
Helicobacter pylori CagA Induces Cortactin Y-470 Phosphorylation-Dependent Gastric Epithelial Cell Scattering via Abl, Vav2 and Rac1 Activation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Bacteria and Inhibitors
2.2. Cell Fractionation, Immunoprecipitation and Western Blotting
2.3. Plasmids, Mutagenesis and Transfection of DNA and siRNA
2.4. Wound Healing Assay
2.5. Rac1 GTPase Activity Assay
2.6. Live Cell Imaging
2.7. Statistical Analysis
3. Results
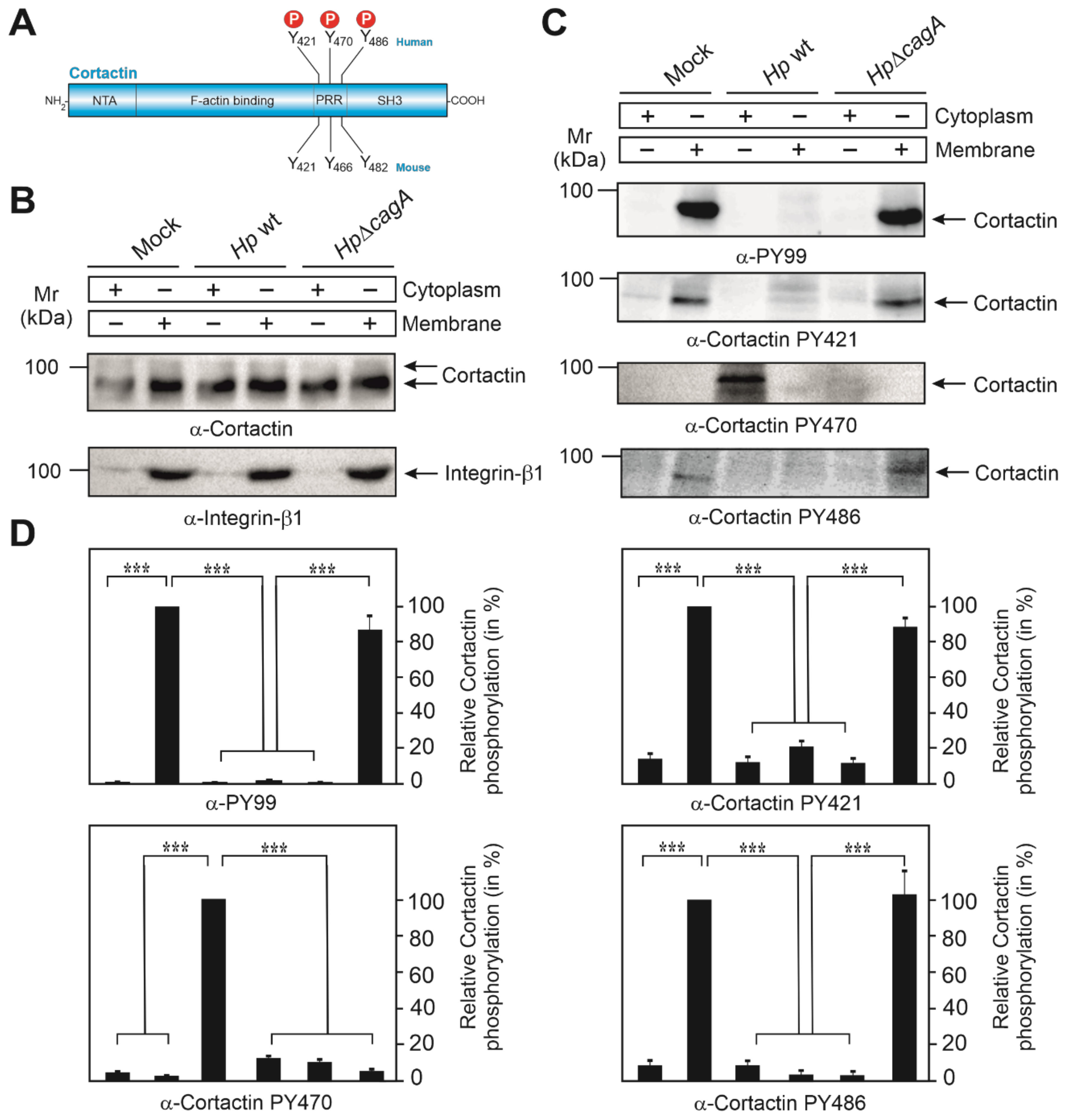
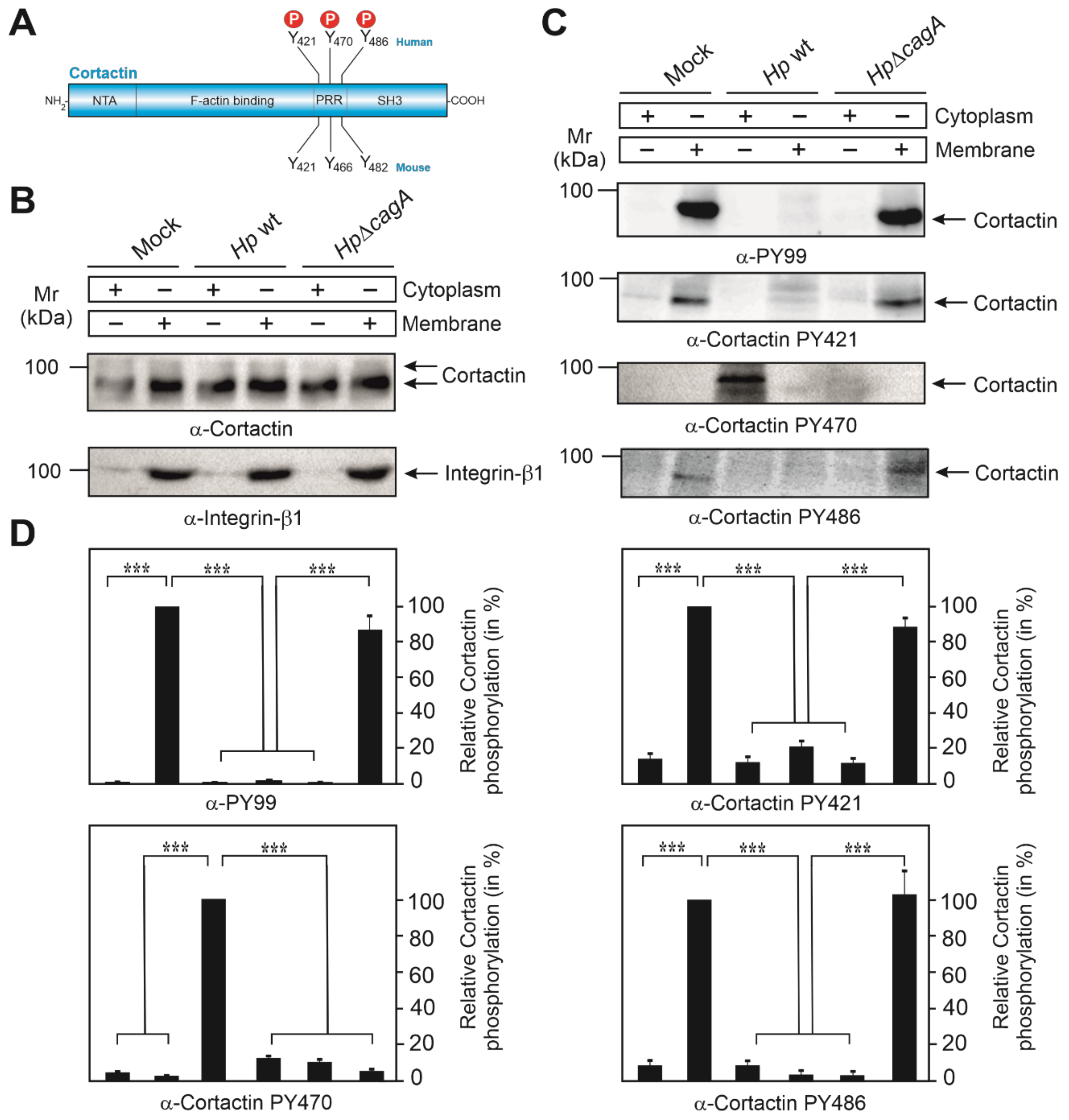
3.1. Subcellular Localization of Phosphotyrosine-Cortactin after Infection with H. pylori
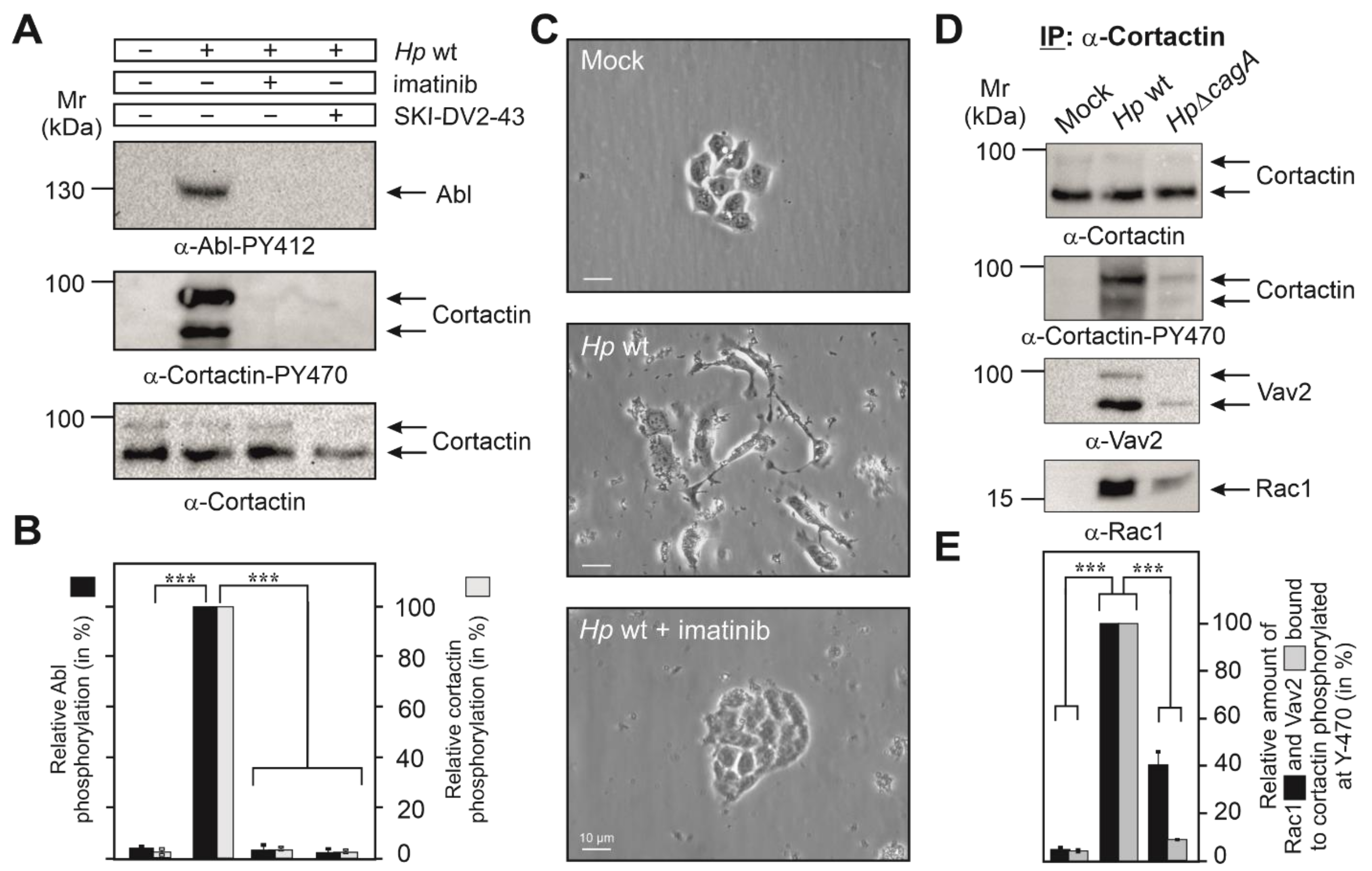
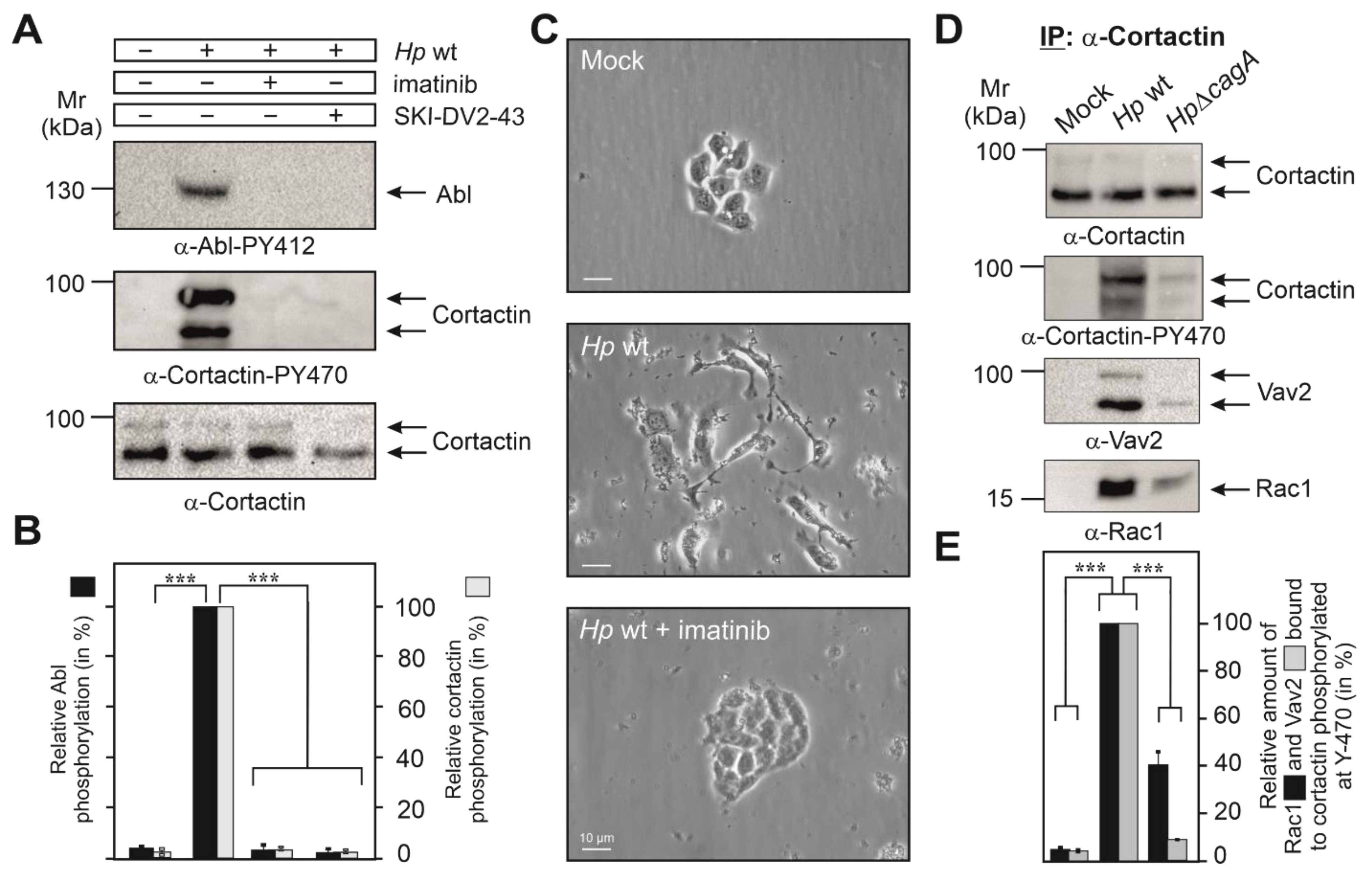
3.2. Cortactin Y-470 Is Phosphorylated by Activated Abl Kinase, Correlating with Cell Scattering
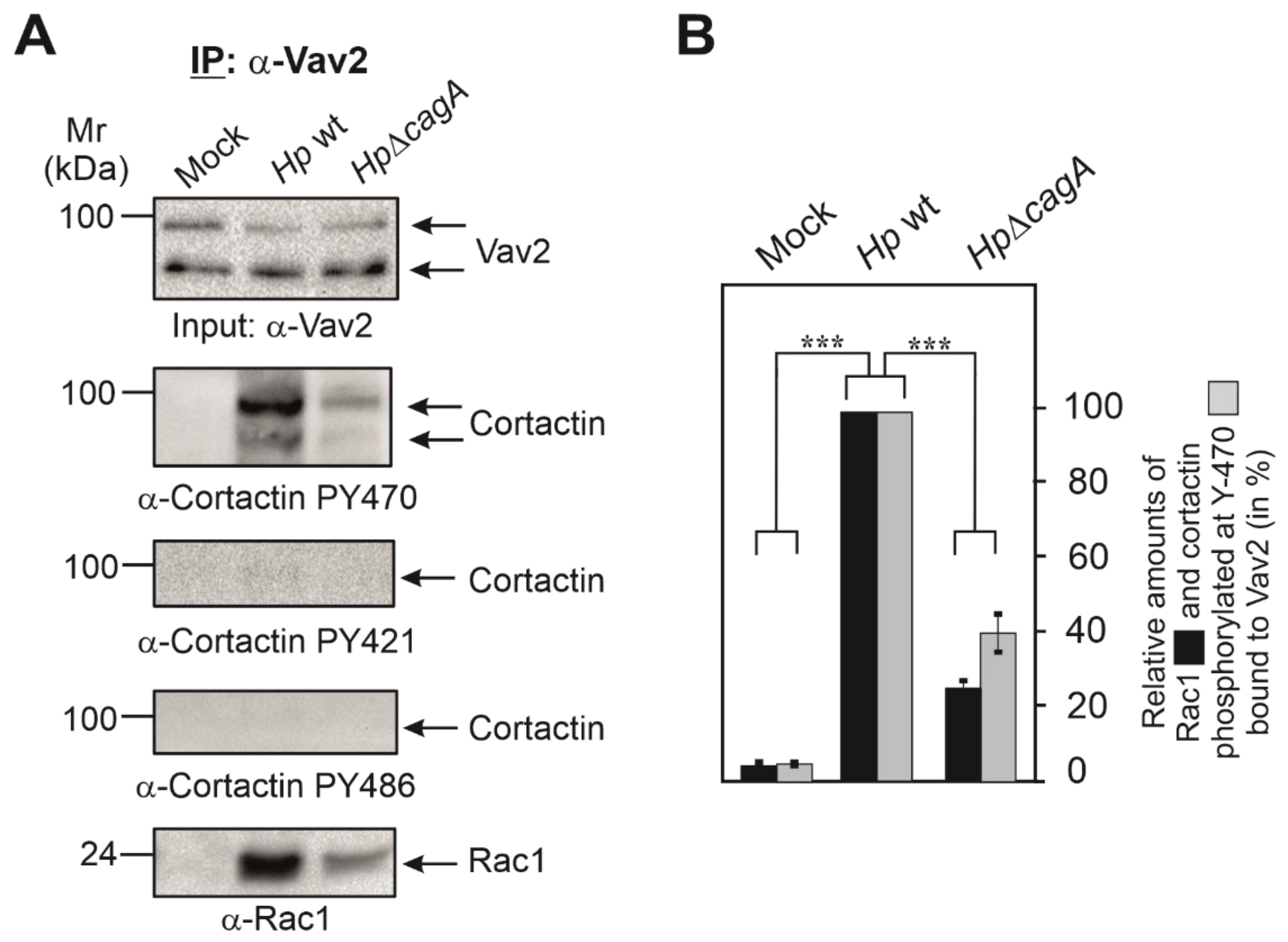
3.3. Cortactin PY-470 Recruits the GEF Vav2 and Small GTPase Rac1 during Infection
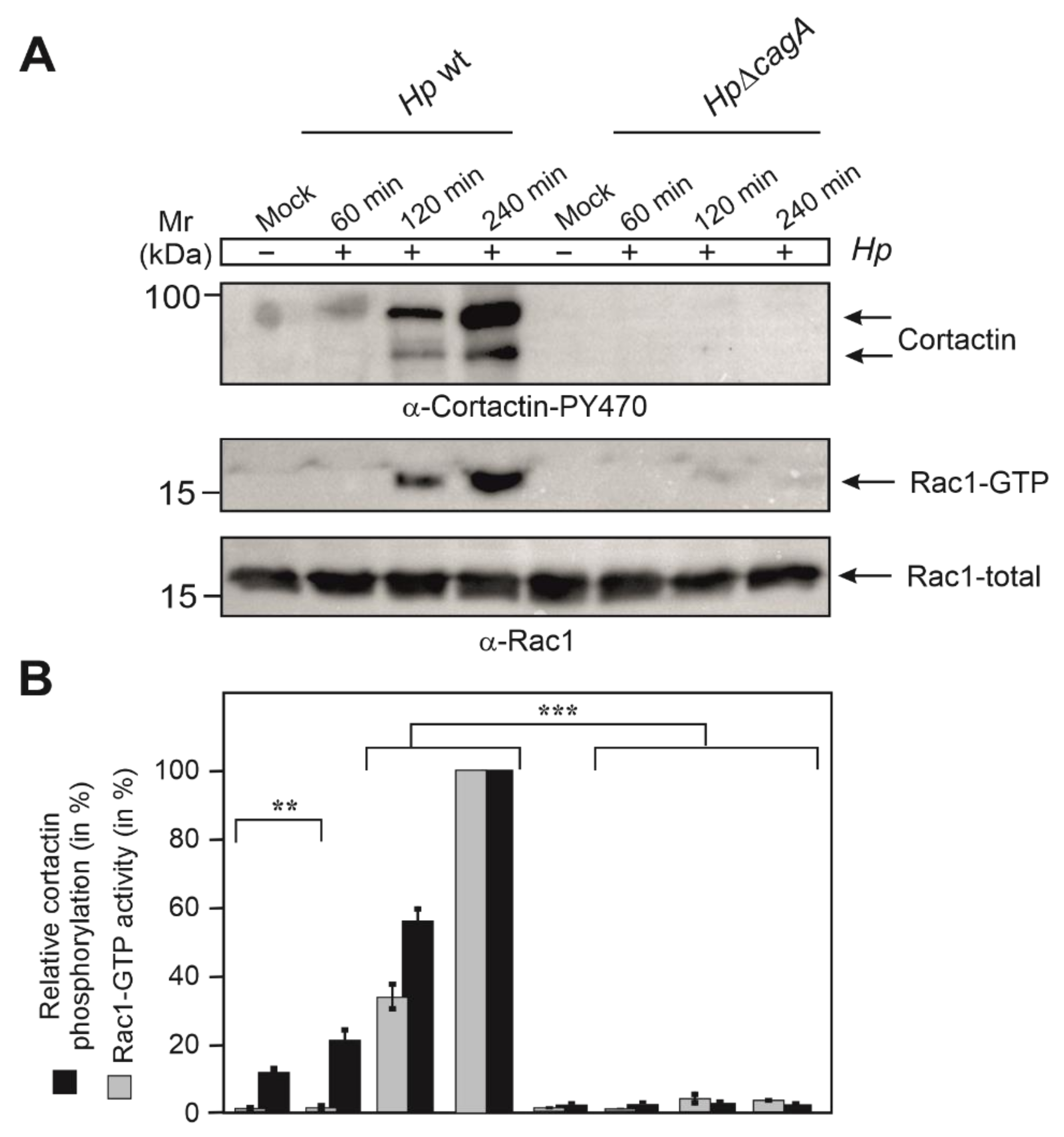
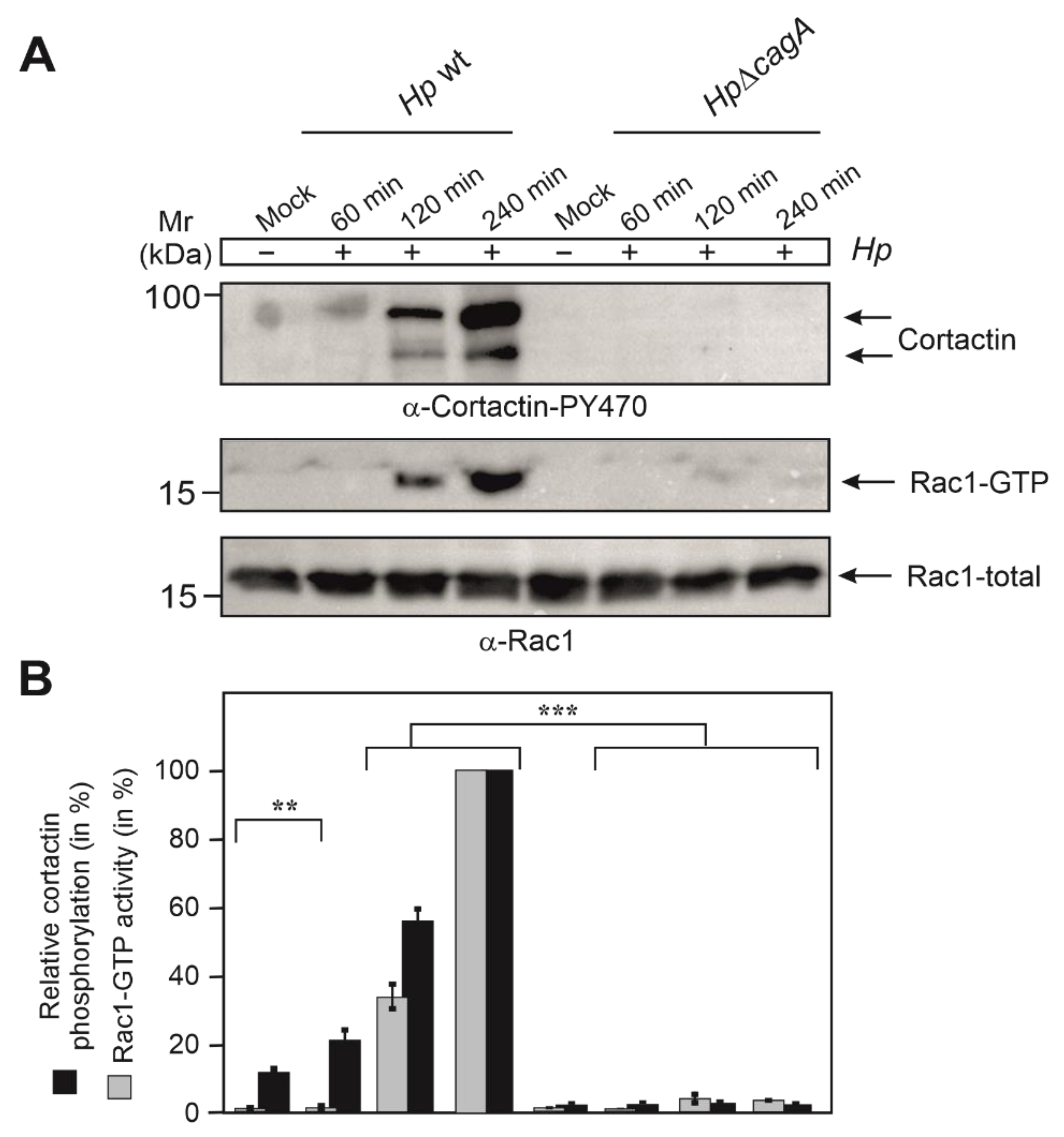
3.4. Phosphorylation of Cortactin at Y-470 Is Associated with Increased Rac1-GTP Levels
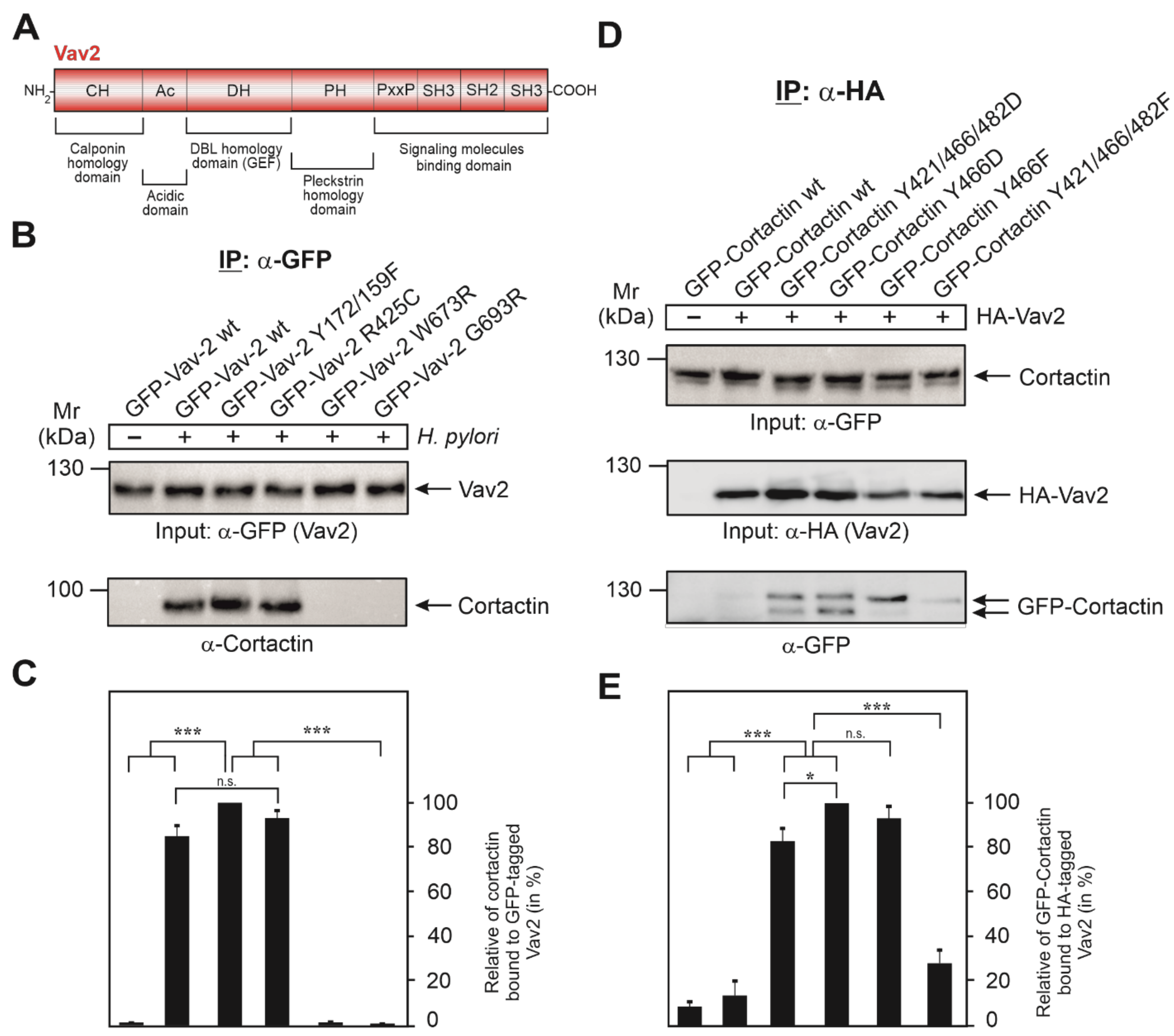
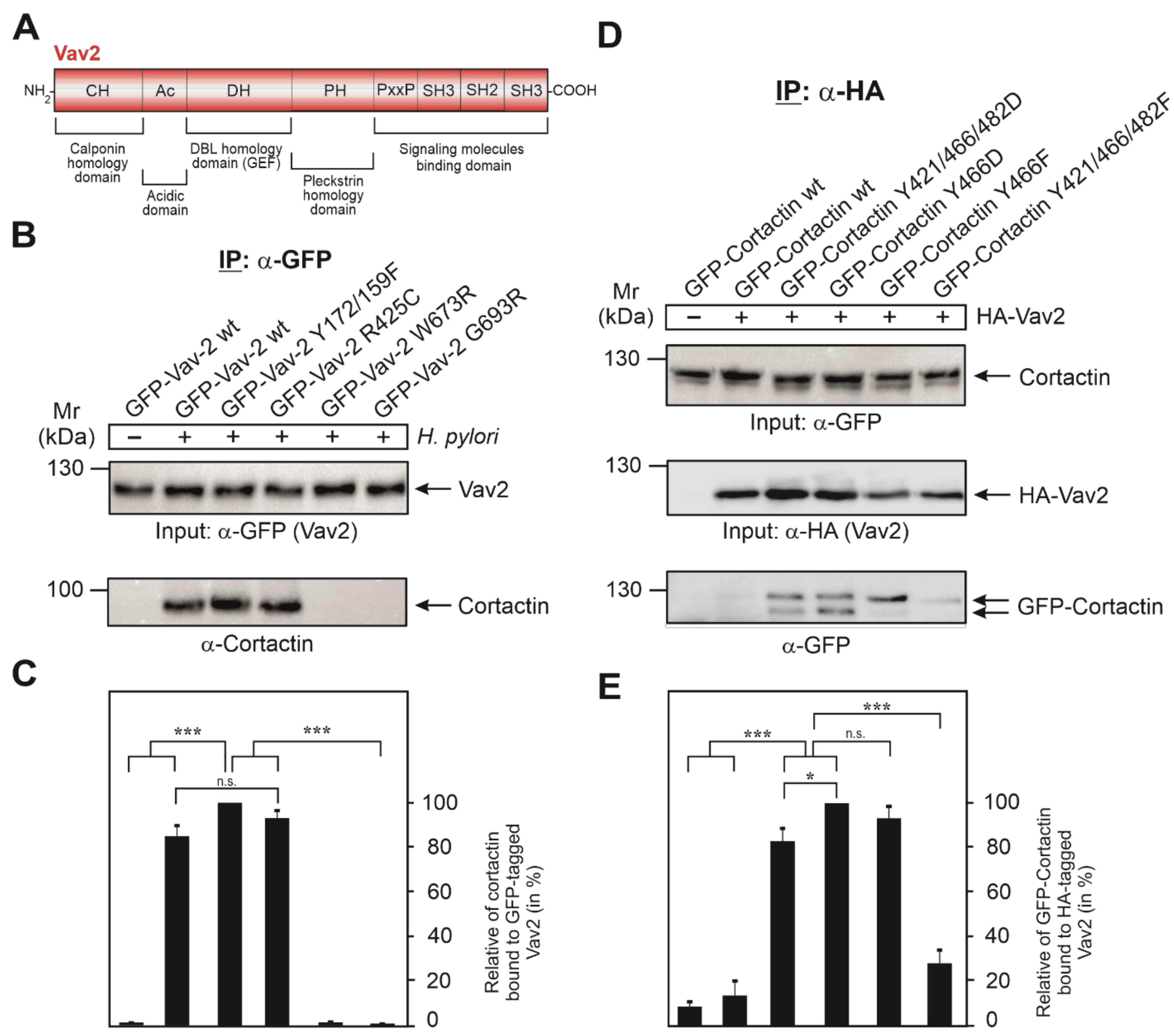
3.5. Vav2 Binds to Cortactin via Its SH2 Domain
3.6. Expression of GFP-Cortactin Point Mutants Confirms Its Requirements for Vav2 Binding
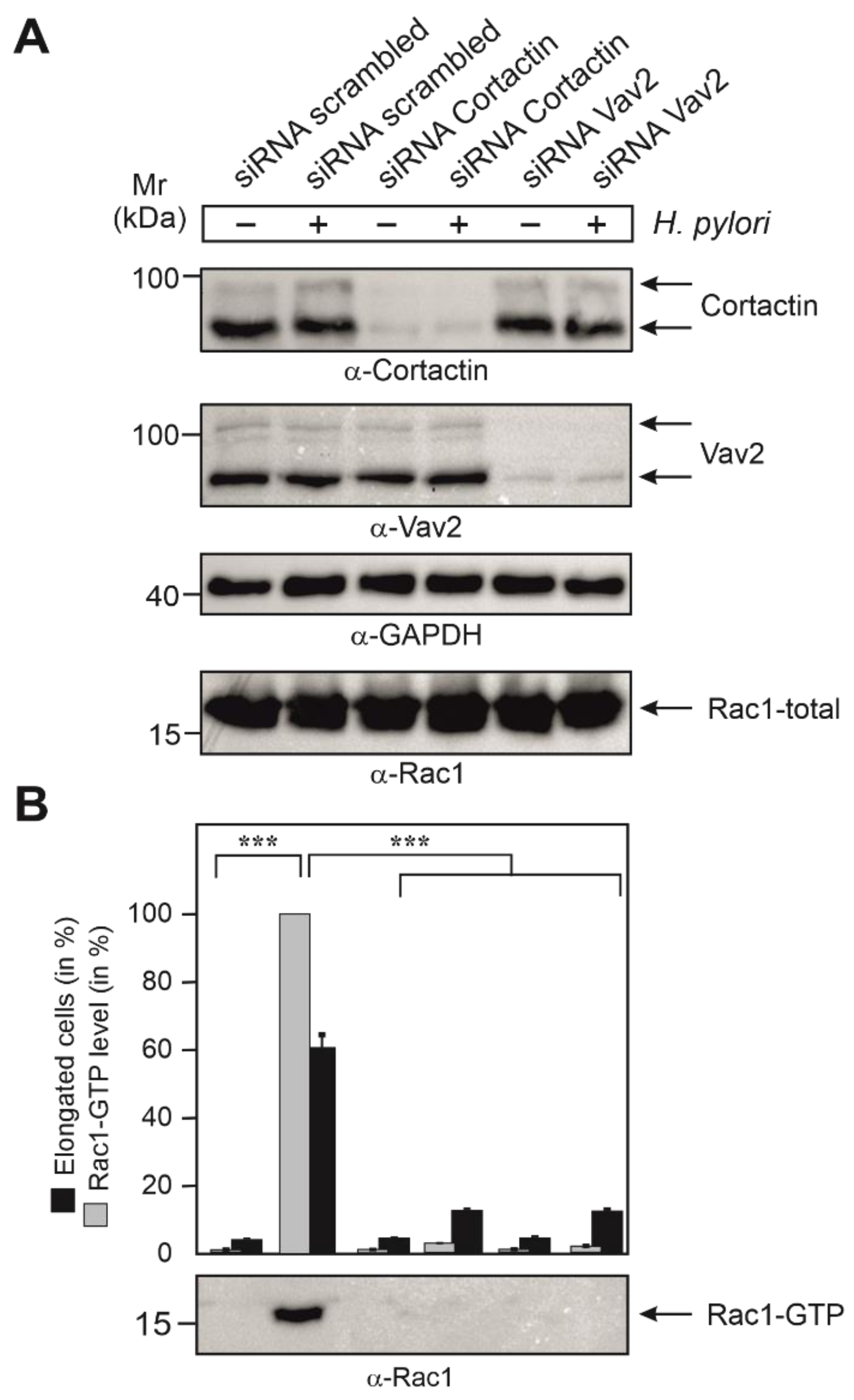
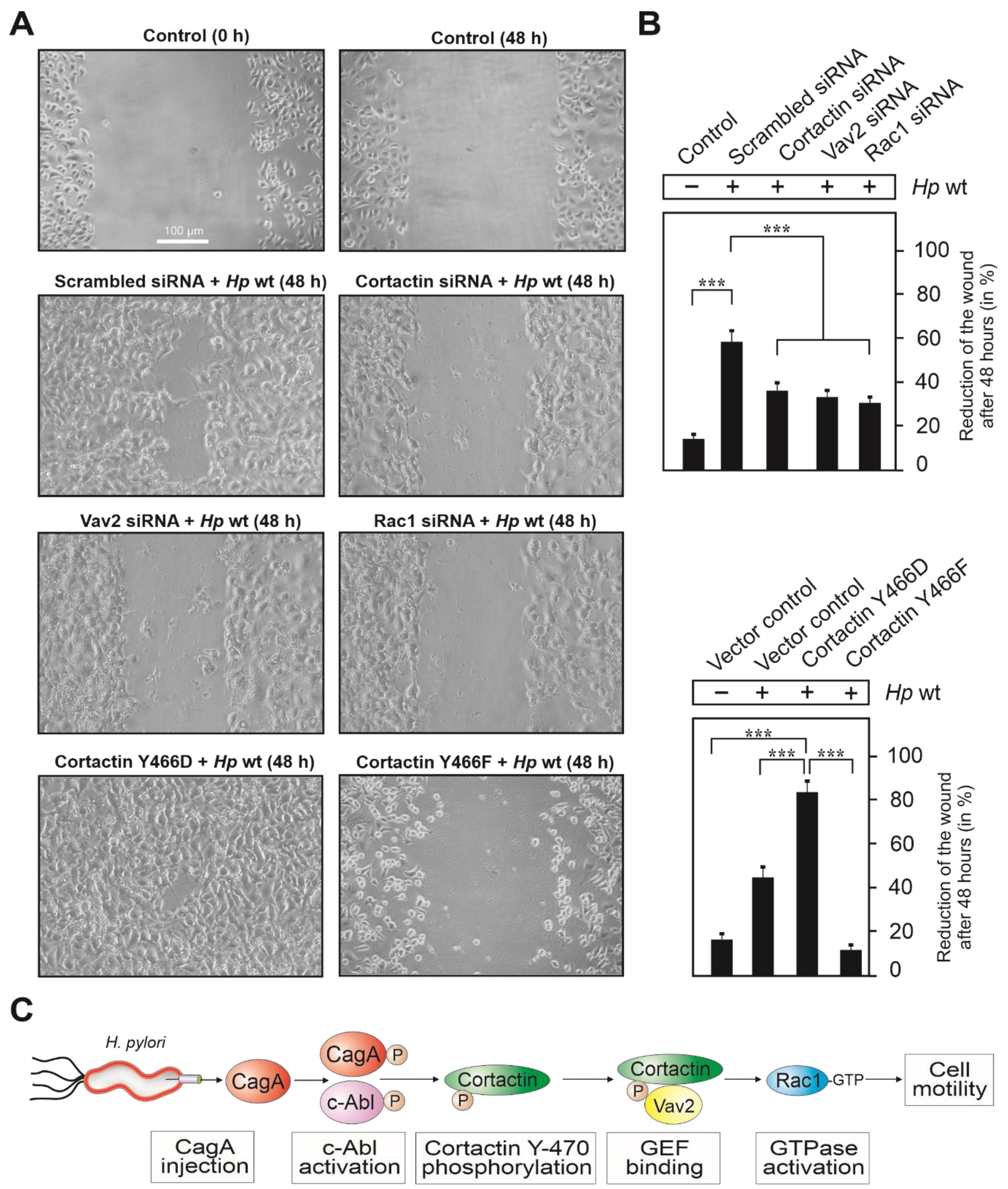
3.7. Cortactin and Vav2 Expression Are Required for Maximal Rac1 Activation
3.8. Expression of Cortactin, Vav2 and Rac1 as Well as Phosphorylation of Cortactin Are Required for Cell Motility Induced by H. pylori
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amieva, M.; Peek, R.M. Pathobiology of H. pylori-Induced Gastric Cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [Green Version]
- Salama, N.R.; Hartung, M.L.; Muller, A. Life in the human stomach: Persistence strategies of the bacterial pathogen H. pylori. Nat. Rev. Micro. 2013, 11, 385–399. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Cordero, J.J.; Hodgson, L.; Condeelis, J. Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol. 2012, 24, 277–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Arcos, J.M.; Chabrier, R.; Deygas, M.; Nader, G.; Barbier, L.; Sáez, P.J.; Mathur, A.; Vargas, P.; Piel, M. Reconstitution of cell migration at a glance. J. Cell Sci. 2019, 132, jcs225565. [Google Scholar] [CrossRef] [Green Version]
- Rottner, K.; Stradal, T.E. Actin dynamics and turnover in cell motility. Curr. Opin. Cell Biol. 2011, 23, 569–578. [Google Scholar] [CrossRef]
- Weaver, A.M. Cortactin in tumor invasiveness. Cancer Lett. 2008, 265, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 2007, 1773, 642–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamason, R.L.; Welch, M.D. Actin-based motility and cell-to-cell spread of bacterial pathogens. Curr. Opin. Microbiol. 2017, 35, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Lemichez, E. New Aspects on Bacterial Effectors Targeting Rho GTPases. In The Actin Cytoskeleton and Bacterial Infection; Current Topics in Microbiology and Immunology; Mannherz, H., Ed.; Springer: Cham, Switzerland, 2016; Volume 399, pp. 155–174. [Google Scholar]
- Stradal, T.E.B.; Schelhaas, M. Actin dynamics in host-pathogen interaction. FEBS Lett. 2018, 592, 3658–3669. [Google Scholar] [CrossRef] [Green Version]
- Mueller, D.; Tegtmeyer, N.; Brandt, S.; Yamaoka, Y.; De Poire, E.; Sgouras, D.; Wessler, S.; Torres, J.; Smolka, A.; Backert, S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J. Clin. Investig. 2012, 122, 1553–1566. [Google Scholar] [CrossRef] [Green Version]
- Backert, S.; Blaser, M.J. The Role of CagA in the Gastric Biology of Helicobacter pylori. Cancer Res. 2016, 76, 4028–4031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammer, I.; Brandt, S.; Hartig, R.; König, W.; Backert, S. Activation of Abl by Helicobacter pylori: A novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 2007, 132, 1309–1319. [Google Scholar] [CrossRef]
- Backert, S.; Feller, S.M.; Wessler, S. Emerging roles of Abl family tyrosine kinases in microbial pathogenesis. Trends Biochem. Sci. 2008, 33, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Posselt, G.; Wiesauer, M.; Chichirau, B.E.; Engler, D.; Krisch, L.M.; Gadermaier, G.; Briza, P.; Schneider, S.; Boccellato, F.; Meyer, T.F.; et al. H. pylori-controlled c-Abl localization promotes cell migration and limits apoptosis. Cell Commun. Signal. 2019, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, N.; Neddermann, M.; Asche, C.I.; Backert, S. Subversion of host kinases: A key network in cellular signaling hijacked by Helicobacter pylori CagA. Mol. Microbiol. 2017, 105, 358–372. [Google Scholar] [CrossRef] [Green Version]
- Churin, Y.; Kardalinou, E.; Meyer, T.F.; Naumann, M. Pathogenicity island-dependent activation of Rho GTPases Rac1 and Cdc42 in H. pylori infection. Mol. Microbiol. 2001, 40, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Shafikhani, S.; Balachandran, P.; Jin, S.; Hartig, R.; König, W.; Engel, J.; Backert, S. Use of a novel coinfection system reveals a role for Rac1, H-Ras, and CrkII phosphorylation in Helicobacter pylori-induced host cell actin cytoskeletal rearrangements. FEMS Immunol. Med. Microbiol. 2007, 50, 190–205. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Higashi, H.; Higuchi, M.; Okada, M.; Hatakeyama, M. Attenuation of H. pylori CagA x SH. PYLORI -2 signaling by interaction between CagA and Csk. J. Biol. Chem. 2003, 278, 3664–3670. [Google Scholar] [CrossRef] [Green Version]
- Selbach, M.; Moese, S.; Hurwitz, R.; Hauck, C.R.; Meyer, T.F.; Backert, S. The Helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c-Src inactivation. EMBO J. 2003, 22, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Schnoor, M.; Stradal, T.E.; Rottner, K. Cortactin: Cell Functions of A Multifaceted Actin-Binding Protein. Trends Cell Biol. 2018, 28, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Ma, W.; An, L. Cortactin in cancer cell migration and invasion. Oncotarget 2017, 8, 88232–88243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uruno, T.; Liu, J.; Zhang, P.; Fan, Y.X.; Egile, C.; Li, R.; Mueller, S.C.; Zhan, X. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell. Biol. 2001, 3, 259–266. [Google Scholar] [CrossRef]
- Daly, R.J. Cortactin signaling and dynamic actin networks. Biochem. J. 2004, 382, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosen-Binker, L.I.; Kapus, A. Cortactin: The gray eminence of the cytoskeleton. Physiology 2006, 21, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Ammer, A.G.; Weed, S.A. Cortactin Branches Out: Roles in Regulating Protrusive Actin Dynamics. Cell Motil. Cytoskelet. 2008, 65, 687–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tegtmeyer, N.; Wittelsberger, R.; Hartig, R.; Wessler, S.; Martinez-Quiles, N.; Backert, S. Serine phosphorylation of cortactin controls focal adhesion kinase activity and cell scattering induced by Helicobacter pylori. Cell Host Microbe 2011, 9, 520–531. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Quiles, N.; Ho, H.Y.; Kirschner, M.W.; Ramesh, N.; Geha, R.S. Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol. Cell Biol. 2004, 24, 5269–5280. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, K.; Miki, H.; He, H.; Maruta, H.; Takenawa, T. Essential role of N-WASP in podosome formation and degradation of extracellular matrix in Src-transformed fibroblasts. Cancer Res. 2002, 62, 669–674. [Google Scholar]
- Kowalski, J.R.; Egile, C.; Gil, S.; Snapper, S.B.; Li, R.; Thomas, S.M. Cortactin regulates cell migration through activation of N-WASP. J. Cell. Sci. 2005, 118, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Hartung, M.L.; Gruber, D.C.; Koch, K.N.; Grüter, L.; Rehrauer, H.; Tegtmeyer, N.; Backert, S.; Müller, A. H. pylori-Induced DNA Strand Breaks Are Introduced by Nucleotide Excision Repair Endonucleases and Promote NF-κB Target Gene Expression. Cell Rep. 2015, 13, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Moese, S.; Selbach, M.; Zimny-Arndt, U.; Jungblut, P.R.; Meyer, T.F.; Backert, S. Identification of a tyrosine-phosphorylated 35 kDa carboxy-terminal fragment (p35CagA) of the Helicobacter pylori CagA protein in phagocytic cells: Processing or breakage? Proteomics 2001, 1, 618–629. [Google Scholar] [CrossRef]
- Blumenthal, B.; Hoffmann, C.; Aktories, K.; Backert, S.; Schmidt, G. The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 2007, 75, 3344–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sason, H.; Milgrom, M.; Weiss, A.M.; Melamed-Book, N.; Balla, T.; Grinstein, S.; Backert, S.; Rosenshine, I.; Aroeti, B. Enteropathogenic Escherichia coli subverts phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate upon epithelial cell infection. Mol. Biol. Cell 2009, 20, 544–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamás, P.; Solti, Z.; Bauer, P.; Illés, A.; Sipeki, S.; Bauer, A.; Downward, J.; Buday, L. Mechanism of epidermal growth factor regulation of Vav2, a guanine nucleotide exchange factor for Rac. J. Biol. Chem. 2003, 278, 5163–5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knauer, O.; Binai, N.A.; Carra, G.; Beckhaus, T.; Hanschmann, K.M.; Renné, T.; Backert, S.; Karas, M.; Wessler, S. Differential phosphoproteome profiling reveals a functional role for VASP in Helicobacter pylori-induced cytoskeleton turnover in gastric epithelial cells. Cell Microbiol. 2008, 10, 2285–2296. [Google Scholar] [CrossRef]
- Conradi, J.; Tegtmeyer, N.; Woźna, M.; Wissbrock, M.; Michalek, C.; Gagell, C.; Cover, T.L.; Frank, R.; Sewald, N.; Backert, S. An RGD helper sequence in CagL of Helicobacter pylori assists in interactions with integrins and injection of CagA. Front. Cell Infect. Microbiol. 2012, 2, 70. [Google Scholar] [CrossRef] [Green Version]
- Brandt, S.; Kenny, B.; Rohde, M.; Martinez-Quiles, N.; Backert, S. Dual infection system identifies a crucial role for PKA-mediated serine phosphorylation of the EPEC-Tir-injected effector protein in regulating Rac1 function. Cell Microbiol. 2009, 11, 1254–1271. [Google Scholar] [CrossRef]
- Krause-Gruszczynska, M.; Boehm, M.; Rohde, M.; Tegtmeyer, N.; Takahashi, S.; Buday, L.; Oyarzabal, O.A.; Backert, S. The signaling pathway of Campylobacter jejuni-induced Cdc42 activation: Role of fibronectin, integrin beta1, tyrosine kinases and guanine exchange factor Vav2. Cell Commun. Signal. 2011, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Nicole Tegtmeyer, N.; (Department of Biology, Division of Microbiology, Friedrich-Alexander University Erlangen-Nuremberg, Erlangen, Germany). Personal Communication, 2021.
- Boehm, M.; Krause-Gruszczynska, M.; Rohde, M.; Tegtmeyer, N.; Takahashi, S.; Oyarzabal, O.A.; Backert, S. Major host factors involved in epithelial cell invasion of Campylobacter jejuni: Role of fibronectin, integrin beta1, FAK, Tiam-1, and DOCK180 in activating Rho GTPase Rac1. Front. Cell Infect. Microbiol. 2011, 1, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devreotes, P.; Horwitz, A.R. Signaling networks that regulate cell migration. Cold Spring Harb. Perspect. Biol. 2015, 7, a005959. [Google Scholar] [CrossRef] [Green Version]
- Schaks, M.; Giannone, G.; Rottner, K. Actin dynamics in cell migration. Essays Biochem. 2019, 63, 483–495. [Google Scholar] [PubMed] [Green Version]
- Selbach, M.; Backert, S. Cortactin: An Achilles’ heel of the actin cytoskeleton targeted by pathogens. Trends Microbiol. 2005, 13, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.J.; Gil-Henn, H.; Halo, T.; Yin, T.; Condeelis, J.; Machida, K.; Wu, Y.I.; Koleske, A.J. Phosphorylated cortactin recruits Vav2 guanine nucleotide exchange factor to activate Rac3 and promote invadopodial function in invasive breast cancer cells. Mol. Biol. Cell 2017, 28, 1347–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarelli, V.V.; Kodama, T.; Nada, S.; Okada, M.; Iida, T.; Honda, T. Tyrosine phosphorylation controls cortactin binding to two EHEC effectors: Tir and EspFu/TccP. Cell Microbiol. 2007, 9, 1782–1795. [Google Scholar] [CrossRef]
- Nieto-Pelegrin, E.; Martinez-Quiles, N. Distinct phosphorylation requirements regulate cortactin activation by TirEPEC and its binding to N-WASP. Cell Commun. Signal. 2009, 7, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, I.; Eugène, E.; Nassif, X.; Couraud, P.O.; Bourdoulous, S. Activation of ErbB2 receptor tyrosine kinase supports invasion of endothelial cells by Neisseria. J. Cell Biol. 2001, 155, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Agerer, F.; Lux, S.; Michel, A.; Rohde, M.; Ohlsen, K.; Hauck, C.R. Cellular invasion by Staphylococcus reveals a functional link between FAK and cortactin in integrin-mediated internalisation. J. Cell Sci. 2005, 118, 2189–2200. [Google Scholar] [CrossRef] [Green Version]
- Bougnères, L.; Girardin, S.E.; Weed, S.A.; Karginov, A.V.; Parsons, J.T.; Sansonetti, P.J.; Van Nhieu, G.T. Cortactin and Crk cooperate to trigger actin polymerization during Shigella invasion of epithelial cells. J. Cell Biol. 2004, 166, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Nhieu, G.T.; Enninga, J.; Sansonetti, P.; Grompone, G. Tyrosine kinase signaling and type III effectors orchestrating Shigella invasion. Curr. Opin. Microbiol. 2005, 8, 16–20. [Google Scholar] [CrossRef]
- Barroso, C.; Rodenbusch, S.E.; Welch, M.D.; Drubin, D.G. A role for cortactin in Listeria monocytogenes invasion of NIH3T3 cells, but not in its intracellular motility. Cell Motil. Cytoskelet. 2006, 63, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Veiga, E.; Cossart, P. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat. Cell Biol. 2005, 7, 894–900. [Google Scholar] [CrossRef]
- Adam, T.; Giry, M.; Boquet, P.; Sansonetti, P. Rho-dependent membrane folding causes Shigella entry into epithelial cells. EMBO J. 1996, 15, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Hauck, C.R.; Meyer, T.F.; Lang, F.; Gulbins, E. CD66-mediated phagocytosis of Opa52 Neisseria gonorrhoeae requires a Src-like tyrosine kinase- and Rac1-dependent signalling pathway. EMBO J. 1998, 17, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mounier, J.; Laurent, V.; Hall, A.; Fort, P.; Carlier, M.F.; Sansonetti, P.J.; Egile, C. Rho family GTPases control entry of Shigella flexneri into epithelial cells but not intracellular motility. J. Cell Sci. 1999, 112, 2069–2080. [Google Scholar] [CrossRef]
- Ireton, K.; Rigano, L.A.; Dowd, G.C. Role of host GTPases in infection by Listeria. Cell Microbiol. 2014, 16, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, K.; Murata-Kamiya, N.; Kondo, S.; Hatakeyama, M. H. pylori stimulates epithelial cell migration via CagA-mediated perturbation of host cell signaling. Microbes Infect. 2012, 14, 470–476. [Google Scholar] [CrossRef]
- Schuuring, E.; Verhoeven, E.; Litvinov, S.; Michalides, R.J. The product of the EMS1 gene, amplified and overexpressed in human carcinomas, is homologous to a v-src substrate and is located in cell-substratum contact sites. Mol. Cell Biol. 1993, 13, 2891–2898. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tegtmeyer, N.; Harrer, A.; Rottner, K.; Backert, S. Helicobacter pylori CagA Induces Cortactin Y-470 Phosphorylation-Dependent Gastric Epithelial Cell Scattering via Abl, Vav2 and Rac1 Activation. Cancers 2021, 13, 4241. https://doi.org/10.3390/cancers13164241
Tegtmeyer N, Harrer A, Rottner K, Backert S. Helicobacter pylori CagA Induces Cortactin Y-470 Phosphorylation-Dependent Gastric Epithelial Cell Scattering via Abl, Vav2 and Rac1 Activation. Cancers. 2021; 13(16):4241. https://doi.org/10.3390/cancers13164241
Chicago/Turabian StyleTegtmeyer, Nicole, Aileen Harrer, Klemens Rottner, and Steffen Backert. 2021. "Helicobacter pylori CagA Induces Cortactin Y-470 Phosphorylation-Dependent Gastric Epithelial Cell Scattering via Abl, Vav2 and Rac1 Activation" Cancers 13, no. 16: 4241. https://doi.org/10.3390/cancers13164241
APA StyleTegtmeyer, N., Harrer, A., Rottner, K., & Backert, S. (2021). Helicobacter pylori CagA Induces Cortactin Y-470 Phosphorylation-Dependent Gastric Epithelial Cell Scattering via Abl, Vav2 and Rac1 Activation. Cancers, 13(16), 4241. https://doi.org/10.3390/cancers13164241