterations could be unspecific for HCC, such as TERT mutations that were present in plasma for 9% of patients with cirrhosis and without evidence of HCC on imaging [70].
When using NGS techniques with panel of frequently altered genes in HCC, ctDNA detection rate reached 63% in a prospective cohort of 30 patients, with two thirds of patients with stage A according to the Barcelona Clinic Liver Cancer score (BCLC A). In this study, the concordance rate between plasma and tissue biopsy was 81% [73].
Despite the utility of gene point mutations, DNA methylation seems to be more broadly informative in HCC. In a recent study, a combination of five aberrant methylation biomarkers was able to distinguish HCC samples from control cirrhotic and not cirrhotic tissue samples, with a specificity of 95% [74].
Some single aberrant methylation genes have shown high concordance rates between plasma and tissue in HCC [75,76,77]. Among patients with hypermethylation of CDKN2A, which is described in up to 73% of HCC patients, Wong et al., reported a concordance rate of 81% between plasma and tissue biopsy with a specificity of 100% among control patients [75]. Hypermethylation of RASSF1A promoter could also to be a candidate and was found in up to 90% of HCC tissues [78,79,80,81,82]. However, it seems to be also detected in patients with non-malignant liver tumor, such as liver cirrhosis, chronic hepatitis B or in healthy controls, with a lower rate (13%, 4%, and 4%, respectively) [82]. Other single hypermethylated candidates, such as SEPT9, VIM, FBLN1, TFPI2, TGR5, MT1M, MT1G, APC, SPINT2, SFRP1, GSTP1, or hypomethylated candidates such as LINE-1 showed promising results for HCC screening [79,83,84,85,86,87,88].
More recently, whole methylome analysis allowed discovering novel methylated DNA markers in HCC. Creation of a new panel with 6 methylated biomarkers (HOXA1, EMX1, AK055957, ECE1, PFKP and CLEC11A) was able to detect 75% of BLCL 0 and 93% of BCLC B HCC patients meeting Milan criteria and was superior to AFP [74].
3.5. Other GI Cancers
Molecular landscape of cholangiocarcinoma (CC) has been widely studied in the past few years trying to detect therapeutic targets [89,90,91]. The cholangiocarcinoma (CC) is usually separated between intrahepatic CC (IHCC) and extra hepatic CC (EHCC). Some mutations such as KRAS, BRAF or TP53 are more frequent in EHCC but remain rare, whereas others, such as FGFR1-3 fusions and IDH1/2 mutations are preferentially detected in IHCC and occur in around 15–20% of tumors [89,90,91]. A recent study including 24 CC patients has reported a concordance rate of 74% between mutations in tumor tissue and ctDNA. When stratifying on tumor localization, concordance rate was 92% for IHCC, but only 55% for EHCC [92].
In squamous cell carcinomas of the anal canal (SCCA), Human papillomavirus (HPV) is found in 90% [93]. Therefore HPV DNA appears as the best candidate to assess the presence of ctDNA in SCCA and can be detected in plasma by ddPCR with sensitivity up to 93% in HPV positive-cancers [94]. In a recent study enrolling 8 SCCA patients, ddPCR demonstrated 100% of specificity for the detection of HVP ctDNA [95].
In gastrointestinal stromal tumors (GIST), mutations of exons 9, 11, 13 and 17 of KIT, and of exons 12, 14 and 18 of PDGFRA are key drivers of oncogenesis and are present in around 85–90% of tumors, whereas the remaining 10–15% of these cancers is referred as KIT/PDGFRA wild-type GISTs. However, other genes, such as BRAF, NF1, and SDH, may be aberrant in this context [96,97,98]. Therefore, the majority of the studies evaluating the utility of ctDNA in GIST were focused on KIT alterations. The ctDNA detection rate varies from 45% to 55% across studies [99,100]. Among 102 archival tumor tissue samples and 163 plasma samples at baseline from patients included in the phase III GRID study in patients with GIST treated by regorafenib versus placebo, following failure of at least imatinib and sunitinib, Demetri et al. first reported a concordance rate of 100% and 71% between plasma and tissue biopsies for primary KIT exon 9 and 11 mutations, respectively [101]. Concordance rate also depended on the primary or secondary character of the mutation. For primary KIT mutations concordance rate between tissue and plasma was 84% whereas secondary KIT mutations were more often detected in plasma (47%) than in tissue (12%) [101]. More recently, Arshad et al. revealed a concordance rate for detection of mutations in GISTs with a positive predictive value of 100% among 243 patients [99].
4. Minimal Residual Disease and Detection of Early Recurrence
Detection of minimal residual disease (MRD) is an important challenge as MRD might serve as a surrogate marker for disease free survival (DFS) and could therefore guide further therapeutic interventions for patients after curative treatment. In GI cancers, assessment of MRD through the analysis of ctDNA is not standardized yet but has already been evaluated in different studies. The most relevant of them are summarized in Table 1 and Table 2.

Table 1.
Main Minimal Residual Disease assessment studies for circulating tumor DNA in colorectal cancers.
Table 2.
Main Minimal Residual Disease assessment studies for circulating tumor DNA in gastro-intestinal cancers except colorectal.
4.1. Colorectal Cancer
Around 50% of patients with localized (stage II–III) and resected CRC will further develop metastasis [113]. The addition of adjuvant chemotherapy significantly lowers the risk of relapse [114]. The clinical utility of tracking ctDNA to detect MRD and stratifying patients based on their risk of developing relapse has now been well established in CRC [102,103,104,105,106,115,116].
For stage II colon cancer, Tie et al. described a correlation between disease recurrence and the levels of ctDNA in post-surgery setting. In a prospective cohort of 230 patients, ctDNA was detected in 7.9% (14/178) after surgery. Among them, after a median follow –up of 27 months, 79% (11/14) presented a tumor relapse. However, relapse occurred with a very lower frequency of 9.8% (16/164) in patients with negative ctDNA. For patients who completed adjuvant chemotherapy, the recurrence-free survival (RFS) was less frequent when ctDNA was undetectable treatment. This data present ctDNA as a relevant method to assess presence of residual disease after stage II colon treatment. It could therefore be used to identify patients at higher risk of recurrence [102].
In stage III colon cancer, the same team reported that ctDNA detection in 21% (20/96) of postsurgical samples was also associated with inferior RFS. The ctDNA was detected in 17% (15/88) of post-chemotherapy samples. The estimated 3-year recurrence free interval (RFI) was significantly lower when ctDNA was detectable. Postsurgical ctDNA status was an independent prognostic factor of RFI [103]. Taieb et al. also worked on the predictive value of ctDNA in adjuvant setting in stage III colon cancer in a large series from patients of the IDEA-FRANCE trial (NCT-00958737) [105]. The aim of this analysis was to determine the prognostic and predictive value of ctDNA for adjuvant treatment duration lasting 3 versus 6 months of oxaliplatin-based adjuvant chemotherapy. The samples of 1017 patients were fully analyzed for ctDNA detection. Among them, 140 patients (13.8%) had ctDNA-positive samples after surgery. The 3-years DFS rates were 66.4% for positive versus 76.7% for negative-ctDNA samples, respectively. Multivariate analysis confirmed that the presence of ctDNA was as an independent prognostic marker. In this series, 6 months of chemotherapy showed better results than 3 months in both ctDNA-positive and –negative groups. Interestingly, ctDNA-positive patients treated for 6 months and ctDNA-negative patients treated for 3 months had a similar prognosis. [105].
In locally advanced rectal cancer (T3/T4 and/or N+), Tie et al. reported in another prospective study that ctDNA could detect MRD after chemoradiotherapy or surgery. The ctDNA detection after these treatments were correlated with an increased risk of recurrence, and a shorter 3-years RFS (33% versus 87% for patients with positive and negative post-operative ctDNA, respectively) [106].
In a larger series, Tarazona et al. recently evaluated the detection and longitudinal monitoring of ctDNA in CRC patients pre- and post-operatively, during and after adjuvant chemotherapy in a prospective multi-centric study on 193 patients with resected stage I-III tumors. Among the 14 out of 152 (9.2%) patients with post-operative ctDNA before adjuvant chemotherapy (identified to be MRD-positive), 78.5% (11/14) relapsed. In contrast, 10.1% (14/138) of MRD-negative patients relapsed. In the multivariable analysis, longitudinal ctDNA status was the only significant prognostic factor associated with RFS. Serial ctDNA analysis also allowed detecting MRD up to a median of 9.08 months before radiologic relapse, with a sensitivity of 79.1% and specificity of 99% [104].
Conventional surveillance strategy might therefore be completed by ctDNA analysis to stratify the risk of recurrence and guide therapeutic interventions in CRC. Interventional trials to assess the clinical benefit of the monitoring of ctDNA in adjuvant setting are currently ongoing in several countries, such as PRODIGE 70-CIRCULATE trial (NCT-04120701). The aim of this French multicentric study is to identify, through the detection of ctDNA, a group of patients with higher risk of relapse among patients with stage II operated colon cancer and to test the benefits of adjuvant chemotherapy (FOLFOX6m-5fluorouracile, leuvocorin and oxaliplatin) in this population.
Recently, ctDNA was also used to detect MRD in post-operative setting in patients with oligometastatic CRC. Among 100 patients in this study, MRD-positive status was associated with a higher level of relapse and also with an inferior overall survival (OS) [107].
4.2. Pancreatic Cancer
In early-stage PC, there is currently no biomarker to guide adjuvant treatment. Several studies and meta-analysis already described the negative prognostic value of ctDNA in localized PC at baseline or in post-operative setting [41,44,108,117,118,119,120,121]. In pre-operative setting, a recent meta-analysis of 5 retrospective studies including 375 patients reported that pre-operative ctDNA detection was significantly associated with poor OS and with a trend to higher risk for disease recurrence [120]. After surgery, immediate post-operative ctDNA detection was associated with a trend for poorer RFS and with a significant poorer OS [120]. These data are in line with the results of some prospective studies including patients with early stage PC [44,119,121].
However, ctDNA is currently unlikely to become a routine tool to avoid some adjuvant treatment in PC due to the high recurrence rate after surgical resection. However, it may help to detect early relapse and therefore shorten the time to treatment. In some resected PC, longitudinal ctDNA monitoring allowed detecting MRD up to a median from around 2.7 to 6.5 months before radiologic relapse [109,122]. However, the potential clinical benefit of early-relapse treatment based still need to be evaluated.
4.3. Esophageal and Gastric Cancer
There are few data on the prognostic and predictive value of MRD in context of esophageal and gastric cancer.
Focusing on 29 patients with resectable GC in their cohort, Maron et al. recently found that patients with detectable ctDNA prior to surgery/therapy had a trend to shorter DFS than those with undetectable ctDNA [63]. Interestingly, after surgery, the residual detection of ctDNA was significantly correlated with worse outcome [63]. However, three apparently positive ctDNA after surgery did not relapse and the mutations detected were not present in their tissue analysis and should therefore trend to a cautious interpretation of these results [63].
In a recent prospective study enrolling 35 patients with localized esophageal adenocarcinoma and 10 patients with ESCC, Azad et al. showed that the detection of ctDNA by deep sequencing method after exclusive or preoperative chemo-radiotherapy was associated with disease progression, formation of distant metastases, and shorter DFS. Moreover, detection of ctDNA after exclusive or preoperative chemoradiotherapy anticipated by 2.8 months radiographic tumor progression [110]. These results are in line with those from a former study showing that ctDNA increased approximately 6 months earlier than the detection of tumor recurrence by imaging tests in two patients with ESCC [123]. In another retrospective study among 17 ESCC patients with stage IIA to IIIB tumor, cfDNA was screened pre and post-surgery. Among the 8 patients with somatic mutations detected in plasma, corresponding to ctDNA in pre-surgery, only 2 patients still had these mutations detected in post-surgery setting and with a lower MAF, suggesting that ctDNA could potentially be used to monitor disease load and detect MRD [124].
4.4. Hepatocellular Carcinoma
After surgery in HCC, the detection of ctDNA, proven by the detection of tumor specific alterations and by the use of methylation panel, seems to be correlated with worse prognosis in several small studies [69,70,75,80,110,111,125,126,127,128].
Recently, a larger retrospective series on 81 resectable HCC showed shorter DFS and OS in patients with presence of TERT, CTNNB1 or TP53 mutations in plasma after curative hepatectomy [111]. In a multivariate analysis of this study, detectable ctDNA was the only independent risk factor for postoperative recurrence [111].
Regarding the use of methylation markers, a first study has reported that higher methylation of RASSF1A in plasma at diagnosis or one year after surgery was correlated with poorer DFS in 63 patients with resectable HCC. However, higher methylation rate one month after resection was not correlated with significantly shorter DFS [80]. Similar conclusions have been reported with the detection in plasma of hypermethylation of insulin growth factor binding protein-7 among 155 HCC patients after surgical resection. This detection was shown as an independent prognostic factor for poorer OS and higher tumor recurrence [127]. In advanced HCC, combination of LINE-1 hypomethylation and measurement of RASSF1A hypermethylation were also described as correlated with poorer OS, earlier recurrence and with poorer prognosis upon curative resection [129].
4.5. Other GI Cancers
In locally advanced SCCA, Cabel et al. recently used ddPCR to detect HPV ctDNA in a study of 18 patients. In this series, presence of HPV ctDNA after chemoradiotherapy was associated with a poor prognostic. In most patients, HPV ctDNA was detectable before chemoradiotherapy and became undetectable along treatment. Patients with residual ctDNA after completed treatment had a shorter DFS [112]. A trial evaluating the detection of ctDNA after curative treatment in patients with pelvic stage II–III HPV induced cancer is currently ongoing (Circa HPV study—IC 2017-01).
In localized GISTs, ctDNA might be used as a tumor specific biomarker for early prediction of recurrence, as suggested by Maier et al. who first described that the amount of ctDNA correlates with prognosis [130]. However, few data are currently available in this context. In pre-operative setting, Kang et al. recently demonstrated that ctDNA could be used as a surrogate biomarker for tissue biopsy to determine KIT and PDGFRA mutations among 25 patients with GISTs [131]. However, due to the small effective, these results should be further validated in larger series.
5. Circulating Tumor DNA as Tumor Burden and/or Prognostic Marker in GI Cancers
The probability to detect ctDNA in GI cancers is associated to tumor stage. Bettegowda et al. analyzed the presence of ctDNA in various cancer types among 640 patients. Concerning GI cancers, they reported the presence of ctDNA in 73%, 57% and 48% of patients with localized CCR, esogastric cancers and PC, respectively, whereas ctDNA was detectable in more than 75% of patients with advanced cancers [132].
5.1. Colorectal Cancer
In CRC, ctDNA appears strongly correlated with tumor burden according to several studies [133,134]. Tie et al. described that pretreatment ctDNA levels correlated more strongly with initial tumor burden as estimated from standard RECIST 1.1 criteria than pretreatment CEA [133]. Moreover, ctDNA levels were described to be significantly associated with presence of liver metastasis and sum of the tumor diameter in metastatic sites [32,134]. However, the association between ctDNA levels and lung, lymph node and peritoneal metastasis, tumor markers, primary tumor location, and number of metastatic organs in CRC remains more controversial [133,134,135].
Furthermore, numerous studies have suggested that detectable ctDNA at diagnosis was strongly negatively correlated with progression-free survival (PFS) and OS, regardless of other prognostic factors or detection method and sample type [136,137,138,139,140,141].
5.2. Pancreatic Cancer
In PC, several studies are in line with the results of Bettegowda et al. showing that ctDNA detection increases with tumor stage and reflects tumor burden [44,142]. In locally advanced or metastatic PC, the prognostic role of ctDNA has been largely evaluated. The presence of ctDNA at baseline before first-line treatment appears to be correlated with worse survival in many studies and meta-analysis [42,44,142,143,144]. Relation between quantitative detection of KRAS mutations and prognosis is however still unclear in PC.
5.3. Esophageal and Gastric Cancer
In GC, a recent meta-analysis Gao et al. reported a significant association between ctDNA and tumor stage, presence of lymph node and distant metastasis [62]. The authors also reported that high level of ctDNA in GC was associated with worse OS [62]. The largest ctDNA dataset yet published, analyzed the serum of 1630 patients with GC to detect ctDNA through NGS method. These authors then recently confirmed that the maximal tumor somatic variant allelic frequency, defined as the largest mutated ctDNA clone detected among all cfDNA present in the plasma that is used to estimate overall ctDNA quantity appeared as a surrogate biomarker for disease volume or burden in metastatic disease [63].
In metastatic GC, it seems that a higher amount of ctDNA at baseline is associated with worse outcomes in the cohort of Maron et al. [63]. Because of the lack of data on the prognostic and predictive impact of ctDNA in GC, an ongoing observational prospective study (PLAGAST) is currently evaluating the correlation between the level of ctDNA and prognosis or response to treatment of localized and advanced gastric cancer (NCT-02674373).
In esophageal squamous cell carcinoma (ESCC), one small study described that the MAF in ctDNA changed concomitantly with tumor burden in two patients [123].
5.4. Hepatocellular Carcinoma
In HCC, ctDNA detection rate was higher in metastatic tumors and correlated with tumor burden [73,145]. For patients with unresectable advanced HCC, ctDNA levels showed a significant correlation with the presence of metastases and survival in an exploratory cohort of 13 patients recruited in the context of the SORAMIC trial [146]. Moreover, in a prospective study among 41 patients with HCC, Liao et al. reported that the presence of TERT, CTNNB1 or TP53 mutations in plasma before surgery were significantly associated with shorter RFS [69]. Lastly, the amount of ctDNA detected immediately after local treatment was significantly correlated with the presence of distant metastases, supporting also a potential prognostic value [146].
5.5. Other GI Cancers
In CC, one study recently described a significant correlation between ctDNA MAF in both IHCC and EHCC, with the respective initial tumor load [92]. Furthermore, in the study of 24 CC patients, ctDNA variant allele frequency values at baseline showed a trend for a shorter PFS but this correlation was significant when focusing on the IHCC group [92].
In SCCAs, the median level of ctDNA was higher in metastatic than in localized tumor stage in two recent studies [112,147]. Bernard-Tessier et al. described in an ancillary study of 57 SCCA patients that ctDNA level has a significant impact on clinical outcomes. The PFS was significantly longer for patients with ctDNA level below the cut-off obtained by area under the curve (AUC) [147]. In this study, HPV ctDNA negativity in non-progressing patients after chemotherapy completion was also a strong predictive biomarker of extended response to chemotherapy [147].
In GISTs, the amount of ctDNA was reported as significantly higher in patients with an active disease compared to those in remission [130]. In a recent study among 44 unpretreated GISTs, ctDNA was detected in all patients with metastatic disease whereas it was inconstant in patients with localized tumor. In this study, tumor burden was the most important detection determinant [148].
6. Circulating Tumor DNA to Monitor Treatment Response and Detect Acquired Resistance
The non-invasive nature of ctDNA allows for repeated testing and molecular assessment of tumor during treatment. This dynamic assessment is a clear advantage over traditional tissue biopsy. In the advanced tumor stage, baseline ctDNA could be more helpful to capture the molecular spatial and temporal heterogeneity of the disease which is a particularly important biological issue, at diagnosis or later because of clonal evolution and selection [33]. Differences in molecular characteristics have been described between primary tumor and metastases, especially in metachronous lesions [34].
Moreover, the monitoring of ctDNA may also anticipate the evaluation of treatment efficacy by detecting emergent actionable molecular alterations implicated in therapeutic resistance to ongoing treatment.
6.1. Colorectal Cancer
In mCRC, longitudinal quantification of ctDNA appears to be correlated with tumor evolution in several studies [133,140]. By sequencing a panel of 15 genes with frequent somatic variant in CRC tissue sample at diagnosis of 53 patients with mCRC, Tie et al. evaluated ctDNA as disease monitoring. They reported that a level of reduction in ctDNA concentration during first cycle of chemotherapy was significantly associated with the objective radiologic response rate at 8–10 weeks and with a trend for a better PFS. [133]. Similarly, Garlan et al. showed that early changes of the ctDNA concentration could predict the efficacy of first- or second-line chemotherapy in a prospective cohort of 82 mCRC. They used ctDNA monitoring between the first and second or/and third cycle of chemotherapy to define a composite marker that allowed to separate patients in two groups of “bad” or “good” ctDNA responder. This marker was based on the “normalization” of the ctDNA concentration (thresholds of 0.1 ng/mL) and the slope of ctDNA concentration decrease. The group of better ctDNA responders demonstrated a significantly better tumor response rate, PFS and OS [140]. The changes of ctDNA concentration during treatment therefore appear as a relevant early tool to assess treatment efficacy and this biomarker should be evaluated in larger prospective series.
Furthermore, ctDNA can also be used to track clonal evolution. It has been suggested that CRC presumably contains resistant mutant clones before treatment that emerge under therapeutic pressure [149]. The acquisition of resistance can be accompanied by the emergence of RAS pathway mutations that could allow to anticipate radiologic progression [150,151]. Several studies have already described emergence of mutations detected by ctDNA under anti-EGFR treatment up to 5–10 months before imaging diagnostic [150,151,152]. By monitoring ctDNA, Siravegna et al. also showed in a subset of patients, that the proportion of ctDNA, based on the detection of KRAS mutations, dynamically varied depending on the presence or the absence of anti-EGFR treatment. These possible dynamic clonal evolutions induced by therapeutic pressure justified to re-challenge anti-EGFR based treatment after a withdrawal period in mCRC. [152,153]. Some retrospective analyses of the phase 2 CRICKET and E-Rechallenge studies suggested that ctDNA could guide this re-challenge therapy because only patients without RAS or BRAF circulating mutations detected plasma at the time of re-challenge might achieve clinical benefit from the retreatment with anti-EGFRs [116,153,154] (Table 3).
Table 3.
Studies evaluating circulating tumor DNA as a screening tool to detect patients who could benefit from anti-EGFR re-challenge in metastatic colorectal cancer.
More recently, Sartore-Bianchi et al. presented the results of the CHRONOS study, the first interventional ctDNA guided study in mCRC. 52 patients were screened by liquid biopsy for anti-EGFR re-challenge. Among them, 16 (31%) were mutated in ctDNA for RAS, BRAF or EGFR ectodomain and avoided a useless treatment by anti-EGFR. Of the 36 (39%) triple wild-type patients, 27 were re-challenged by anti-EGFR and obtained an ORR of 30% [155]. Some ongoing studies, such as the prospective RASINTRO study (NCT-03259009) or the randomized FIRE4 trial (NCT02934529) are currently ongoing to confirm the clinical use of liquid biopsy-driven re-challenge and the predictive impact of RAS mutations in ctDNA for the efficacy of anti-EGFR reintroduction treatment in patients with mCRC (Table 3).
Other molecular alteration than KRAS mutations can emerge under therapeutic pressure and can be detected by analysis of ctDNA, such as amplifications of MET and HER2 or EGFR mutations [156,157,158].
6.2. Pancreatic Cancer
In advanced PC, some regimens such as FOLFIRINOX (5-fluorouracil, leucovorin, oxaliplatin, and irinotecan) and gemcitabine plus nab-paclitaxel are effective but are not devoided of toxicities [159,160,161,162]. The monitoring of KRAS mutation through ctDNA has been performed in several studies and suggested that its detection could predict radiological progression, but some results were however discordant [119,163,164]. The clearance of KRAS ctDNA during treatment predicted better PFS than remaining positive ctDNA [164], and increasing levels of KRAS ctDNA were also associated with worse PFS and OS [165,166]. Finally, the decline slope of ctDNA concentration based on mutation of KRAS was associated with OS in another study [167]. Apart from KRAS mutations, evolution of other mutations in plasma, such as TP53, SMAD4, CDKN2A, KRAS, APC, ATM, FBXW7 and others could also be used to reliably reflect response to therapy [47,48].
Unlike other GI cancers, there is currently no targetable molecular alteration for all patients with advanced PC in clinical routine. However, some new treatment could be promising in PC, such as PARP inhibitors in case of germline BRCA1/2 mutations [168]. Moreover, like in other tumors, checkpoints inhibitors seem to be efficient in advanced PC with microsatellite instability [169,170,171]. Molecular alterations could be detected in ctDNA in PC [163] and therefore maybe screen patients for targeted therapies in the future. In this context, Bachet et al. recently confirmed from the data of a randomized phase II trial that the ctDNA could be a predictive biomarker of l-asparaginase encapsulated in erythrocytes (eryaspase) efficacy in advanced PC [172].
6.3. Esophageal and Gastric Cancer
In patients with advanced gastroesophageal adenocarcinoma, the addition of trastuzumab to chemotherapy was associated with improvement of clinical outcomes for tumors with a high level of HER2 expression (IHC3+ or IHC2+ and FISH+) [173]. Some studies have already described the potential for ctDNA to detect HER2 amplification by ddPCR with high concordance with classic immunohistochemistry and fluorescent in situ hybridization on tissue samples [174,175]. However, in the recent cohort of Maron et al. seven patients with advanced disease were tested for HER2 amplification in both primary and metastatic tumor, and in ctDNA. Among them, only 2 patients (28%) were concordant for HER2 amplification detection in the three samples, underlying possible missed detection of HER2 amplification by NGS and then the risk of missed opportunities to use anti-HER2 therapies [63]. Despite its lack of sensitivity, ctDNA could however be used in combination with tissue NGS to define a group of extremely sensitive HER2 amplified patients when treated with trastuzumab [63].
Moreover, some authors already suggested that ctDNA could also be used to monitor response to therapy in GC. In a recent study, tumor responses to lapatinib plus capecitabine were closely related with changes of the level of amplification of HER2 detected in plasma through serial ctDNA sequencing [176]. In the study of Maron et al. dynamic measurements of ctDNA before and during treatment showed that a decrease superior to 50% in MAF was correlated with better OS [63]. The detection of therapeutic resistance to treatment in advanced GC could also be improved by ctDNA. In the cohort of Maron et al. some anti-HER2 therapy acquired resistance mechanisms were detected using ctDNA [63].
6.4. Hepatocellular Carcinoma
In advanced HCC, ctDNA could be used to monitor tumor burden under therapy. A diagnostic prediction model with 10 selected methylation markers through ctDNA was recently developed by Xu et al. and correlated with tumor burden, treatment response, and disease stage [145].
In a study using whole exome sequencing to evaluate ctDNA among HCC patients who underwent surgery, in patients with positive ctDNA after surgery, the levels of serum ctDNA increased with disease progression and responded to the additional treatments [126].
The somatic MAF of ctDNA could also reflect clinical dynamics as demonstrated in one patient with advanced HCC undergoing trans-arterial chemoembolization in whom increasing level of 8 somatic mutations in plasma was detected before imaging diagnosis and the increase of standard biomarker AFP [177].
6.5. Other GI Cancers
In CC, until past years, chemotherapy was the only validated treatment for advanced disease [91,178]. Recently, some targeted therapies emerged in the therapeutic arsenal. Ivosidenib, a first-in-class oral IDH1 inhibitor, has demonstrated an improvement of PFS over placebo in advanced CC with IDH1 mutations in the phase III ClarIDHy study [179]. In another phase II study (NCT-02150967), BGJ398, an orally bioavailable, selective pan-FGFR kinase inhibitor demonstrated clinical activity against chemotherapy-refractory CC with FGFR2 fusions [180]. Lastly, the phase II study FIGHT-202 also supported the efficiency of pemigatinib, an oral inhibitor of FGFR1, 2, and 3 in previously treated patients with cholangiocarcinoma with FGFR2 fusions or rearrangements [181]. Therefore, the interest in monitoring ctDNA in CC is increasing. Goyal et al. already monitored 9 patients with FGFR2 fusions and detected de novo point mutations that conferred resistance to BGJ298 in all patients (n = 3) who underwent progression [182]. Ettrich et al. recently demonstrated that 63% of treatment naïve patients with advanced CC had changes in their mutational profile during chemotherapy. They evaluated and identified a set of 76 potential progression driver genes among a large-scale panel sequencing of 710 cancer-related genes [92]. These data suggest that ctDNA could be used to track disease progression.
In SCCA, few data suggest that HPV ctDNA could be used to monitor the efficacy of immunotherapy as suggested in a recent case report [183].
In GIST, one main application of ctDNA seems to be monitoring response to therapy and tracking therapeutic resistance to tyrosine kinase inhibitors (TKI) [99,131,148,184,185]. Indeed, despite the revolution in GIST management through the contribution of first line TKI such as imatinib targeting KIT or PDGFRA molecular drivers, the majority of GIST will progress with the acquisition of secondary KIT or PDGFRA mutations. In this context, second and third line TKI have been used in some refractory GIST patients [186,187,188,189]. Maier et al. first described a dynamic change in MAF in plasma of advanced GIST under treatment. A decrease or a disappearance of ctDNA occurred in patients responding to TKIs [130]. In other studies, the usefulness of ctDNA for the identification of TKI resistance mutations and their prognostic utility was demonstrated [184,185]. In a phase II study patients with secondary KIT mutations had significantly worse OS than those with no detectable secondary mutations [184]. ctDNA can also be used to detect resistance mutations in other gene than KIT, as demonstrated in a prospective study that collected 30 plasma samples from 22 patients with metastatic GIST [190]. Monitoring ctDNA using NGS patients with GIST under TKI treatment detected primary but also secondary mutations emerging in patients who had a progressive disease whereas only primary mutations were detected in patients with stable disease. These resistance mutations in ctDNA could represent early biomarkers for treatment response [148,191].
7. Conclusions
CtDNA harbors great potential to improve the management of patients with GI cancers. However, the level of advancement of its development in the different tumor types is still inhomogeneous. It seems that the level of data for colorectal cancer might soon allow the use the ctDNA in clinical routine both in adjuvant and in metastatic setting. In pancreatic cancer, the level of proof could be soon reliable but the applications remain more limited for the moment, partially due to the dark prognosis of these tumors and the lack of efficient therapeutic arsenal. In GIST as for CRC, due to the amount of resistance mutations and the frequent use of tyrosine kinase inhibitors, ctDNA appears as a very promising tool. For most other gastro-intestinal cancers, ctDNA shows promising preliminary data but further studies are still needed, and some of them are currently ongoing, to help specifying the exact role of ctDNA in clinical routine.
Lastly, the ctDNA might also be used as a surrogate marker to predict a response to immunotherapy. The mismatch repair deficient (dMMR) or microsatellite instability (MSI) status cannot be directly assessed by liquid biopsy. However, the tumor mutation burden can be determined on plasma and appears to be strongly correlated with dMMR/MSI high status and tumor response to immune checkpoint inhibitors. It is strongly relevant as ICI are becoming a key factor in management of GI cancer and particularly in metastatic CRC cancer in which pembrolizumab recently became the first line reference treatment in MSI-high metastatic CRC.
Author Contributions
Conceptualization, E.M. and A.Z.; Methodology, E.M., A.Z.; Software, E.M.; Validation, E.M., V.T., S.G., A.D., J.T., P.L.-P. and A.Z.; Formal analysis, E.M.; Investigation, E.M.; Resources, E.M., V.T., S.G., A.D., J.T., P.L.-P. and A.Z.; Writing—original draft preparation, E.M., S.G. and A.Z.; Writing—review and editing, E.M., V.T., S.G., A.D., J.T., P.L.-P. and A.Z.; Visualization, E.M.; Supervision, A.Z.; Project administration, A.Z.; Funding acquisition, This research received no external funding. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
E.M. declares a consulting and/or advisory boards for Servier; V.T. declares a consulting and/or advisory boards and co-founder of Emulseo, research Funding for Servier, Fondation Roche; S.G. declares a consulting and/or advisory boards for Lilly and Boehringer Ingelheim; A.D. declares no conflict of interest; J.T. declares a consulting and/or advisory board for Roche, Merck, KGaA, Amgen Lilly, Baxalta, Servier and Sirtex Medical; P.L.-P. declares a personnel fees for a consulting and/or advisory board for Astra Zeneca, Amgen, Biocartis, Pierre Fabre, Sanofi, Roche, Servier, Lilly, research Funding for Biocartis, Servier, Fondation Roche; A.Z. declares a consulting and/or advisory boards for Amgen, Lilly, Merck, Roche, Sanofi, Servier, Baxter, MSD, Pierre Fabre, Havas Life, Alira Health, Zymeworks.
References
- Chan, A.K.C.; Chiu, R.W.K.; Lo, Y.M.D. Clinical Sciences Reviews Committee of the Association of Clinical Biochemists Cell-Free Nucleic Acids in Plasma, Serum and Urine: A New Tool in Molecular Diagnosis. Ann. Clin. Biochem. 2003, 40, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-Free Nucleic Acids as Biomarkers in Cancer Patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the Serum of Cancer Patients and the Effect of Therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Haber, D.A.; Velculescu, V.E. Blood-Based Analyses of Cancer: Circulating Tumor Cells and Circulating Tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct Detection of Early-Stage Cancers Using Circulating Tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef]
- Pekin, D.; Skhiri, Y.; Baret, J.-C.; Le Corre, D.; Mazutis, L.; Salem, C.B.; Millot, F.; El Harrak, A.; Hutchison, J.B.; Larson, J.W.; et al. Quantitative and Sensitive Detection of Rare Mutations Using Droplet-Based Microfluidics. Lab. Chip. 2011, 11, 2156–2166. [Google Scholar] [CrossRef]
- Manoharan, A.; Sambandam, R.; Bhat, V. Recent Technologies Enhancing the Clinical Utility of Circulating Tumor DNA. Clin. Chim. Acta. 2020, 510, 498–506. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Suzuki, T.; Suzuki, T.; Yoshimura, Y.; Yahata, M.; Yew, P.Y.; Nakamura, T.; Nakamura, Y.; Park, J.-H.; Matsuo, R. Detection of Circulating Tumor DNA in Patients of Operative Colorectal and Gastric Cancers. Oncotarget 2020, 11, 3198–3207. [Google Scholar] [CrossRef]
- Huerta, M.; Roselló, S.; Sabater, L.; Ferrer, A.; Tarazona, N.; Roda, D.; Gambardella, V.; Alfaro-Cervelló, C.; Garcés-Albir, M.; Cervantes, A.; et al. Circulating Tumor DNA Detection by Digital-Droplet PCR in Pancreatic Ductal Adenocarcinoma: A Systematic Review. Cancers 2021, 13, 994. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of Oncogenic KRAS in the Diagnosis, Prognosis and Treatment of Pancreatic Cancer. Nat. Rev. Gastroenterol Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Pécuchet, N.; Rozenholc, Y.; Zonta, E.; Pietrasz, D.; Didelot, A.; Combe, P.; Gibault, L.; Bachet, J.-B.; Taly, V.; Fabre, E.; et al. Analysis of Base-Position Error Rate of Next-Generation Sequencing to Detect Tumor Mutations in Circulating DNA. Clin. Chem. 2016, 62, 1492–1503. [Google Scholar] [CrossRef]
- Kou, R.; Lam, H.; Duan, H.; Ye, L.; Jongkam, N.; Chen, W.; Zhang, S.; Li, S. Benefits and Challenges with Applying Unique Molecular Identifiers in Next Generation Sequencing to Detect Low Frequency Mutations. PLoS ONE 2016, 11, e0146638. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of Using Circulating Tumor DNA as a Liquid Biopsy Component in Cancer Management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Lam, S.N.; Zhou, Y.C.; Chan, Y.M.; Foo, C.M.; Lee, P.Y.; Mok, W.Y.; Wong, W.S.; Fung, Y.Y.; Wong, K.Y.; Huang, J.Y.; et al. Comparison of Target Enrichment Platforms for Circulating Tumor DNA Detection. Sci. Rep. 2020, 10, 4124. [Google Scholar] [CrossRef]
- Verma, S.; Moore, M.W.; Ringler, R.; Ghosal, A.; Horvath, K.; Naef, T.; Anvari, S.; Cotter, P.D.; Gunn, S. Analytical Performance Evaluation of a Commercial next Generation Sequencing Liquid Biopsy Platform Using Plasma CtDNA, Reference Standards, and Synthetic Serial Dilution Samples Derived from Normal Plasma. BMC Cancer 2020, 20, 945. [Google Scholar] [CrossRef]
- Thakur, K.; Singh, M.S.; Feldstein-Davydova, S.; Hannes, V.; Hershkovitz, D.; Tsuriel, S. Extracellular Vesicle-Derived DNA vs. CfDNA as a Biomarker for the Detection of Colon Cancer. Genes 2021, 12, 1171. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive Tumour Detection and Classification Using Plasma Cell-Free DNA Methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical Validation of a Targeted Methylation-Based Multi-Cancer Early Detection Test Using an Independent Validation Set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Mathios, D.; Johansen, J.S.; Cristiano, S.; Medina, J.E.; Phallen, J.; Larsen, K.R.; Bruhm, D.C.; Niknafs, N.; Ferreira, L.; Adleff, V.; et al. Detection and Characterization of Lung Cancer Using Cell-Free DNA Fragmentomes. Nat. Commun. 2021, 12, 5060. [Google Scholar] [CrossRef]
- Lamy, P.-J.; van der Leest, P.; Lozano, N.; Becht, C.; Duboeuf, F.; Groen, H.J.M.; Hilgers, W.; Pourel, N.; Rifaela, N.; Schuuring, E.; et al. Mass Spectrometry as a Highly Sensitive Method for Specific Circulating Tumor DNA Analysis in NSCLC: A Comparison Study. Cancers 2020, 12, 3002. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, Y.; Wu, L.; Li, J.; Ji, J.; Yu, Q.; Dai, W.; Feng, J.; Wu, J.; Guo, C. Current Status of CtDNA in Precision Oncology for Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 140. [Google Scholar] [CrossRef]
- Campuzano, S.; Serafín, V.; Gamella, M.; Pedrero, M.; Yáñez-Sedeño, P.; Pingarrón, J.M. Opportunities, Challenges, and Prospects in Electrochemical Biosensing of Circulating Tumor DNA and Its Specific Features. Sensors 2019, 19, 3762. [Google Scholar] [CrossRef] [PubMed]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-Invasive Early Detection of Cancer Four Years before Conventional Diagnosis Using a Blood Test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef]
- Garrigou, S.; Perkins, G.; Garlan, F.; Normand, C.; Didelot, A.; Le Corre, D.; Peyvandi, S.; Mulot, C.; Niarra, R.; Aucouturier, P.; et al. A Study of Hypermethylated Circulating Tumor DNA as a Universal Colorectal Cancer Biomarker. Clin. Chem. 2016, 62, 1129–1139. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 Treatment and RAS Mutations in Colorectal Cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical Validation of the Detection of KRAS and BRAF Mutations from Circulating Tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; Le Corre, D.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex Picodroplet Digital PCR to Detect KRAS Mutations in Circulating DNA from the Plasma of Colorectal Cancer Patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef]
- Bachet, J.B.; Bouché, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS Mutation Analysis in Circulating Tumor DNA from Patients with Metastatic Colorectal Cancer: The AGEO RASANC Prospective Multicenter Study. Ann. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Kleppe, M.; Levine, R.L. Tumor Heterogeneity Confounds and Illuminates: Assessing the Implications. Nat. Med. 2014, 20, 342–344. [Google Scholar] [CrossRef]
- Spindler, K.-L.G.; Pallisgaard, N.; Vogelius, I.; Jakobsen, A. Quantitative Cell-Free DNA, KRAS, and BRAF Mutations in Plasma from Patients with Metastatic Colorectal Cancer during Treatment with Cetuximab and Irinotecan. Clin. Cancer Res. 2012, 18, 1177–1185. [Google Scholar] [CrossRef]
- Spindler, K.-L.G.; Pallisgaard, N.; Appelt, A.L.; Andersen, R.F.; Schou, J.V.; Nielsen, D.; Pfeiffer, P.; Yilmaz, M.; Johansen, J.S.; Hoegdall, E.V.; et al. Clinical Utility of KRAS Status in Circulating Plasma DNA Compared to Archival Tumour Tissue from Patients with Metastatic Colorectal Cancer Treated with Anti-Epidermal Growth Factor Receptor Therapy. Eur. J. Cancer 2015, 51, 2678–2685. [Google Scholar] [CrossRef]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most Human Carcinomas of the Exocrine Pancreas Contain Mutant C-K-Ras Genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef]
- Hruban, R.H.; van Mansfeld, A.D.; Offerhaus, G.J.; van Weering, D.H.; Allison, D.C.; Goodman, S.N.; Kensler, T.W.; Bose, K.K.; Cameron, J.L.; Bos, J.L. K-Ras Oncogene Activation in Adenocarcinoma of the Human Pancreas. A Study of 82 Carcinomas Using a Combination of Mutant-Enriched Polymerase Chain Reaction Analysis and Allele-Specific Oligonucleotide Hybridization. Am. J. Pathol. 1993, 143, 545–554. [Google Scholar]
- Algül, H.; Treiber, M.; Lesina, M.; Schmid, R.M. Mechanisms of Disease: Chronic Inflammation and Cancer in the Pancreas—A Potential Role for Pancreatic Stellate Cells? Nat. Clin. Pract. Gastroenterol Hepatol 2007, 4, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Kim, M.K.; Woo, S.M.; Park, B.; Yoon, K.-A.; Kim, Y.-H.; Joo, J.; Lee, W.J.; Han, S.-S.; Park, S.-J.; Kong, S.-Y. Prognostic Implications of Multiplex Detection of KRAS Mutations in Cell-Free DNA from Patients with Pancreatic Ductal Adenocarcinoma. Clin. Chem. 2018, 64, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.; Cheng, Y.; Zhang, D.; Zhu, S.; Ma, X. Prognostic Value of Circulating Cell-Free DNA in Patients with Pancreatic Cancer: A Systemic Review and Meta-Analysis. Gene 2018, 679, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gupta, S.; Pandey, R.M.; Chauhan, S.S.; Saraya, A. High Levels of Cell-Free Circulating Nucleic Acids in Pancreatic Cancer Are Associated with Vascular Encasement, Metastasis and Poor Survival. Cancer Invest. 2015, 33, 78–85. [Google Scholar] [CrossRef]
- Pietrasz, D.; Pécuchet, N.; Garlan, F.; Didelot, A.; Dubreuil, O.; Doat, S.; Imbert-Bismut, F.; Karoui, M.; Vaillant, J.-C.; Taly, V.; et al. Plasma Circulating Tumor DNA in Pancreatic Cancer Patients Is a Prognostic Marker. Clin. Cancer Res. 2017, 23, 116–123. [Google Scholar] [CrossRef]
- Luchini, C.; Veronese, N.; Nottegar, A.; Cappelletti, V.; Daidone, M.G.; Smith, L.; Parris, C.; Brosens, L.A.A.; Caruso, M.G.; Cheng, L.; et al. Liquid Biopsy as Surrogate for Tissue for Molecular Profiling in Pancreatic Cancer: A Meta-Analysis Towards Precision Medicine. Cancers 2019, 11, 1152. [Google Scholar] [CrossRef]
- Adamo, P.; Cowley, C.M.; Neal, C.P.; Mistry, V.; Page, K.; Dennison, A.R.; Isherwood, J.; Hastings, R.; Luo, J.; Moore, D.A.; et al. Profiling Tumour Heterogeneity through Circulating Tumour DNA in Patients with Pancreatic Cancer. Oncotarget 2017, 8, 87221–87233. [Google Scholar] [CrossRef]
- Berger, A.W.; Schwerdel, D.; Ettrich, T.J.; Hann, A.; Schmidt, S.A.; Kleger, A.; Marienfeld, R.; Seufferlein, T. Targeted Deep Sequencing of Circulating Tumor DNA in Metastatic Pancreatic Cancer. Oncotarget 2018, 9, 2076–2085. [Google Scholar] [CrossRef]
- Park, G.; Park, J.K.; Son, D.-S.; Shin, S.-H.; Kim, Y.J.; Jeon, H.-J.; Lee, J.; Park, W.-Y.; Lee, K.H.; Park, D. Utility of Targeted Deep Sequencing for Detecting Circulating Tumor DNA in Pancreatic Cancer Patients. Sci. Rep. 2018, 8, 11631. [Google Scholar] [CrossRef]
- Eissa, M.A.L.; Lerner, L.; Abdelfatah, E.; Shankar, N.; Canner, J.K.; Hasan, N.M.; Yaghoobi, V.; Huang, B.; Kerner, Z.; Takaesu, F.; et al. Promoter Methylation of ADAMTS1 and BNC1 as Potential Biomarkers for Early Detection of Pancreatic Cancer in Blood. Clin. Epigenetics 2019, 11, 59. [Google Scholar] [CrossRef]
- Rashid, S.; Singh, N.; Gupta, S.; Rashid, S.; Nalika, N.; Sachdev, V.; Bal, C.S.; Datta Gupta, S.; Chauhan, S.S.; Saraya, A. Progression of Chronic Pancreatitis to Pancreatic Cancer: Is There a Role of Gene Mutations as a Screening Tool? Pancreas 2018, 47, 227–232. [Google Scholar] [CrossRef]
- Berger, A.W.; Schwerdel, D.; Costa, I.G.; Hackert, T.; Strobel, O.; Lam, S.; Barth, T.F.; Schröppel, B.; Meining, A.; Büchler, M.W.; et al. Detection of Hot-Spot Mutations in Circulating Cell-Free DNA From Patients with Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology 2016, 151, 267–270. [Google Scholar] [CrossRef]
- Sefrioui, D.; Blanchard, F.; Toure, E.; Basile, P.; Beaussire, L.; Dolfus, C.; Perdrix, A.; Paresy, M.; Antonietti, M.; Iwanicki-Caron, I.; et al. Diagnostic Value of CA19.9, Circulating Tumour DNA and Circulating Tumour Cells in Patients with Solid Pancreatic Tumours. Br. J. Cancer 2017, 117, 1017–1025. [Google Scholar] [CrossRef]
- Berger, A.W.; Schwerdel, D.; Reinacher-Schick, A.; Uhl, W.; Algül, H.; Friess, H.; Janssen, K.-P.; König, A.; Ghadimi, M.; Gallmeier, E.; et al. A Blood-Based Multi Marker Assay Supports the Differential Diagnosis of Early-Stage Pancreatic Cancer. Theranostics 2019, 9, 1280–1287. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Ding, X.-Q.; Zhu, H.; Wang, R.-X.; Pan, X.-R.; Tong, J.-H. KRAS Mutant Allele Fraction in Circulating Cell-Free DNA Correlates with Clinical Stage in Pancreatic Cancer Patients. Front Oncol. 2019, 9, 1295. [Google Scholar] [CrossRef] [PubMed]
- Pectasides, E.; Stachler, M.D.; Derks, S.; Liu, Y.; Maron, S.; Islam, M.; Alpert, L.; Kwak, H.; Kindler, H.; Polite, B.; et al. Genomic Heterogeneity as a Barrier to Precision Medicine in Gastroesophageal Adenocarcinoma. Cancer Discov. 2018, 8, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Frankell, A.M.; Jammula, S.; Li, X.; Contino, G.; Killcoyne, S.; Abbas, S.; Perner, J.; Bower, L.; Devonshire, G.; Ococks, E.; et al. The Landscape of Selection in 551 Esophageal Adenocarcinomas Defines Genomic Biomarkers for the Clinic. Nat. Genet. 2019, 51, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Zang, Z.J.; Ong, C.K.; Cutcutache, I.; Yu, W.; Zhang, S.L.; Huang, D.; Ler, L.D.; Dykema, K.; Gan, A.; Tao, J.; et al. Genetic and Structural Variation in the Gastric Cancer Kinome Revealed through Targeted Deep Sequencing. Cancer Res. 2011, 71, 29–39. [Google Scholar] [CrossRef]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A Comprehensive Survey of Genomic Alterations in Gastric Cancer Reveals Systematic Patterns of Molecular Exclusivity and Co-Occurrence among Distinct Therapeutic Targets. Gut. 2012, 61, 673–684. [Google Scholar] [CrossRef]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired Exome Analysis of Barrett’s Esophagus and Adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef]
- Wong, G.S.; Zhou, J.; Liu, J.B.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J.; et al. Targeting Wild-Type KRAS-Amplified Gastroesophageal Cancer through Combined MEK and SHP2 Inhibition. Nat. Med. 2018, 24, 968–977. [Google Scholar] [CrossRef]
- Kato, S.; Okamura, R.; Baumgartner, J.M.; Patel, H.; Leichman, L.; Kelly, K.; Sicklick, J.K.; Fanta, P.T.; Lippman, S.M.; Kurzrock, R. Analysis of Circulating Tumor DNA and Clinical Correlates in Patients with Esophageal, Gastroesophageal Junction, and Gastric Adenocarcinoma. Clin. Cancer Res. 2018, 24, 6248–6256. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, K.; Xi, H.; Cai, A.; Wu, X.; Cui, J.; Li, J.; Qiao, Z.; Wei, B.; Chen, L. Diagnostic and Prognostic Value of Circulating Tumor DNA in Gastric Cancer: A Meta-Analysis. Oncotarget 2017, 8, 6330–6340. [Google Scholar] [CrossRef]
- Maron, S.B.; Chase, L.M.; Lomnicki, S.; Kochanny, S.; Moore, K.L.; Joshi, S.S.; Landron, S.; Johnson, J.; Kiedrowski, L.A.; Nagy, R.J.; et al. Circulating Tumor DNA Sequencing Analysis of Gastroesophageal Adenocarcinoma. Clin. Cancer Res. 2019, 25, 7098–7112. [Google Scholar] [CrossRef]
- Saluja, H.; Karapetis, C.S.; Pedersen, S.K.; Young, G.P.; Symonds, E.L. The Use of Circulating Tumor DNA for Prognosis of Gastrointestinal Cancers. Front. Oncol. 2018, 8, 275. [Google Scholar] [CrossRef]
- Balgkouranidou, I.; Matthaios, D.; Karayiannakis, A.; Bolanaki, H.; Michailidis, P.; Xenidis, N.; Amarantidis, K.; Chelis, L.; Trypsianis, G.; Chatzaki, E.; et al. Prognostic Role of APC and RASSF1A Promoter Methylation Status in Cell Free Circulating DNA of Operable Gastric Cancer Patients. Mutat. Res. 2015, 778, 46–51. [Google Scholar] [CrossRef]
- Luo, H.; Li, H.; Hu, Z.; Wu, H.; Liu, C.; Li, Y.; Zhang, X.; Lin, P.; Hou, Q.; Ding, G.; et al. Noninvasive Diagnosis and Monitoring of Mutations by Deep Sequencing of Circulating Tumor DNA in Esophageal Squamous Cell Carcinoma. Biochem. Biophys. Res. Commun. 2016, 471, 596–602. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Huang, A.; Zhang, X.; Zhou, S.-L.; Cao, Y.; Huang, X.-W.; Fan, J.; Yang, X.-R.; Zhou, J. Detecting Circulating Tumor DNA in Hepatocellular Carcinoma Patients Using Droplet Digital PCR Is Feasible and Reflects Intratumoral Heterogeneity. J. Cancer 2016, 7, 1907–1914. [Google Scholar] [CrossRef]
- Liao, W.; Yang, H.; Xu, H.; Wang, Y.; Ge, P.; Ren, J.; Xu, W.; Lu, X.; Sang, X.; Zhong, S.; et al. Noninvasive Detection of Tumor-Associated Mutations from Circulating Cell-Free DNA in Hepatocellular Carcinoma Patients by Targeted Deep Sequencing. Oncotarget 2016, 7, 40481–40490. [Google Scholar] [CrossRef]
- Jiao, J.; Watt, G.P.; Stevenson, H.L.; Calderone, T.L.; Fisher-Hoch, S.P.; Ye, Y.; Wu, X.; Vierling, J.M.; Beretta, L. Telomerase Reverse Transcriptase Mutations in Plasma DNA in Patients with Hepatocellular Carcinoma or Cirrhosis: Prevalence and Risk Factors. Hepatol Commun. 2018, 2, 718–731. [Google Scholar] [CrossRef]
- Marchio, A.; Amougou Atsama, M.; Béré, A.; Komas, N.-P.; Noah Noah, D.; Atangana, P.J.A.; Camengo-Police, S.-M.; Njouom, R.; Bekondi, C.; Pineau, P. Droplet Digital PCR Detects High Rate of TP53 R249S Mutants in Cell-Free DNA of Middle African Patients with Hepatocellular Carcinoma. Clin. Exp. Med. 2018, 18, 421–431. [Google Scholar] [CrossRef]
- Jiao, J.; Niu, W.; Wang, Y.; Baggerly, K.; Ye, Y.; Wu, X.; Davenport, D.; Almeda, J.L.; Betancourt-Garcia, M.M.; Forse, R.A.; et al. Prevalence of Aflatoxin-Associated TP53R249S Mutation in Hepatocellular Carcinoma in Hispanics in South Texas. Cancer Prev. Res. 2018, 11, 103–112. [Google Scholar] [CrossRef]
- Ng, C.K.Y.; Di Costanzo, G.G.; Tosti, N.; Paradiso, V.; Coto-Llerena, M.; Roscigno, G.; Perrina, V.; Quintavalle, C.; Boldanova, T.; Wieland, S.; et al. Genetic Profiling Using Plasma-Derived Cell-Free DNA in Therapy-Naïve Hepatocellular Carcinoma Patients: A Pilot Study. Ann. Oncol. 2018, 29, 1286–1291. [Google Scholar] [CrossRef]
- Kisiel, J.B.; Dukek, B.A.; Kanipakam, V.S.R.; Ghoz, H.M.; Yab, T.C.; Berger, C.K.; Taylor, W.R.; Foote, P.H.; Giama, N.H.; Onyirioha, K.; et al. Hepatocellular Carcinoma Detection by Plasma Methylated DNA: Discovery, Phase I Pilot, and Phase II Clinical Validation. Hepatology 2019, 69, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.H.; Lo, Y.M.; Zhang, J.; Liew, C.T.; Ng, M.H.; Wong, N.; Lai, P.B.; Lau, W.Y.; Hjelm, N.M.; Johnson, P.J. Detection of Aberrant P16 Methylation in the Plasma and Serum of Liver Cancer Patients. Cancer Res. 1999, 59, 71–73. [Google Scholar] [PubMed]
- Wong, I.H.; Lo, Y.M.; Yeo, W.; Lau, W.Y.; Johnson, P.J. Frequent P15 Promoter Methylation in Tumor and Peripheral Blood from Hepatocellular Carcinoma Patients. Clin. Cancer Res. 2000, 6, 3516–3521. [Google Scholar] [PubMed]
- Wang, J.; Qin, Y.; Li, B.; Sun, Z.; Yang, B. Detection of Aberrant Promoter Methylation of GSTP1 in the Tumor and Serum of Chinese Human Primary Hepatocellular Carcinoma Patients. Clin. Biochem. 2006, 39, 344–348. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Wu, H.-C.; Shen, J.; Ahsan, H.; Tsai, W.Y.; Yang, H.-I.; Wang, L.-Y.; Chen, S.-Y.; Chen, C.-J.; Santella, R.M. Predicting Hepatocellular Carcinoma by Detection of Aberrant Promoter Methylation in Serum DNA. Clin Cancer Res. 2007, 13, 2378–2384. [Google Scholar] [CrossRef]
- Zhang, C.; Li, J.; Huang, T.; Duan, S.; Dai, D.; Jiang, D.; Sui, X.; Li, D.; Chen, Y.; Ding, F.; et al. Meta-Analysis of DNA Methylation Biomarkers in Hepatocellular Carcinoma. Oncotarget 2016, 7, 81255–81267. [Google Scholar] [CrossRef]
- Chan, K.C.A.; Lai, P.B.S.; Mok, T.S.K.; Chan, H.L.Y.; Ding, C.; Yeung, S.W.; Lo, Y.M.D. Quantitative Analysis of Circulating Methylated DNA as a Biomarker for Hepatocellular Carcinoma. Clin. Chem. 2008, 54, 1528–1536. [Google Scholar] [CrossRef]
- Yeo, W.; Wong, N.; Wong, W.-L.; Lai, P.B.S.; Zhong, S.; Johnson, P.J. High Frequency of Promoter Hypermethylation of RASSF1A in Tumor and Plasma of Patients with Hepatocellular Carcinoma. Liver Int. 2005, 25, 266–272. [Google Scholar] [CrossRef]
- Dong, X.; Hou, Q.; Chen, Y.; Wang, X. Diagnostic Value of the Methylation of Multiple Gene Promoters in Serum in Hepatitis B Virus-Related Hepatocellular Carcinoma. Dis. Markers 2017, 2017, 977. [Google Scholar] [CrossRef]
- Holmila, R.; Sklias, A.; Muller, D.C.; Degli Esposti, D.; Guilloreau, P.; Mckay, J.; Sangrajrang, S.; Srivatanakul, P.; Hainaut, P.; Merle, P.; et al. Targeted Deep Sequencing of Plasma Circulating Cell-Free DNA Reveals Vimentin and Fibulin 1 as Potential Epigenetic Biomarkers for Hepatocellular Carcinoma. PLoS ONE 2017, 12, e0174265. [Google Scholar] [CrossRef]
- Oussalah, A.; Rischer, S.; Bensenane, M.; Conroy, G.; Filhine-Tresarrieu, P.; Debard, R.; Forest-Tramoy, D.; Josse, T.; Reinicke, D.; Garcia, M.; et al. Plasma MSEPT9: A Novel Circulating Cell-Free DNA-Based Epigenetic Biomarker to Diagnose Hepatocellular Carcinoma. EBioMedicine 2018, 30, 138–147. [Google Scholar] [CrossRef]
- Hlady, R.A.; Zhao, X.; Pan, X.; Yang, J.D.; Ahmed, F.; Antwi, S.O.; Giama, N.H.; Patel, T.; Roberts, L.R.; Liu, C.; et al. Genome-Wide Discovery and Validation of Diagnostic DNA Methylation-Based Biomarkers for Hepatocellular Cancer Detection in Circulating Cell Free DNA. Theranostics 2019, 9, 7239–7250. [Google Scholar] [CrossRef]
- Sun, F.-K.; Fan, Y.-C.; Zhao, J.; Zhang, F.; Gao, S.; Zhao, Z.-H.; Sun, Q.; Wang, K. Detection of TFPI2 Methylation in the Serum of Hepatocellular Carcinoma Patients. Dig. Dis. Sci. 2013, 58, 1010–1015. [Google Scholar] [CrossRef]
- Han, L.-Y.; Fan, Y.-C.; Mu, N.-N.; Gao, S.; Li, F.; Ji, X.-F.; Dou, C.-Y.; Wang, K. Aberrant DNA Methylation of G-Protein-Coupled Bile Acid Receptor Gpbar1 (TGR5) Is a Potential Biomarker for Hepatitis B Virus Associated Hepatocellular Carcinoma. Int. J. Med. Sci. 2014, 11, 164–171. [Google Scholar] [CrossRef]
- Ji, X.-F.; Fan, Y.-C.; Gao, S.; Yang, Y.; Zhang, J.-J.; Wang, K. MT1M and MT1G Promoter Methylation as Biomarkers for Hepatocellular Carcinoma. World J. Gastroenterol 2014, 20, 4723–4729. [Google Scholar] [CrossRef]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.C.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational Landscape of Intrahepatic Cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.-C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell. Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef]
- Ettrich, T.J.; Schwerdel, D.; Dolnik, A.; Beuter, F.; Blätte, T.J.; Schmidt, S.A.; Stanescu-Siegmund, N.; Steinacker, J.; Marienfeld, R.; Kleger, A.; et al. Genotyping of Circulating Tumor DNA in Cholangiocarcinoma Reveals Diagnostic and Prognostic Information. Sci. Rep. 2019, 9, 13261. [Google Scholar] [CrossRef]
- Vincent-Salomon, A.; de la Rochefordière, A.; Salmon, R.; Validire, P.; Zafrani, B.; Sastre-Garau, X. Frequent Association of Human Papillomavirus 16 and 18 DNA with Anal Squamous Cell and Basaloid Carcinoma. Mod. Pathol. 1996, 9, 614–620. [Google Scholar]
- Jeannot, E.; Becette, V.; Campitelli, M.; Calméjane, M.-A.; Lappartient, E.; Ruff, E.; Saada, S.; Holmes, A.; Bellet, D.; Sastre-Garau, X. Circulating Human Papillomavirus DNA Detected Using Droplet Digital PCR in the Serum of Patients Diagnosed with Early Stage Human Papillomavirus-Associated Invasive Carcinoma. J. Pathol. Clin. Res. 2016, 2, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Damerla, R.R.; Lee, N.Y.; You, D.; Soni, R.; Shah, R.; Reyngold, M.; Katabi, N.; Wu, V.; McBride, S.M.; Tsai, C.J.; et al. Detection of Early Human Papillomavirus-Associated Cancers by Liquid Biopsy. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef] [PubMed]
- Maleddu, A.; Pantaleo, M.A.; Nannini, M.; Biasco, G. The Role of Mutational Analysis of KIT and PDGFRA in Gastrointestinal Stromal Tumors in a Clinical Setting. J. Transl. Med. 2011, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Fletcher, J.A.; Heinrich, M.C. Biology of Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2004, 22, 3813–3825. [Google Scholar] [CrossRef] [PubMed]
- Angelini, S.; Ravegnini, G.; Fletcher, J.A.; Maffei, F.; Hrelia, P. Clinical Relevance of Pharmacogenetics in Gastrointestinal Stromal Tumor Treatment in the Era of Personalized Therapy. Pharmacy 2013, 14, 941–956. [Google Scholar] [CrossRef]
- Arshad, J.; Roberts, A.; Ahmed, J.; Cotta, J.; Pico, B.A.; Kwon, D.; Trent, J.C. Utility of Circulating Tumor DNA in the Management of Patients With GI Stromal Tumor: Analysis of 243 Patients. JCO Precis. Oncol. 2020. [Google Scholar] [CrossRef]
- Xu, H.; Chen, L.; Shao, Y.; Zhu, D.; Zhi, X.; Zhang, Q.; Li, F.; Xu, J.; Liu, X.; Xu, Z. Clinical Application of Circulating Tumor DNA in the Genetic Analysis of Patients with Advanced GIST. Mol. Cancer Ther. 2018, 17, 290–296. [Google Scholar] [CrossRef]
- Demetri, G.D.; Jeffers, M.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.G.; von Mehren, M.; et al. Mutational Analysis of Plasma DNA from Patients (Pts) in the Phase III GRID Study of Regorafenib (REG) versus Placebo (PL) in Tyrosine Kinase Inhibitor (TKI)-Refractory GIST: Correlating Genotype with Clinical Outcomes. JCO 2013, 31, 10503. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.-L.; Christie, M.; et al. Circulating Tumor DNA Analysis Detects Minimal Residual Disease and Predicts Recurrence in Patients with Stage II Colon Cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Tarazona, N.; Henriksen, T.V.; Carbonell-Asins, J.A.; Reinert, T.; Sharma, S.; Roda, D.; Shchegrova, S.; Huerta, M.; Roselló, S.; Renner, D.; et al. Circulating Tumor DNA to Detect Minimal Residual Disease, Response to Adjuvant Therapy, and Identify Patients at High Risk of Recurrence in Patients with Stage I-III CRC. JCO 2020, 38, 4009. [Google Scholar] [CrossRef]
- Taieb, J.; Taly, V.; Henriques, J.; Bourreau, C.; Mineur, L.; Bennouna, J.; Desrame, J.; Louvet, C.; Lepere, C.; Mabro, M.; et al. Prognostic Value and Relation with Adjuvant Treatment Duration of CtDNA in Stage III Colon Cancer: A Post-Hoc Analysis of the PRODIGE-GERCOR IDEA-France Trial. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Li, L.; Christie, M.; Simons, K.; Elsaleh, H.; Kosmider, S.; Wong, R.; Yip, D.; et al. Serial Circulating Tumour DNA Analysis during Multimodality Treatment of Locally Advanced Rectal Cancer: A Prospective Biomarker Study. Gut 2019, 68, 663–671. [Google Scholar] [CrossRef]
- Loupakis, F.; Derouazi, M.; Murgioni, S.; Rizzato, M.D.; Sharma, S.; Renner, D.; Shchegrova, S.; Sethi, H.; Zimmermann, B.; Aleshin, A.; et al. 405MO Personalized Circulating Tumour DNA Assay for the Detection of Minimal Residual Disease in CRC Patients after Resection of Metastases. Ann. Oncol. 2020, 31, S413. [Google Scholar] [CrossRef]
- Nakano, Y.; Kitago, M.; Matsuda, S.; Nakamura, Y.; Fujita, Y.; Imai, S.; Shinoda, M.; Yagi, H.; Abe, Y.; Hibi, T.; et al. KRAS Mutations in Cell-Free DNA from Preoperative and Postoperative Sera as a Pancreatic Cancer Marker: A Retrospective Study. Br. J. Cancer 2018, 118, 662–669. [Google Scholar] [CrossRef]
- Groot, V.P.; Mosier, S.; Javed, A.A.; Teinor, J.A.; Gemenetzis, G.; Ding, D.; Haley, L.M.; Yu, J.; Burkhart, R.A.; Hasanain, A.; et al. Circulating Tumor DNA as a Clinical Test in Resected Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 4973–4984. [Google Scholar] [CrossRef]
- Azad, T.D.; Chaudhuri, A.A.; Fang, P.; Qiao, Y.; Esfahani, M.S.; Chabon, J.J.; Hamilton, E.G.; Yang, Y.D.; Lovejoy, A.; Newman, A.M.; et al. Circulating Tumor DNA Analysis for Detection of Minimal Residual Disease After Chemoradiotherapy for Localized Esophageal Cancer. Gastroenterology 2020, 158, 494–505. [Google Scholar] [CrossRef]
- Wang, J.; Huang, A.; Wang, Y.-P.; Yin, Y.; Fu, P.-Y.; Zhang, X.; Zhou, J. Circulating Tumor DNA Correlates with Microvascular Invasion and Predicts Tumor Recurrence of Hepatocellular Carcinoma. Ann. Transl. Med. 2020, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Cabel, L.; Jeannot, E.; Bieche, I.; Vacher, S.; Callens, C.; Bazire, L.; Morel, A.; Bernard-Tessier, A.; Chemlali, W.; Schnitzler, A.; et al. Prognostic Impact of Residual HPV CtDNA Detection after Chemoradiotherapy for Anal Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 5767–5771. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- André, T.; de Gramont, A.; Vernerey, D.; Chibaudel, B.; Bonnetain, F.; Tijeras-Raballand, A.; Scriva, A.; Hickish, T.; Tabernero, J.; Van Laethem, J.L.; et al. Adjuvant Fluorouracil, Leucovorin, and Oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF Mutation and Mismatch Repair Status of the MOSAIC Study. J. Clin. Oncol. 2015, 33, 4176–4187. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Hsieh, J.-S.; Chang, M.-Y.; Huang, T.-J.; Chen, F.-M.; Cheng, T.-L.; Alexandersen, K.; Huang, Y.-S.; Tzou, W.-S.; Lin, S.-R. Molecular Detection of APC, K- Ras, and P53 Mutations in the Serum of Colorectal Cancer Patients as Circulating Biomarkers. World J. Surg. 2004, 28, 721–726. [Google Scholar] [CrossRef]
- Parseghian, C.M.; Loree, J.M.; Morris, V.K.; Liu, X.; Clifton, K.K.; Napolitano, S.; Henry, J.T.; Pereira, A.A.; Vilar, E.; Johnson, B.; et al. Anti-EGFR-Resistant Clones Decay Exponentially after Progression: Implications for Anti-EGFR Re-Challenge. Ann. Oncol. 2019, 30, 243–249. [Google Scholar] [CrossRef]
- Takai, E.; Totoki, Y.; Nakamura, H.; Morizane, C.; Nara, S.; Hama, N.; Suzuki, M.; Furukawa, E.; Kato, M.; Hayashi, H.; et al. Clinical Utility of Circulating Tumor DNA for Molecular Assessment in Pancreatic Cancer. Sci. Rep. 2015, 5, 18425. [Google Scholar] [CrossRef]
- Hadano, N.; Murakami, Y.; Uemura, K.; Hashimoto, Y.; Kondo, N.; Nakagawa, N.; Sueda, T.; Hiyama, E. Prognostic Value of Circulating Tumour DNA in Patients Undergoing Curative Resection for Pancreatic Cancer. Br. J. Cancer 2016, 115, 59–65. [Google Scholar] [CrossRef]
- Watanabe, F.; Suzuki, K.; Tamaki, S.; Abe, I.; Endo, Y.; Takayama, Y.; Ishikawa, H.; Kakizawa, N.; Saito, M.; Futsuhara, K.; et al. Longitudinal Monitoring of KRAS-Mutated Circulating Tumor DNA Enables the Prediction of Prognosis and Therapeutic Responses in Patients with Pancreatic Cancer. PLoS ONE 2019, 14, e0227366. [Google Scholar] [CrossRef]
- Lee, J.-S.; Rhee, T.-M.; Pietrasz, D.; Bachet, J.-B.; Laurent-Puig, P.; Kong, S.-Y.; Takai, E.; Yachida, S.; Shibata, T.; Lee, J.W.; et al. Circulating Tumor DNA as a Prognostic Indicator in Resectable Pancreatic Ductal Adenocarcinoma: A Systematic Review and Meta-Analysis. Sci. Rep. 2019, 9, 16971. [Google Scholar] [CrossRef]
- Lee, B.; Lipton, L.; Cohen, J.; Tie, J.; Javed, A.A.; Li, L.; Goldstein, D.; Burge, M.; Cooray, P.; Nagrial, A.; et al. Circulating Tumor DNA as a Potential Marker of Adjuvant Chemotherapy Benefit Following Surgery for Localized Pancreatic Cancer. Ann. Oncol. 2019, 30, 1472–1478. [Google Scholar] [CrossRef]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical Implications of Genomic Alterations in the Tumour and Circulation of Pancreatic Cancer Patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef]
- Ueda, M.; Iguchi, T.; Masuda, T.; Nakahara, Y.; Hirata, H.; Uchi, R.; Niida, A.; Momose, K.; Sakimura, S.; Chiba, K.; et al. Somatic Mutations in Plasma Cell-Free DNA Are Diagnostic Markers for Esophageal Squamous Cell Carcinoma Recurrence. Oncotarget 2016, 7, 62280–62291. [Google Scholar] [CrossRef]
- Meng, P.; Wei, J.; Geng, Y.; Chen, S.; Terpstra, M.M.; Huang, Q.; Zhang, Q.; Su, Z.; Yu, W.; Su, M.; et al. Targeted Sequencing of Circulating Cell-Free DNA in Stage II-III Resectable Oesophageal Squamous Cell Carcinoma Patients. BMC Cancer 2019, 19, 818. [Google Scholar] [CrossRef]
- Tangkijvanich, P.; Hourpai, N.; Rattanatanyong, P.; Wisedopas, N.; Mahachai, V.; Mutirangura, A. Serum LINE-1 Hypomethylation as a Potential Prognostic Marker for Hepatocellular Carcinoma. Clin. Chim. Acta 2007, 379, 127–133. [Google Scholar] [CrossRef]
- Ono, A.; Fujimoto, A.; Yamamoto, Y.; Akamatsu, S.; Hiraga, N.; Imamura, M.; Kawaoka, T.; Tsuge, M.; Abe, H.; Hayes, C.N.; et al. Circulating Tumor DNA Analysis for Liver Cancers and Its Usefulness as a Liquid Biopsy. Cell Mol. Gastroenterol Hepatol 2015, 1, 516–534. [Google Scholar] [CrossRef]
- Li, F.; Qiao, C.-Y.; Gao, S.; Fan, Y.-C.; Chen, L.-Y.; Wang, K. Circulating Cell-Free DNA of Methylated Insulin-like Growth Factor-Binding Protein 7 Predicts a Poor Prognosis in Hepatitis B Virus-Associated Hepatocellular Carcinoma after Hepatectomy. Free Radic. Res. 2018, 52, 455–464. [Google Scholar] [CrossRef]
- An, Y.; Guan, Y.; Xu, Y.; Han, Y.; Wu, C.; Bao, C.; Zhou, B.; Wang, H.; Zhang, M.; Liu, W.; et al. The Diagnostic and Prognostic Usage of Circulating Tumor DNA in Operable Hepatocellular Carcinoma. Am. J. Transl. Res. 2019, 11, 6462–6474. [Google Scholar]
- Liu, X.Y.; Fan, Y.C.; Gao, S.; Zhao, J.; Chen, L.Y.; Li, F.; Wang, K. Methylation of SOX1 and VIM Promoters in Serum as Potential Biomarkers for Hepatocellular Carcinoma. Neoplasma 2017, 64, 745–753. [Google Scholar] [CrossRef]
- Maier, J.; Lange, T.; Kerle, I.; Specht, K.; Bruegel, M.; Wickenhauser, C.; Jost, P.; Niederwieser, D.; Peschel, C.; Duyster, J.; et al. Detection of Mutant Free Circulating Tumor DNA in the Plasma of Patients with Gastrointestinal Stromal Tumor Harboring Activating Mutations of CKIT or PDGFRA. Clin. Cancer Res. 2013, 19, 4854–4867. [Google Scholar] [CrossRef]
- Kang, G.; Sohn, B.S.; Pyo, J.-S.; Kim, J.Y.; Lee, B.; Kim, K.-M. Detecting Primary KIT Mutations in Presurgical Plasma of Patients with Gastrointestinal Stromal Tumor. Mol. Diagn. Ther. 2016, 20, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating Tumor DNA as an Early Marker of Therapeutic Response in Patients with Metastatic Colorectal Cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Osumi, H.; Shinozaki, E.; Takeda, Y.; Wakatsuki, T.; Ichimura, T.; Saiura, A.; Yamaguchi, K.; Takahashi, S.; Noda, T.; Zembutsu, H. Clinical Relevance of Circulating Tumor DNA Assessed through Deep Sequencing in Patients with Metastatic Colorectal Cancer. Cancer Med. 2019, 8, 408–417. [Google Scholar] [CrossRef]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma CtDNA RAS Mutation Analysis for the Diagnosis and Treatment Monitoring of Metastatic Colorectal Cancer Patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef]
- Spindler, K.G.; Appelt, A.L.; Pallisgaard, N.; Andersen, R.F.; Jakobsen, A. KRAS-Mutated Plasma DNA as Predictor of Outcome from Irinotecan Monotherapy in Metastatic Colorectal Cancer. Br. J. Cancer 2013, 109, 3067–3072. [Google Scholar] [CrossRef]
- El Messaoudi, S.; Mouliere, F.; Du Manoir, S.; Bascoul-Mollevi, C.; Gillet, B.; Nouaille, M.; Fiess, C.; Crapez, E.; Bibeau, F.; Theillet, C.; et al. Circulating DNA as a Strong Multimarker Prognostic Tool for Metastatic Colorectal Cancer Patient Management Care. Clin. Cancer Res. 2016, 22, 3067–3077. [Google Scholar] [CrossRef]
- Tabernero, J.; Lenz, H.-J.; Siena, S.; Sobrero, A.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Analysis of Circulating DNA and Protein Biomarkers to Predict the Clinical Activity of Regorafenib and Assess Prognosis in Patients with Metastatic Colorectal Cancer: A Retrospective, Exploratory Analysis of the CORRECT Trial. Lancet Oncol. 2015, 16, 937–948. [Google Scholar] [CrossRef]
- Wong, A.L.A.; Lim, J.S.J.; Sinha, A.; Gopinathan, A.; Lim, R.; Tan, C.-S.; Soh, T.; Venkatesh, S.; Titin, C.; Sapari, N.S.; et al. Tumour Pharmacodynamics and Circulating Cell Free DNA in Patients with Refractory Colorectal Carcinoma Treated with Regorafenib. J. Transl. Med. 2015, 13, 57. [Google Scholar] [CrossRef]
- Garlan, F.; Laurent-Puig, P.; Sefrioui, D.; Siauve, N.; Didelot, A.; Sarafan-Vasseur, N.; Michel, P.; Perkins, G.; Mulot, C.; Blons, H.; et al. Early Evaluation of Circulating Tumor DNA as Marker of Therapeutic Efficacy in Metastatic Colorectal Cancer Patients (PLACOL Study). Clin. Cancer Res. 2017, 23, 5416–5425. [Google Scholar] [CrossRef]
- Basnet, S.; Zhang, Z.-Y.; Liao, W.-Q.; Li, S.-H.; Li, P.-S.; Ge, H.-Y. The Prognostic Value of Circulating Cell-Free DNA in Colorectal Cancer: A Meta-Analysis. J. Cancer 2016, 7, 1105–1113. [Google Scholar] [CrossRef]
- Strijker, M.; Soer, E.C.; de Pastena, M.; Creemers, A.; Balduzzi, A.; Beagan, J.J.; Busch, O.R.; van Delden, O.M.; Halfwerk, H.; van Hooft, J.E.; et al. Circulating Tumor DNA Quantity Is Related to Tumor Volume and Both Predict Survival in Metastatic Pancreatic Ductal Adenocarcinoma. Int. J. Cancer 2020, 146, 1445–1456. [Google Scholar] [CrossRef]
- Chen, H.; Tu, H.; Meng, Z.Q.; Chen, Z.; Wang, P.; Liu, L.M. K-Ras Mutational Status Predicts Poor Prognosis in Unresectable Pancreatic Cancer. Eur. J. Surg. Oncol. 2010, 36, 657–662. [Google Scholar] [CrossRef]
- Tjensvoll, K.; Lapin, M.; Buhl, T.; Oltedal, S.; Steen-Ottosen Berry, K.; Gilje, B.; Søreide, J.A.; Javle, M.; Nordgård, O.; Smaaland, R. Clinical Relevance of Circulating KRAS Mutated DNA in Plasma from Patients with Advanced Pancreatic Cancer. Mol. Oncol. 2016, 10, 635–643. [Google Scholar] [CrossRef]
- Xu, R.-H.; Wei, W.; Krawczyk, M.; Wang, W.; Luo, H.; Flagg, K.; Yi, S.; Shi, W.; Quan, Q.; Li, K.; et al. Circulating Tumour DNA Methylation Markers for Diagnosis and Prognosis of Hepatocellular Carcinoma. Nat. Mater. 2017, 16, 1155–1161. [Google Scholar] [CrossRef]
- Alunni-Fabbroni, M.; Rönsch, K.; Huber, T.; Cyran, C.C.; Seidensticker, M.; Mayerle, J.; Pech, M.; Basu, B.; Verslype, C.; Benckert, J.; et al. Circulating DNA as Prognostic Biomarker in Patients with Advanced Hepatocellular Carcinoma: A Translational Exploratory Study from the SORAMIC Trial. J. Transl. Med. 2019, 17, 328. [Google Scholar] [CrossRef]
- Bernard-Tessier, A.; Jeannot, E.; Guenat, D.; Debernardi, A.; Michel, M.; Proudhon, C.; Vincent-Salomon, A.; Bièche, I.; Pierga, J.-Y.; Buecher, B.; et al. Clinical Validity of HPV Circulating Tumor DNA in Advanced Anal Carcinoma: An Ancillary Study to the Epitopes-HPV02 Trial. Clin. Cancer Res. 2019, 25, 2109–2115. [Google Scholar] [CrossRef]
- Namløs, H.M.; Boye, K.; Mishkin, S.J.; Barøy, T.; Lorenz, S.; Bjerkehagen, B.; Stratford, E.W.; Munthe, E.; Kudlow, B.A.; Myklebost, O.; et al. Noninvasive Detection of CtDNA Reveals Intratumor Heterogeneity and Is Associated with Tumor Burden in Gastrointestinal Stromal Tumor. Mol. Cancer Ther. 2018, 17, 2473–2480. [Google Scholar] [CrossRef]
- Santini, D.; Fratto, M.E.; Spoto, C.; Russo, A.; Galluzzo, S.; Zoccoli, A.; Vincenzi, B.; Tonini, G. Cetuximab in Small Bowel Adenocarcinoma: A New Friend? Br. J. Cancer 2010, 103, 1305. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS Mutations and Acquired Resistance to Anti-EGFR Therapy in Colorectal Cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef]
- Diaz, L.A.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The Molecular Evolution of Acquired Resistance to Targeted EGFR Blockade in Colorectal Cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal Evolution and Resistance to EGFR Blockade in the Blood of Colorectal Cancer Patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for Patients with RAS and BRAF Wild-Type Metastatic Colorectal Cancer With Acquired Resistance to First-Line Cetuximab and Irinotecan. JAMA Oncol 2019, 5, 343–350. [Google Scholar] [CrossRef]
- Nakamura, M. MO3-12-5—Phase II Study of Cetuximab Rechallenge in Patients with RAS Wild-Type Metastatic Colorectal Cancer: E-Rechallenge Trial. Ann. Oncol. 2019, 30, 343–350. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Fenocchio, E.; Amatu, A.; Corallo, S.; Manai, C.; Tosi, F.; et al. Phase II Study of Anti-EGFR Rechallenge Therapy with Panitumumab Driven by Circulating Tumor DNA Molecular Selection in Metastatic Colorectal Cancer: The CHRONOS Trial. JCO 2021, 39, 3506. [Google Scholar] [CrossRef]
- Mohan, S.; Heitzer, E.; Ulz, P.; Lafer, I.; Lax, S.; Auer, M.; Pichler, M.; Gerger, A.; Eisner, F.; Hoefler, G.; et al. Changes in Colorectal Carcinoma Genomes under Anti-EGFR Therapy Identified by Whole-Genome Plasma DNA Sequencing. PLoS Genet. 2014, 10, e1004271. [Google Scholar] [CrossRef]
- Valtorta, E.; Misale, S.; Sartore-Bianchi, A.; Nagtegaal, I.D.; Paraf, F.; Lauricella, C.; Dimartino, V.; Hobor, S.; Jacobs, B.; Ercolani, C.; et al. KRAS Gene Amplification in Colorectal Cancer and Impact on Response to EGFR-Targeted Therapy. Int. J. Cancer 2013, 133, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET Receptor Drives Resistance to Anti-EGFR Therapies in Colorectal Cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Castillo, C.F.F.-D. A Changing Landscape in Pancreatic Cancer. Ann. Surg. 2018, 268, 9–10. [Google Scholar] [CrossRef]
- Cheng, H.; Liu, C.; Jiang, J.; Luo, G.; Lu, Y.; Jin, K.; Guo, M.; Zhang, Z.; Xu, J.; Liu, L.; et al. Analysis of CtDNA to Predict Prognosis and Monitor Treatment Responses in Metastatic Pancreatic Cancer Patients. Int. J. Cancer 2017, 140, 2344–2350. [Google Scholar] [CrossRef]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated With Outcomes of Patients With Pancreatic Cancer. Gastroenterol 2019, 156, 108–118.e4. [Google Scholar] [CrossRef]
- Del Re, M.; Vivaldi, C.; Rofi, E.; Vasile, E.; Miccoli, M.; Caparello, C.; d’Arienzo, P.D.; Fornaro, L.; Falcone, A.; Danesi, R. Early Changes in Plasma DNA Levels of Mutant KRAS as a Sensitive Marker of Response to Chemotherapy in Pancreatic Cancer. Sci. Rep. 2017, 7, 7931. [Google Scholar] [CrossRef]
- Kruger, S.; Heinemann, V.; Ross, C.; Diehl, F.; Nagel, D.; Ormanns, S.; Liebmann, S.; Prinz-Bravin, I.; Westphalen, C.B.; Haas, M.; et al. Repeated MutKRAS CtDNA Measurements Represent a Novel and Promising Tool for Early Response Prediction and Therapy Monitoring in Advanced Pancreatic Cancer. Ann. Oncol. 2018, 29, 2348–2355. [Google Scholar] [CrossRef]
- Perets, R.; Greenberg, O.; Shentzer, T.; Semenisty, V.; Epelbaum, R.; Bick, T.; Sarji, S.; Ben-Izhak, O.; Sabo, E.; Hershkovitz, D. Mutant KRAS Circulating Tumor DNA Is an Accurate Tool for Pancreatic Cancer Monitoring. Oncologist 2018, 23, 566–572. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef]
- Bachet, J.-B.; Blons, H.; Hammel, P.; Hariry, I.E.; Portales, F.; Mineur, L.; Metges, J.-P.; Mulot, C.; Bourreau, C.; Cain, J.; et al. Circulating Tumor DNA Is Prognostic and Potentially Predictive of Eryaspase Efficacy in Second-Line in Patients with Advanced Pancreatic Adenocarcinoma. Clin. Cancer Res. 2020, 26, 5208–5216. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in Combination with Chemotherapy versus Chemotherapy Alone for Treatment of HER2-Positive Advanced Gastric or Gastro-Oesophageal Junction Cancer (ToGA): A Phase 3, Open-Label, Randomised Controlled Trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Shoda, K.; Masuda, K.; Ichikawa, D.; Arita, T.; Miyakami, Y.; Watanabe, M.; Konishi, H.; Imoto, I.; Otsuji, E. HER2 Amplification Detected in the Circulating DNA of Patients with Gastric Cancer: A Retrospective Pilot Study. Gastric Cancer 2015, 18, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Shoda, K.; Ichikawa, D.; Fujita, Y.; Masuda, K.; Hiramoto, H.; Hamada, J.; Arita, T.; Konishi, H.; Komatsu, S.; Shiozaki, A.; et al. Monitoring the HER2 Copy Number Status in Circulating Tumor DNA by Droplet Digital PCR in Patients with Gastric Cancer. Gastric Cancer 2017, 20, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Banks, K.C.; Pectasides, E.; Kim, S.Y.; Kim, K.; Lanman, R.B.; Talasaz, A.; An, J.; Choi, M.G.; Lee, J.H.; et al. Impact of Genomic Alterations on Lapatinib Treatment Outcome and Cell-Free Genomic Landscape during HER2 Therapy in HER2+ Gastric Cancer Patients. Ann. Oncol. 2018, 29, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.-X.; Chen, G.; Zeng, Y.-Y.; Dong, X.-Q.; Lin, M.-J.; Huang, X.-H.; Zhang, D.; Liu, X.-L.; Liu, J.-F. Circulating Tumor DNA Profiling Reveals Clonal Evolution and Real-Time Disease Progression in Advanced Hepatocellular Carcinoma. Int. J. Cancer 2017, 141, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. ABC-06 | A Randomised Phase III, Multi-Centre, Open-Label Study of Active Symptom Control (ASC) Alone or ASC with Oxaliplatin / 5-FU Chemotherapy (ASC+mFOLFOX) for Patients (Pts) with Locally Advanced / Metastatic Biliary Tract Cancers (ABC) Previously-Treated with Cisplatin/Gemcitabine (CisGem) Chemotherapy. JCO 2019, 37, 4003. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-Mutant, Chemotherapy-Refractory Cholangiocarcinoma (ClarIDHy): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Study. Lancet Oncol 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for Previously Treated, Locally Advanced or Metastatic Cholangiocarcinoma: A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef]
- Cabel, L.; Bidard, F.-C.; Servois, V.; Cacheux, W.; Mariani, P.; Romano, E.; Minsat, M.; Bieche, I.; Farkhondeh, F.; Jeannot, E.; et al. HPV Circulating Tumor DNA to Monitor the Efficacy of Anti-PD-1 Therapy in Metastatic Squamous Cell Carcinoma of the Anal Canal: A Case Report. Int. J. Cancer 2017, 141, 1667–1670. [Google Scholar] [CrossRef]
- Yoo, C.; Ryu, M.-H.; Na, Y.S.; Ryoo, B.-Y.; Park, S.R.; Kang, Y.-K. Analysis of Serum Protein Biomarkers, Circulating Tumor DNA, and Dovitinib Activity in Patients with Tyrosine Kinase Inhibitor-Refractory Gastrointestinal Stromal Tumors. Ann. Oncol. 2014, 25, 2272–2277. [Google Scholar] [CrossRef]
- Wada, N.; Kurokawa, Y.; Takahashi, T.; Hamakawa, T.; Hirota, S.; Naka, T.; Miyazaki, Y.; Makino, T.; Yamasaki, M.; Nakajima, K.; et al. Detecting Secondary C-KIT Mutations in the Peripheral Blood of Patients with Imatinib-Resistant Gastrointestinal Stromal Tumor. Oncology 2016, 90, 112–117. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and Safety of Sunitinib in Patients with Advanced Gastrointestinal Stromal Tumour after Failure of Imatinib: A Randomised Controlled Trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Ravegnini, G.; Nannini, M.; Zenesini, C.; Simeon, V.; Sammarini, G.; Urbini, M.; Gatto, L.; Saponara, M.; Biasco, G.; Pantaleo, M.A.; et al. An Exploratory Association of Polymorphisms in Angiogenesis-Related Genes with Susceptibility, Clinical Response and Toxicity in Gastrointestinal Stromal Tumors Receiving Sunitinib after Imatinib Failure. Angiogenesis 2017, 20, 139–148. [Google Scholar] [CrossRef]
- George, S.; Wang, Q.; Heinrich, M.C.; Corless, C.L.; Zhu, M.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; Choy, E.; Tap, W.D.; et al. Efficacy and Safety of Regorafenib in Patients with Metastatic and/or Unresectable GI Stromal Tumor after Failure of Imatinib and Sunitinib: A Multicenter Phase II Trial. J. Clin. Oncol. 2012, 30, 2401–2407. [Google Scholar] [CrossRef]
- Ravegnini, G.; Nannini, M.; Sammarini, G.; Astolfi, A.; Biasco, G.; Pantaleo, M.A.; Hrelia, P.; Angelini, S. Personalized Medicine in Gastrointestinal Stromal Tumor (GIST): Clinical Implications of the Somatic and Germline DNA Analysis. Int. J. Mol. Sci. 2015, 16, 15592–15608. [Google Scholar] [CrossRef]
- Bauer, S.; Herold, T.; Mühlenberg, T.; Reis, A.-C.; Falkenhorst, J.; Backs, M.; Ketzer, J.; Breitenbuecher, F.; Schuler, M.H.; Grunewald, S. Plasma Sequencing to Detect a Multitude of Secondary KIT Resistance Mutations in Metastatic Gastrointestinal Stromal Tumors (GIST). JCO 2015, 33, 10518. [Google Scholar] [CrossRef]
- Kang, G.; Bae, B.N.; Sohn, B.S.; Pyo, J.-S.; Kang, G.H.; Kim, K.-M. Detection of KIT and PDGFRA Mutations in the Plasma of Patients with Gastrointestinal Stromal Tumor. Target Oncol. 2015, 10, 597–601. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).