
Exploring the Interplay of Telomerase Reverse Transcriptase and β-Catenin in Hepatocellular Carcinoma



Abstract
Simple Summary
Abstract
1. Introduction
2. Telomeres, Telomerase, and TERT
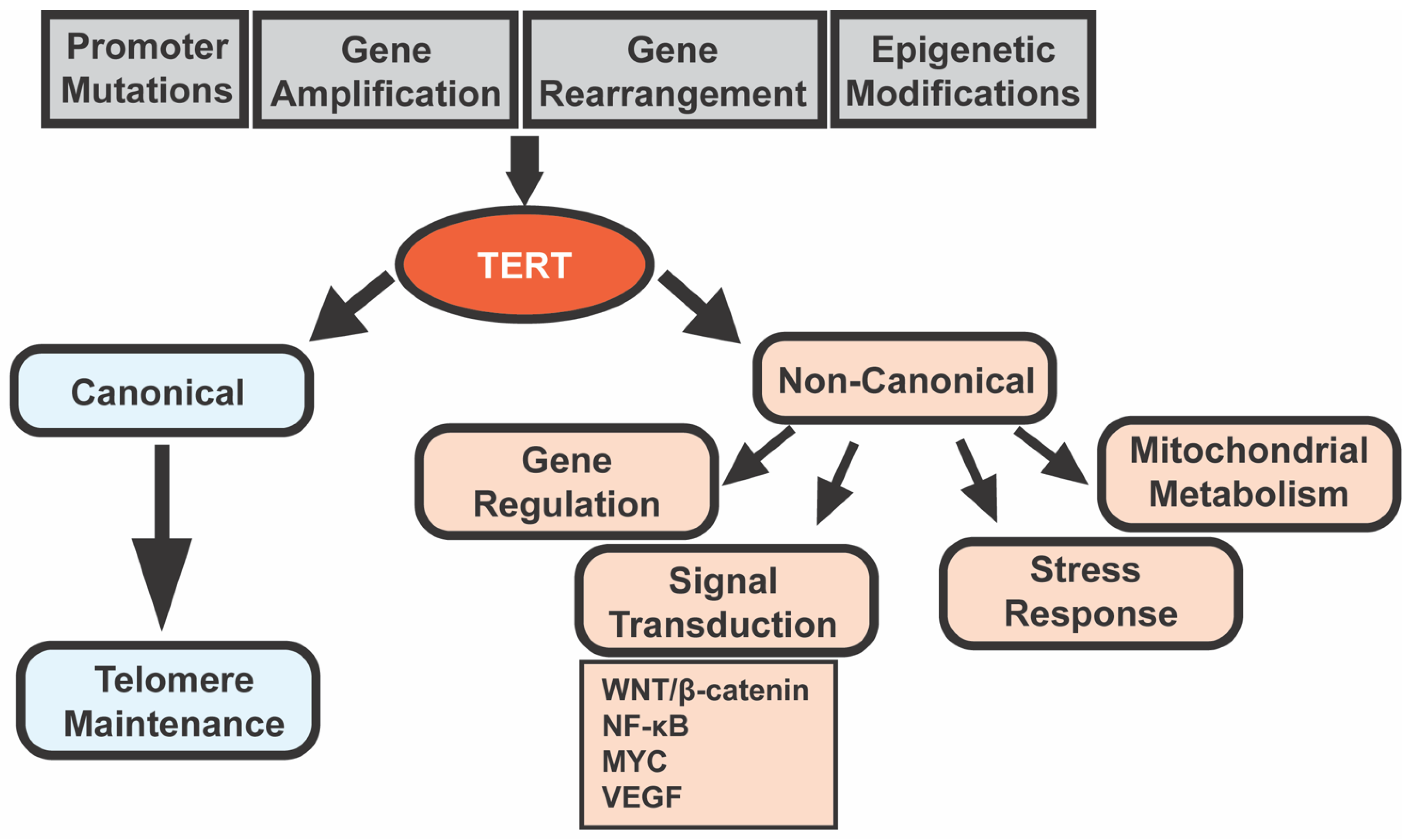
3. Telomerase and Cancer

{kind=link}
{kind=link}
{kind=link}
| Gene (Animal) | Site Specificity | Expression * | Result ** | Ref. |
|---|---|---|---|---|
| Tert (mouse) | Thymocytes and peripheral T cells | + | ↑ T-Cell Lymphomas | [16] |
| Tert (mouse) | Basal keratinocytes | + | ↑ skin papillomas (DMBA + TPA induction) | [17] |
| Tert (mouse) | Whole body | + | ↑ mammary tumors in aged females | [18] |
| tert and terc (zebrafish) | Neural progenitor cells | + | ↓ aggressiveness of RAS-mediated brain tumors | [27] |
| Tert (mouse) | Whole body | − | Delayed onset of lymphomas in EμMYC mice | [20] |
| Tert (mouse) | Whole body | − | Delayed onset of mammary tumors in PyMT mice | [19] |
| Tert (mouse) | Whole body | − | ↓ HCC “initiation foci” (CCl4 induction) | [21] |
| Terc (mouse) | Whole body | − | ↑ HCC “initiation foci” but ↓ HCC progression (uPA, CCl4 or DEN induction) | [22] |
| Terc (mouse) | Whole body | − | ↑ tumors (lymphomas, teratocarcinomas, HCC, squamous cell carcinoma) | [26] |
| Terc (mouse) | Whole body | − | ↓ skin papillomas (DMBA + TPA induction) | [28] |
| Terc (mouse) | Whole body | − | ↑ epithelial cancers in TP53−/− mice | [29] |
| Terc (mouse) | Whole body | − | ↑ adenoma initiation but ↓ progression in ApcMin mice | [30] |
| terthu3430 (zebrafish) | Whole body | − | Earlier onset of tumors (germ cell tumors, hematopoietic neoplasms, HCA, etc.) | [23] |
| terthu3430 (zebrafish) | Whole body | − | ↑ tumor incidence and aggressiveness of melanoma model *** | [31] |
4. TERT Promoter Mutations in HCC
| Region | Etiology (N) | C228T Incidence % (N) | C250T Incidence % (N) | Association * | Ref. |
|---|---|---|---|---|---|
| (1) Asia (2) Africa (3) Europe | 52.3% HBV, rest unknown (regions 1–3) (44) | (1) 16% (3) (2) 33% (5) (3) 20% (2) | (1) 5% (1) (2) 20% (3) (3) 10% (1) | ↑ frequency in Africans vs. non-Africans (p = 0.056) and in HBV− vs. HBV+ (p = 0.295) (regions 1–3) | [48] |
| Southern Italy | (67) | 41.8% (28) | 0% (0) | ↑ TERT expression in mutated tumors vs. control tissue (p < 0.0001) | [41] |
| Southern Italy | 7.9% HBV+, 86.6% HCV+ (127) | 48.8% (62) | 1.6% (2) | ↑ frequency in HCV+ vs. HBV+ (p < 0.001) | [43] |
| Germany | (78) | 47.4% (37) | [49] | ||
| France, Italy and Spain | 14% HBV+, 26% HCV+ (243) | 54.3% (132) | 2% (5) | [6] | |
| France | 22% HBV+, 26% HCV+ (305) | 55% (168) | 3.6% (11) | ↑ frequency in males vs. females (p = 0.001) and in HBV− vs. HBV+ (p < 0.0001); ↑ TERT expression in mutated HCC vs. normal liver, cirrhosis, and HCA (p = 0.0007) | [5] |
| USA | 24.6% HBV+, 26% HCV+ (61) | 42.6% (26) | 1.6% (1) | no correlation with etiology, sex, age, or ethnicity | [50] |
| (1) USA (2) Japan | (1) 14.6% HBV+, 57% HCV+ (89) (2) 28.6% HBV+, 37% HCV+ (374) | (1) 34.8% (31) (2) 55.6% (208) | (1) 2.2% (2) (2) 2.4% (9) | ↑ frequency in HCV+ vs. HCV− (p = 0.0016) | [7,42] |
| China | 94% HBV+ (276) | 30.5% (84) | 0.36% (1) | ↑ frequency in older vs. younger age (p = 0.04); no correlation with sex or etiology | [51] |
| China | (35) | 25.7% (9) | 5.7% (2) | [52] | |
| China | 83% HBV+ (190) | 26.3% (50) | 3.7% (7) | no correlation with age, sex, etiology, or tumor status | [53] |
| Japan | (11) | 81.8% (9) | [54] | ||
| Japan | 23% HBV+, 61.6% HCV+ (125) | 66.4% (83) | 1.6% (2) | ↑ frequency in HCV+ vs. HCV− (p = 0.0007) and in viral vs. non-viral (p = 0.0282) | [45] |
| South Korea | 36% HBV+, 3% HCV+ (160) | 20% (32) | 8.75% (14) | ↑ frequency in males vs. females (p = 0.027) and in HCV+ vs. HCV− (p = 0.285); no association with telomere length or HCC prognosis | [35] |
| South Korea | 74% HBV+, 5.7% HCV+ (105) | 37% (39) | 1.9% (2) | ↑ frequency in HCV+ vs. HCV− (p = 0.001) | [46] |
| Taiwan | 63% HBV+, 40.2% HCV+ (195) | 27.7% (54) | 1.5% (3) | ↑ frequency in HCV+ vs. HCV− (p = 0.0048), older vs. younger age (p = 0.0122), and HBV− vs. HBV+ (p = 0.0007) | [34] |
| USA, Canada, South Korea, Vietnam, and Russia | 22.4% HBV+, 17.8% HCV+ (196) | 40.8% (80) | 3.6% (7) | ↑ frequency in older vs. younger age (p = 0.0006), males vs. females (p = 0.006), HCV+ vs. HCV− (p = 0.04), and HBV− vs. HBV+ (p = 0.02); no association with TERT expression | [44] |
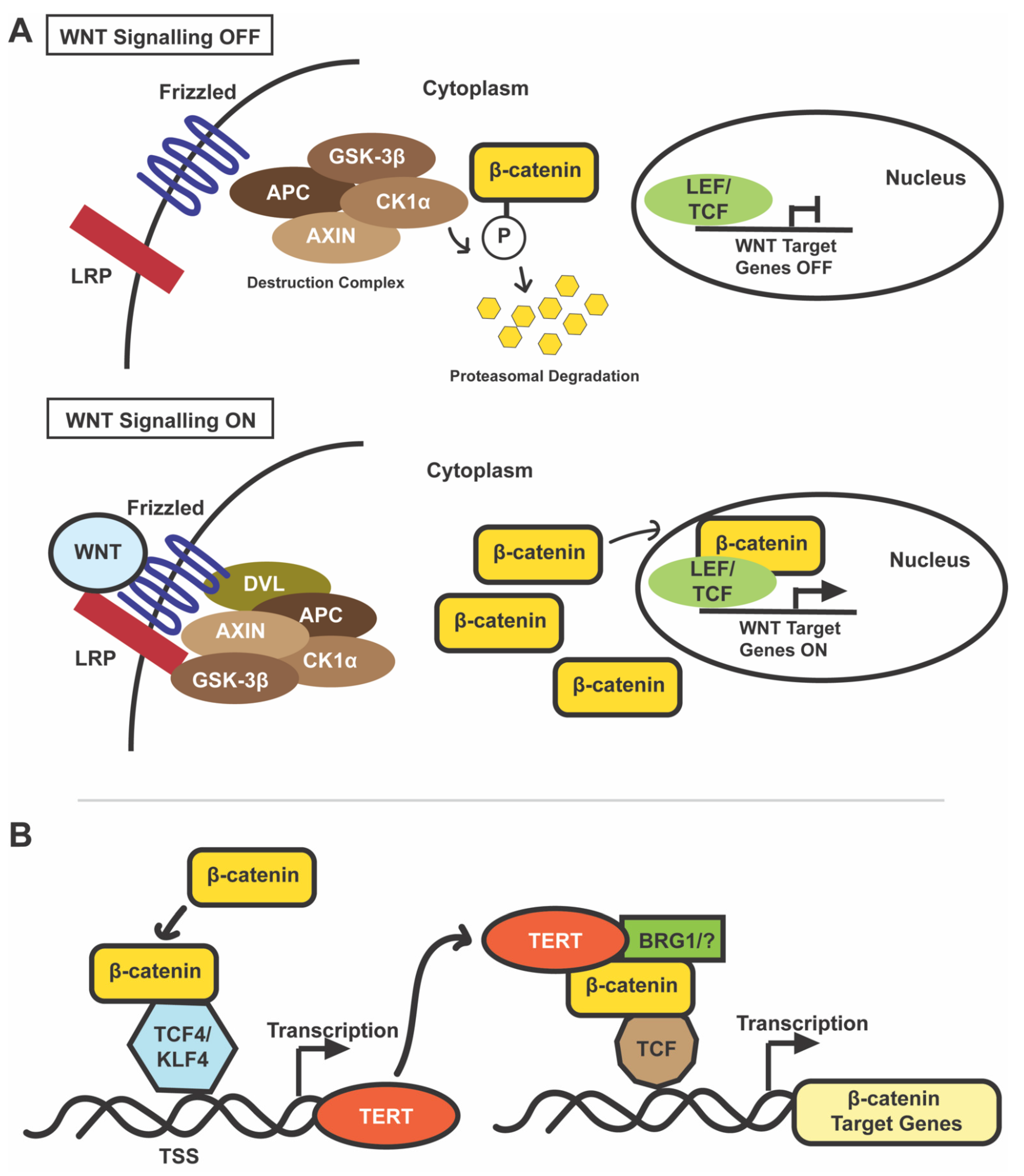
5. WNT/β-Catenin Signaling in Normal and Cancer Cells

6. WNT/β-Catenin in HCC
| Animal | Method * | Expression ** | CTNNB1 Status *** | Results | Ref. |
|---|---|---|---|---|---|
| Mice | Alb promoter | + | Wildtype | Transgenic mice develop hepatomegaly but no tumors | [71] |
| Alb promoter | + | Ser45 mutated | Transgenic mice develop hepatomegaly (only in younger mice) but no tumors; Increased HCC (DEN induction) | [72] | |
| Fabp: Cre | + | Exon 3 deleted | Transgenic mice develop hepatomegaly but no tumors | [73] | |
| CaBP9K promoter | + | N131 deleted | Transgenic mice develop hepatomegaly but no tumors | [74] | |
| Cited1: CreER | + | Exon 3 deleted | Transgenic mice develop HCCs, hepatoblastomas, and lung metastases | [79] | |
| AdCMV-Cre | + | Exon 3 deleted (and H-RAS) | Double transgenic mice develop HCC, but no HCC develops with expression of mutated CTNNB1 alone | [78] | |
| SB-HDT | + | S45 or S33 mutated (and MET) | Mice expressing MET and either form of mutated CTNNB1, but not mutated CTNNB1 alone, develop HCC | [75] | |
| SB-HDT | + | N90 deleted (and activated YAP) | Mice expressing activated YAP and mutated CTNNB1, but not mutated CTNNB1 alone, develop HCC | [76] | |
| SB-HDT | + | S45 or S33 mutated (and K-RAS) | Mice expressing K-RAS and either form of mutated CTNNB1, but not mutated CTNNB1 alone, develop HCC | [77] | |
| siRNA | − | S45 mutated (HDT induction) | Decreased HCC (K-RAS plus S45-mutated CTNNB1 HDT induction) | [77] | |
| Alb:Cre | − | Wildtype (endogenous) | Increased HCC (DEN induction) | [82] | |
| Alb:Cre | − | Wildtype (endogenous) | Increased HCC (MET and N90-deleted CTNNB1 induction) | [83] | |
| Zebrafish | fabp10a promoter | + | S33A, S37A, T41A, and S45A mutated | Transgenic zebrafish develop HCC as adults (78% by 6 months) | [81] |
| fabp: CreERT2 | + | Transgenic zebrafish develop HCC as adults (13% by 6 months) | [80] |
7. Interactions between TERT and β-Catenin in Cultured Cells and Animal Models
8. TERT and β-Catenin in Human Cancer
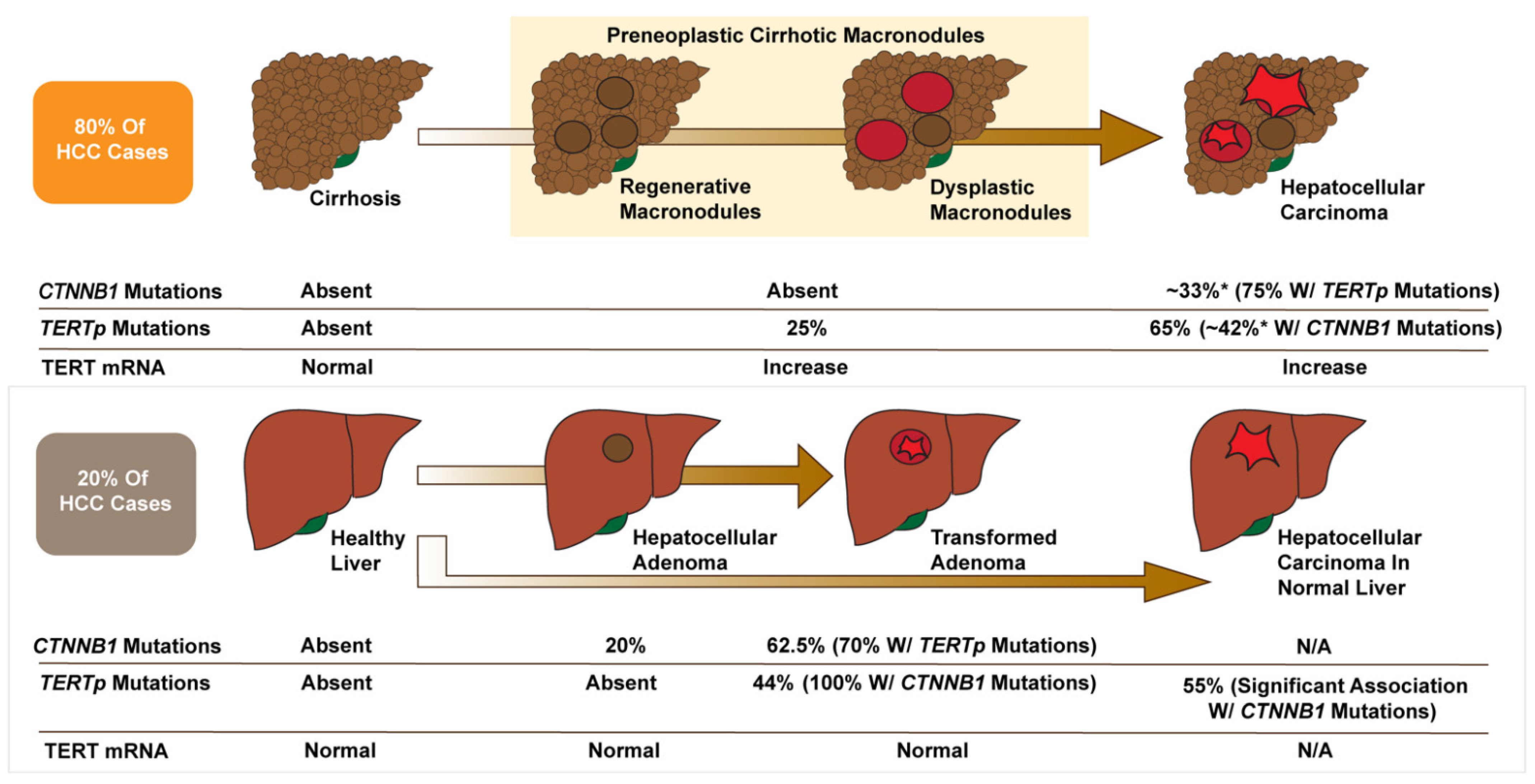
9. TERT and β-Catenin in HCC Patients
10. TERT and β-Catenin in Vertebrate HCC Models
11. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Lee, J.-S. The mutational landscape of hepatocellular carcinoma. Clin. Mol. Hepatol. 2015, 21, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/β-Catenin Signaling Regulates Telomerase in Stem Cells and Cancer Cells. Science 2012, 336, 1549. [Google Scholar] [CrossRef]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, T.B.; Sá, A.; Lopes, J.M.; Sobrinho-Simões, M.; Soares, P.; Vinagre, J. Telomere Maintenance Mechanisms in Cancer. Genes 2018, 9, 241. [Google Scholar] [CrossRef]
- Heidenreich, B.; Kumar, R. TERT promoter mutations in telomere biology. Mutat. Res. Rev. Mutat. Res. 2017, 771, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Kim, N.-K.; Feigon, J. Architecture of human telomerase RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20325–20332. [Google Scholar] [CrossRef]
- Ségal-Bendirdjian, E.; Geli, V. Non-canonical Roles of Telomerase: Unraveling the Imbroglio. Front. Cell Dev. Biol. 2019, 7, 332. [Google Scholar] [CrossRef]
- Romaniuk, A.; Paszel-Jaworska, A.; Totoń, E.; Lisiak, N.; Hołysz, H.; Królak, A.; Grodecka-Gazdecka, S.; Rubiś, B. The non-canonical functions of telomerase: To turn off or not to turn off. Mol. Biol. Rep. 2019, 46, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhou, Y.; Chen, D.; Li, L.; Yang, X.; You, Y.; Ling, X. Impact of mitochondrial telomerase over-expression on drug resistance of hepatocellular carcinoma. Am. J. Transl. Res. 2015, 7, 88–99. [Google Scholar]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- Yuan, X.; Dai, M.; Xu, D. TERT promoter mutations and GABP transcription factors in carcinogenesis: More foes than friends. Cancer Lett. 2020, 493, 1–9. [Google Scholar] [CrossRef]
- Canela, A.; Martín-Caballero, J.; Flores, J.M.; Blasco, M.A. Constitutive expression of tert in thymocytes leads to increased incidence and dissemination of T-cell lymphoma in Lck-Tert mice. Mol. Cell Biol. 2004, 24, 4275–4293. [Google Scholar] [CrossRef]
- González-Suárez, E.; Samper, E.; Ramírez, A.; Flores, J.M.; Martín-Caballero, J.; Jorcano, J.L.; Blasco, M.A. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J. 2001, 20, 2619–2630. [Google Scholar] [CrossRef] [PubMed]
- Artandi, S.E.; Alson, S.; Tietze, M.K.; Sharpless, N.E.; Ye, S.; Greenberg, R.A.; Castrillon, D.H.; Horner, J.W.; Weiler, S.R.; Carrasco, R.D.; et al. Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8191–8196. [Google Scholar] [CrossRef]
- Khattar, E.; Kumar, P.; Liu, C.Y.; Akıncılar, S.C.; Raju, A.; Lakshmanan, M.; Maury, J.J.P.; Qiang, Y.; Li, S.; Tan, E.Y.; et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J. Clin. Investig. 2016, 126, 4045–4060. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Khattar, E.; Leow, S.C.; Liu, C.Y.; Muller, J.; Ang, W.X.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E.; et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Investig. 2015, 125, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; Glickman, J.; Horner, J.; DePinho, R.A. Cooperative Interactions of p53 Mutation, Telomere Dysfunction, and Chronic Liver Damage in Hepatocellular Carcinoma Progression. Cancer Res. 2006, 66, 4766. [Google Scholar] [CrossRef]
- Farazi, P.A.; Glickman, J.; Jiang, S.; Yu, A.; Rudolph, K.L.; DePinho, R.A. Differential Impact of Telomere Dysfunction on Initiation and Progression of Hepatocellular Carcinoma. Cancer Res. 2003, 63, 5021. [Google Scholar] [PubMed]
- Carneiro, M.C.; Henriques, C.M.; Nabais, J.; Ferreira, T.; Carvalho, T.; Ferreira, M.G. Short Telomeres in Key Tissues Initiate Local and Systemic Aging in Zebrafish. PLoS Genet. 2016, 12, e1005798. [Google Scholar] [CrossRef] [PubMed]
- Marzec, P.; Armenise, C.; Pérot, G.; Roumelioti, F.-M.; Basyuk, E.; Gagos, S.; Chibon, F.; Déjardin, J. Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell 2015, 160, 913–927. [Google Scholar] [CrossRef]
- Cayuela, M.L.; Claes, K.B.M.; Ferreira, M.G.; Henriques, C.M.; van Eeden, F.; Varga, M.; Vierstraete, J.; Mione, M.C. The Zebrafish as an Emerging Model to Study DNA Damage in Aging, Cancer and Other Diseases. Front. Cell Dev. Biol. 2019, 6, 178. [Google Scholar] [CrossRef]
- Rudolph, K.L.; Chang, S.; Lee, H.-W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef]
- Idilli, A.I.; Cusanelli, E.; Pagani, F.; Berardinelli, F.; Bernabé, M.; Cayuela, M.L.; Poliani, P.L.; Mione, M.C. Expression of tert Prevents ALT in Zebrafish Brain Tumors. Front. Cell Dev. Biol. 2020, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- González-Suárez, E.; Samper, E.; Flores, J.M.; Blasco, M.A. Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat. Genet. 2000, 26, 114–117. [Google Scholar] [CrossRef]
- Artandi, S.E.; Chang, S.; Lee, S.-L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L.; Millard, M.; Bosenberg, M.W.; DePinho, R.A. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat. Genet. 2001, 28, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Lex, K.; Maia Gil, M.; Lopes-Bastos, B.; Figueira, M.; Marzullo, M.; Giannetti, K.; Carvalho, T.; Ferreira, M.G. Telomere shortening produces an inflammatory environment that increases tumor incidence in zebrafish. Proc. Natl. Acad. Sci. USA 2020, 117, 15066. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Zhu, Y.-J.; Wang, H.-Y.; Chen, L. Gender disparity in hepatocellular carcinoma (HCC): Multiple underlying mechanisms. Sci. China Life Sci. 2017, 60, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Jeng, Y.-M.; Chang, C.-N.; Lee, H.-J.; Hsu, H.-C.; Lai, P.-L.; Yuan, R.-H. TERT promoter mutation in resectable hepatocellular carcinomas: A strong association with hepatitis C infection and absence of hepatitis B infection. Int. J. Surg. 2014, 12, 659–665. [Google Scholar] [CrossRef]
- Lee, H.W.; Park, T.I.; Jang, S.Y.; Park, S.Y.; Park, W.-J.; Jung, S.-J.; Lee, J.-H. Clinicopathological characteristics of TERT promoter mutation and telomere length in hepatocellular carcinoma. Medicine 2017, 96, e5766. [Google Scholar] [CrossRef]
- Qu, Y.; Dang, S.; Wu, K.; Shao, Y.; Yang, Q.; Ji, M.; Shi, B.; Hou, P. TERT promoter mutations predict worse survival in laryngeal cancer patients. Int. J. Cancer 2014, 135, 1008–1010. [Google Scholar] [CrossRef]
- Bell, R.J.A.; Rube, H.T.; Xavier-Magalhães, A.; Costa, B.M.; Mancini, A.; Song, J.S.; Costello, J.F. Understanding TERT Promoter Mutations: A Common Path to Immortality. Mol. Cancer Res. 2016, 14, 315. [Google Scholar] [CrossRef]
- Liu, T.; Yuan, X.; Xu, D. Cancer-Specific Telomerase Reverse Transcriptase (TERT) Promoter Mutations: Biological and Clinical Implications. Genes 2016, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Tallet, A.; Nault, J.-C.; Renier, A.; Hysi, I.; Galateau-Sallé, F.; Cazes, A.; Copin, M.-C.; Hofman, P.; Andujar, P.; Le Pimpec-Barthes, F.; et al. Overexpression and promoter mutation of the TERT gene in malignant pleural mesothelioma. Oncogene 2014, 33, 3748–3752. [Google Scholar] [CrossRef]
- Melo, M.; Da Rocha, A.G.; Vinagre, J.; Batista, R.; Peixoto, J.; Tavares, C.; Celestino, R.; Almeida, A.; Salgado, C.; Eloy, C.; et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E754–E765. [Google Scholar] [CrossRef]
- Lombardo, D.; Saitta, C.; Giosa, D.; Di Tocco, F.C.; Musolino, C.; Caminiti, G.; Chines, V.; Franzè, M.S.; Alibrandi, A.; Navarra, G.; et al. Frequency of somatic mutations in TERT promoter, TP53 and CTNNB1 genes in patients with hepatocellular carcinoma from Southern Italy. Oncol. Lett. 2020, 19, 2368–2374. [Google Scholar] [PubMed]
- Pezzuto, F.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Frequency and geographic distribution of TERT promoter mutations in primary hepatocellular carcinoma. Infect. Agent Cancer 2017, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, F.; Izzo, F.; Buonaguro, L.; Annunziata, C.; Tatangelo, F.; Botti, G.; Buonaguro, F.M.; Tornesello, M.L. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget 2016, 7, 54253–54262. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address: Wheeler@bcm.edu; Cancer Genome Atlas Research Network Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341 e23. [Google Scholar] [CrossRef]
- Nishida, N.; Nishimura, T.; Kaido, T.; Minaga, K.; Yamao, K.; Kamata, K.; Takenaka, M.; Ida, H.; Hagiwara, S.; Minami, Y.; et al. Molecular Scoring of Hepatocellular Carcinoma for Predicting Metastatic Recurrence and Requirements of Systemic Chemotherapy. Cancers 2018, 10, 367. [Google Scholar] [CrossRef]
- Lee, S.E.; Chang, S.-H.; Kim, W.Y.; Lim, S.D.; Kim, W.S.; Hwang, T.S.; Han, H.S. Frequent somatic TERT promoter mutations and CTNNB1 mutations in hepatocellular carcinoma. Oncotarget 2016, 7, 69267–69275. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, W.; Kang, W.; Wong, S.H.; Wang, M.; Zhou, Y.; Fang, X.; Zhang, X.; Yang, H.; Wong, C.H.; et al. Genomic analysis of liver cancer unveils novel driver genes and distinct prognostic features. Theranostics 2018, 8, 1740–1751. [Google Scholar] [CrossRef]
- Cevik, D.; Yildiz, G.; Ozturk, M. Common telomerase reverse transcriptase promoter mutations in hepatocellular carcinomas from different geographical locations. World J. Gastroenterol. 2015, 21, 311–317. [Google Scholar] [CrossRef]
- Quaas, A.; Oldopp, T.; Tharun, L.; Klingenfeld, C.; Krech, T.; Sauter, G.; Grob, T.J. Frequency of TERT promoter mutations in primary tumors of the liver. Virchows Arch. 2014, 465, 673–677. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Yang, X.; Guo, X.; Chen, Y.; Chen, G.; Ma, Y.; Huang, K.; Zhang, Y.; Zhao, Q.; Winkler, C.A.; An, P.; et al. Telomerase reverse transcriptase promoter mutations in hepatitis B virus-associated hepatocellular carcinoma. Oncotarget 2016, 7, 27838–27847. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.-S.; Wang, Z.; He, X.-J.; Diplas, B.H.; Yang, R.; Killela, P.J.; Meng, Q.; Ye, Z.-Y.; Wang, W.; Jiang, X.-T.; et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur. J. Cancer 2015, 51, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Cheng, G.; Yu, J.; Zheng, S.; Sun, C.; Sun, Q.; Li, K.; Lin, Z.; Liu, T.; Li, P.; et al. The TERT promoter mutation incidence is modified by germline TERT rs2736098 and rs2736100 polymorphisms in hepatocellular carcinoma. Oncotarget 2017, 8, 23120–23129. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Ueda, Y.; Hatano, E.; Kakiuchi, N.; Takeda, H.; Goto, T.; Shimizu, T.; Yoshida, K.; Ikura, Y.; Shiraishi, Y.; et al. TERT promoter mutations and chromosome 8p loss are characteristic of nonalcoholic fatty liver disease-related hepatocellular carcinoma. Int. J. Cancer 2016, 139, 2512–2518. [Google Scholar] [CrossRef]
- Park, C.-K.; Lee, S.-H.; Kim, J.Y.; Kim, J.E.; Kim, T.M.; Lee, S.-T.; Choi, S.H.; Park, S.-H.; Kim, I.H. Expression level of hTERT is regulated by somatic mutation and common single nucleotide polymorphism at promoter region in glioblastoma. Oncotarget 2014, 5, 3399–3407. [Google Scholar] [CrossRef][Green Version]
- Ko, E.; Seo, H.-W.; Jung, E.S.; Kim, B.; Jung, G. The TERT promoter SNP rs2853669 decreases E2F1 transcription factor binding and increases mortality and recurrence risks in liver cancer. Oncotarget 2016, 7, 684–699. [Google Scholar] [CrossRef]
- Kim, Y.J.; Yoo, J.E.; Jeon, Y. Suppression of PROX1-mediated TERT expression in hepatitis B viral hepatocellular carcinoma. Int. J. Cancer 2018, 143, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Russell, J.O.; Monga, S.P. Wnt/β-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu. Rev. Pathol. 2018, 13, 351–378. [Google Scholar] [CrossRef]
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 mutations of CTNNB1 drive tumorigenesis: A review. Oncotarget 2017, 9, 5492–5508. [Google Scholar] [CrossRef]
- Mohammed, M.K.; Shao, C.; Wang, J.; Wei, Q.; Wang, X.; Collier, Z.; Tang, S.; Liu, H.; Zhang, F.; Huang, J.; et al. Wnt/β-catenin signaling plays an ever-expanding role in stem cell self-renewal, tumorigenesis and cancer chemoresistance. Genes Dis. 2016, 3, 11–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Toh, L.; Lau, P.; Wang, X. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/β-catenin pathway in human cancer. J. Biol. Chem. 2012, 287, 32494–32511. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pan, Q.; Fuhler, G.M.; Smits, R.; Peppelenbosch, M.P. Action and function of Wnt/β-catenin signaling in the progression from chronic hepatitis C to hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, A.M.; Fuentes, D.; Morshid, A.I.; Burke, M.R.; Kaseb, A.O.; Hassan, M.; Hazle, J.D.; Elsayes, K.M. Role of Wnt/β-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J. Hepatocell. Carcinoma 2018, 5, 61–73. [Google Scholar] [CrossRef]
- Wang, Z.; Sheng, Y.-Y.; Gao, X.-M.; Wang, C.-Q.; Wang, X.-Y.; Lu, X.U.; Wei, J.-W.; Zhang, K.-L.; Dong, Q.-Z.; Qin, L.-X. β-catenin mutation is correlated with a favorable prognosis in patients with hepatocellular carcinoma. Mol. Clin. Oncol. 2015, 3, 936–940. [Google Scholar] [CrossRef]
- Mao, T.-L.; Chu, J.-S.; Jeng, Y.-M.; Lai, P.-L.; Hsu, H.-C. Expression of mutant nuclear β-catenin correlates with non-invasive hepatocellular carcinoma, absence of portal vein spread, and good prognosis. J. Pathol. 2001, 193, 95–101. [Google Scholar] [CrossRef]
- Yuan, R.-H.; Jeng, Y.-M.; Hu, R.-H.; Lai, P.-L.; Lee, P.-H.; Cheng, C.-C.; Hsu, H.-C. Role of p53 and β-catenin Mutations in Conjunction with CK19 Expression on Early Tumor Recurrence and Prognosis of Hepatocellular Carcinoma. J. Gastrointest. Surg. 2011, 15, 321–329. [Google Scholar] [CrossRef]
- Peng, S.-Y.; Chen, W.J.; Lai, P.-L.; Jeng, Y.-M.; Sheu, J.-C.; Hsu, H.-C. High α-fetoprotein level correlates with high stage, early recurrence and poor prognosis of hepatocellular carcinoma: Significance of hepatitis virus infection, age, p53 and β-catenin mutations. Int. J. Cancer 2004, 112, 44–50. [Google Scholar] [CrossRef]
- Kitao, A.; Matsui, O.; Yoneda, N.; Kozaka, K.; Kobayashi, S.; Sanada, J.; Koda, W.; Minami, T.; Inoue, D.; Yoshida, K.; et al. Hepatocellular Carcinoma with β-Catenin Mutation: Imaging and Pathologic Characteristics. Radiology 2015, 275, 708–717. [Google Scholar] [CrossRef]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouzé, E.; Imbeaud, S.; Pilati, C.; Nault, J.-C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ß-catenin activity associated with liver tumor progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef]
- Tan, X.; Apte, U.; Micsenyi, A.; Kotsagrelos, E.; Luo, J.-H.; Ranganathan, S.; Monga, D.K.; Bell, A.; Michalopoulos, G.K.; Monga, S.P.S. Epidermal growth factor receptor: A novel target of the Wnt/beta-catenin pathway in liver. Gastroenterology 2005, 129, 285–302. [Google Scholar] [CrossRef]
- Nejak-Bowen, K.N.; Thompson, M.D.; Singh, S.; Bowen, W.C.; Dar, M.J.; Khillan, J.; Dai, C.; Monga, S.P.S. Accelerated Liver Regeneration And Hepatocarcinogenesis In Mice Overexpressing Serine-45 Mutant Beta-Catenin. Hepatology 2010, 51, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Miyoshi, H.; Murai, N.; Oshima, H.; Tamai, Y.; Oshima, M.; Taketo, M.M. Lack of Tumorigenesis in the Mouse Liver after Adenovirus-mediated Expression of a Dominant Stable Mutant of β-Catenin. Cancer Res. 2002, 62, 1971. [Google Scholar] [PubMed]
- Cadoret, A.; Ovejero, C.; Saadi-Kheddouci, S.; Souil, E.; Fabre, M.; Romagnolo, B.; Kahn, A.; Perret, C. Hepatomegaly in Transgenic Mice Expressing an Oncogenic Form of β-Catenin. Cancer Res. 2001, 61, 3245. [Google Scholar]
- Tao, J.; Xu, E.; Zhao, Y.; Singh, S.; Li, X.; Couchy, G.; Chen, X.; Zucman-Rossi, J.; Chikina, M.; Monga, S.P.S. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant β-catenin. Hepatology 2016, 64, 1587–1605. [Google Scholar] [CrossRef]
- Tao, J.; Calvisi, D.F.; Ranganathan, S.; Cigliano, A.; Zhou, L.; Singh, S.; Jiang, L.; Fan, B.; Terracciano, L.; Armeanu-Ebinger, S.; et al. Activation of β-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 2014, 147, 690–701. [Google Scholar] [CrossRef]
- Tao, J.; Zhang, R.; Singh, S.; Poddar, M.; Xu, E.; Oertel, M.; Chen, X.; Ganesh, S.; Abrams, M.; Monga, S.P. Targeting β-catenin in hepatocellular cancers induced by coexpression of mutant β-catenin and K-Ras in mice. Hepatology 2017, 65, 1581–1599. [Google Scholar] [CrossRef]
- Harada, N.; Oshima, H.; Katoh, M.; Tamai, Y.; Oshima, M.; Taketo, M.M. Hepatocarcinogenesis in Mice with β-Catenin and Ha-Ras Gene Mutations. Cancer Res. 2004, 64, 48. [Google Scholar] [CrossRef] [PubMed]
- Mokkapati, S.; Niopek, K.; Huang, L.; Cunniff, K.J.; Ruteshouser, E.C.; deCaestecker, M.; Finegold, M.J.; Huff, V. β-catenin activation in a novel liver progenitor cell type is sufficient to cause hepatocellular carcinoma and hepatoblastoma. Cancer Res. 2014, 74, 4515–4525. [Google Scholar] [CrossRef] [PubMed]
- Kalasekar, S.M.; Kotiyal, S.; Conley, C.; Phan, C.; Young, A.; Evason, K.J. Heterogeneous beta-catenin activation is sufficient to cause hepatocellular carcinoma in zebrafish. Biol. Open 2019, 8, bio047829. [Google Scholar] [CrossRef] [PubMed]
- Evason, K.J.; Francisco, M.T.; Juric, V.; Balakrishnan, S.; Pazmino, M.d.P.L.; Gordan, J.D.; Kakar, S.; Spitsbergen, J.; Goga, A.; Stainier, D.Y.R. Identification of Chemical Inhibitors of β-Catenin-Driven Liver Tumorigenesis in Zebrafish. PLoS Genet. 2015, 11, e1005305. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Tan, X.; Zeng, G.; Misse, A.; Singh, S.; Kim, Y.; Klaunig, J.E.; Monga, S.P.S. Conditional beta-catenin loss in mice promotes chemical hepatocarcinogenesis: Role of oxidative stress and platelet-derived growth factor receptor alpha/phosphoinositide 3-kinase signaling. Hepatology 2010, 52, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Feng, Y.; Zong, M.; Wei, X.-F.; Lee, J.; Feng, Y.; Li, H.; Yang, G.-S.; Wu, Z.-J.; Fu, X.-D.; et al. β-catenin deficiency in hepatocytes aggravates hepatocarcinogenesis driven by oncogenic β-catenin and MET. Hepatology 2018, 67, 1807–1822. [Google Scholar] [CrossRef] [PubMed]
- Sarin, K.Y.; Cheung, P.; Gilison, D.; Lee, E.; Tennen, R.I.; Wang, E.; Artandi, M.K.; Oro, A.E.; Artandi, S.E. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature 2005, 436, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Gat, U.; DasGupta, R.; Degenstein, L.; Fuchs, E. De Novo Hair Follicle Morphogenesis and Hair Tumors in Mice Expressing a Truncated β-Catenin in Skin. Cell 1998, 95, 605–614. [Google Scholar] [CrossRef]
- Andl, T.; Reddy, S.T.; Gaddapara, T. Millar SE WNT signals are required for the initiation of hair follicle development. Dev. Cell. 2002, 2, 643–653. [Google Scholar] [CrossRef]
- Lowry, W.E.; Blanpain, C.; Nowak, J.A.; Guasch, G.; Lewis, L.; Fuchs, E. Defining the impact of beta-catenin/Tcf transactivation on epithelial stem cells. Genes Dev. 2005, 19, 1596–1611. [Google Scholar] [CrossRef]
- Huelsken, J.; Vogel, R.; Erdmann, B.; Cotsarelis, G.; Birchmeier, W. Beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell 2001, 105, 533–545. [Google Scholar] [CrossRef]
- Choi, J.; Southworth, L.K.; Sarin, K.Y.; Venteicher, A.S.; Ma, W.; Chang, W.; Cheung, P.; Jun, S.; Artandi, M.K.; Shah, N.; et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008, 4, e10. [Google Scholar] [CrossRef]
- Hrdličková, R.; Nehyba, J.; Bose, H.R. Alternatively Spliced Telomerase Reverse Transcriptase Variants Lacking Telomerase Activity Stimulate Cell Proliferation. Mol. Cell. Biol. 2012, 32, 4283. [Google Scholar] [CrossRef]
- Khattar, E.; Tergaonkar, V. Transcriptional Regulation of Telomerase Reverse Transcriptase (TERT) by MYC. Front. Cell Dev. Biol 2017, 5, 1. [Google Scholar] [CrossRef]
- Wu, K.-J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef]
- Jaitner, S.; Reiche, J.A.; Schäffauer, A.J.; Hiendlmeyer, E.; Herbst, H.; Brabletz, T.; Kirchner, T.; Jung, A. Human telomerase reverse transcriptase (hTERT) is a target gene of β-catenin in human colorectal tumors. Null 2012, 11, 3331–3338. [Google Scholar] [CrossRef]
- Listerman, I.; Gazzaniga, F.S.; Blackburn, E.H. An Investigation of the Effects of the Core Protein Telomerase Reverse Transcriptase on Wnt Signaling in Breast Cancer Cells. Mol. Cell. Biol. 2014, 34, 280. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.A.; Vidal-Cardenas, S.L.; Karim, B.; Yu, H.; Guo, N.; Greider, C.W. Phenotypes in mTERT+/− and mTERT−/− mice are due to short telomeres, not telomere-independent functions of telomerase reverse transcriptase. Mol. Cell Biol. 2011, 31, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Cardenas, S.L.; Greider, C.W. Comparing effects of mTR and mTERT deletion on gene expression and DNA damage response: A critical examination of telomere length maintenance-independent roles of telomerase. Nucleic Acids Res. 2010, 38, 60–71. [Google Scholar] [CrossRef]
- Yang, T.-L.B.; Chen, Q.; Deng, J.T.; Jagannathan, G.; Tobias, J.W.; Schultz, D.C.; Wang, S.; Lengner, C.J.; Rustgi, A.K.; Lynch, J.P.; et al. Mutual reinforcement between telomere capping and canonical Wnt signalling in the intestinal stem cell niche. Nat. Commun. 2017, 8, 14766. [Google Scholar] [CrossRef][Green Version]
- Fernandez, R.J., 3rd; Johnson, F.B. A regulatory loop connecting WNT signaling and telomere capping: Possible therapeutic implications for dyskeratosis congenita. Ann. N. Y. Acad. Sci. 2018, 1418, 56–68. [Google Scholar] [CrossRef]
- Singh, V.; Singh, A.P.; Sharma, I. Epigenetic deregulations of Wnt/β-catenin and transforming growth factor beta-Smad pathways in esophageal cancer: Outcome of DNA methylation. J. Cancer Res. Ther. 2019, 15, 192–203. [Google Scholar] [CrossRef]
- Tang, B.; Xie, R.; Qin, Y.; Xiao, Y.-F.; Yong, X.; Zheng, L.; Dong, H.; Yang, S.-M. Human telomerase reverse transcriptase (hTERT) promotes gastric cancer invasion through cooperating with c-Myc to upregulate heparanase expression. Oncotarget 2016, 7, 11364–11379. [Google Scholar] [CrossRef]
- Remke, M.; Ramaswamy, V.; Peacock, J.; Shih, D.J.H.; Koelsche, C.; Northcott, P.A.; Hill, N.; Cavalli, F.M.G.; Kool, M.; Wang, X.; et al. TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol. 2013, 126, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Bernabé-García, M.; Martínez-Balsalobre, E.; García-Moreno, D.; García-Castillo, J.; Revilla-Nuin, B.; Blanco-Alcaina, E.; Mulero, V.; Alcaraz-Pérez, F.; Cayuela, M.L. TERT-mediated induction of MIR500A contributes to tumor invasiveness by targeting Hedgehog pathway. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nault, J.-C.; Martin, Y.; Caruso, S.; Hirsch, T.Z.; Bayard, Q.; Calderaro, J.; Charpy, C.; Copie-Bergman, C.; Ziol, M.; Bioulac-Sage, P.; et al. Clinical Impact of Genomic Diversity From Early to Advanced Hepatocellular Carcinoma. Hepatology 2020, 71, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, S.; Sia, D.; Harrington, A.N.; Zhang, Z.; Cabellos, L.; Cornella, H.; Moeini, A.; Camprecios, G.; Leow, W.-Q.; Fiel, M.I.; et al. Trunk mutational events present minimal intra- and inter-tumoral heterogeneity in hepatocellular carcinoma. J. Hepatol. 2017, 67, 1222–1231. [Google Scholar] [CrossRef]
- Pinyol, R.; Tovar, V.; Llovet, J.M. TERT promoter mutations: Gatekeeper and driver of hepatocellular carcinoma. J. Hepatol. 2014, 61, 685–687. [Google Scholar] [CrossRef]
- Bioulac-Sage, P.; Rebouissou, S.; Thomas, C.; Blanc, J.-F.; Saric, J.; Sa Cunha, A.; Rullier, A.; Cubel, G.; Couchy, G.; Imbeaud, S.; et al. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology 2007, 46, 740–748. [Google Scholar] [CrossRef]
- Nault, J.-C.; Bioulac–Sage, P.; Zucman–Rossi, J. Hepatocellular Benign Tumors—From Molecular Classification to Personalized Clinical Care. Gastroenterology 2013, 144, 888–902. [Google Scholar] [CrossRef]
- Nault, J.-C.; Couchy, G.; Balabaud, C.; Morcrette, G.; Caruso, S.; Blanc, J.-F.; Bacq, Y.; Calderaro, J.; Paradis, V.; Ramos, J.; et al. Molecular Classification of Hepatocellular Adenoma Associates With Risk Factors, Bleeding, and Malignant Transformation. Gastroenterology 2017, 152, 880–894.e6. [Google Scholar] [CrossRef]
- Pilati, C.; Letouzé, E.; Nault, J.-C.; Imbeaud, S.; Boulai, A.; Calderaro, J.; Poussin, K.; Franconi, A.; Couchy, G.; Morcrette, G.; et al. Genomic Profiling of Hepatocellular Adenomas Reveals Recurrent FRK-Activating Mutations and the Mechanisms of Malignant Transformation. Cancer Cell 2014, 25, 428–441. [Google Scholar] [CrossRef]
- Zheng, J.; Sadot, E.; Vigidal, J.A.; Klimstra, D.S.; Balachandran, V.P.; Kingham, T.P.; Allen, P.J.; D’Angelica, M.I.; DeMatteo, R.P.; Jarnagin, W.R.; et al. Characterization of hepatocellular adenoma and carcinoma using microRNA profiling and targeted gene sequencing. PLoS ONE 2018, 13, e0200776. [Google Scholar] [CrossRef]
- Jiao, J.; Watt, G.P.; Stevenson, H.L.; Calderone, T.L.; Fisher-Hoch, S.P.; Ye, Y.; Wu, X.; Vierling, J.M.; Beretta, L. Telomerase reverse transcriptase mutations in plasma DNA in patients with hepatocellular carcinoma or cirrhosis: Prevalence and risk factors. Hepatol. Commun. 2018, 2, 718–731. [Google Scholar] [CrossRef]
- Wege, H.; Heim, D.; Lütgehetmann, M.; Dierlamm, J.; Lohse, A.W.; Brümmendorf, T.H. Forced Activation of β-Catenin Signaling Supports the Transformation of hTERT-Immortalized Human Fetal Hepatocytes. Mol. Cancer Res. 2011, 9, 1222. [Google Scholar] [CrossRef]
- Molina-Sánchez, P.; Ruiz de Galarreta, M.; Yao, M.A.; Lindblad, K.E.; Bresnahan, E.; Bitterman, E.; Martin, T.C.; Rubenstein, T.; Nie, K.; Golas, J.; et al. Cooperation Between Distinct Cancer Driver Genes Underlies Intertumor Heterogeneity in Hepatocellular Carcinoma. Gastroenterology 2020, 159, 2203–2220.e14. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Park, J.-I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef]
- Guterres, A.N.; Villanueva, J. Targeting telomerase for cancer therapy. Oncogene 2020, 39, 5811–5824. [Google Scholar] [CrossRef] [PubMed]
- Trybek, T.; Kowalik, A.; Góźdź, S.; Kowalska, A. Telomeres and telomerase in oncogenesis. Oncol. Lett. 2020, 20, 1015–1027. [Google Scholar] [CrossRef]
- Alessandrini, I.; Recagni, M.; Zaffaroni, N.; Folini, M. On the Road to Fight Cancer: The Potential of G-quadruplex Ligands as Novel Therapeutic Agents. Int. J. Mol. Sci. 2021, 22, 5947. [Google Scholar] [CrossRef] [PubMed]
- Ruden, M.; Puri, N. Novel anticancer therapeutics targeting telomerase. Cancer Treat. Rev. 2013, 39, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, E.; Kaneko, S. Telomerase-Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 1823. [Google Scholar] [CrossRef]


| Region | Etiology (N) | TERTp % Incidence (N) | CTNNB1 % Incidence (N) | Association | Ref. |
|---|---|---|---|---|---|
| France | 22% HBV+, 26% HCV+ (305) | 59% (179) | 33% (101) | Significant, p < 0.0001 | [5] |
| France | 20.7% HBV+, 26.5% HCV+ (801) | 58.1% (441) | 30.7% (229) | Significant, p = 0.0000001 | [103] |
| Southern Italy | 7.9% HBV+, 86.6% HCV+ (127) | 50.4% (64) | 26% (33) | Not significant, p = 0.4192 | [43] |
| USA and Japan | 25.6% HBV+, 42.6% HCV+ (469) | 54.1% (254) | 31.1% (146) | Significant, p < 0.0001 | [7] |
| Japan | NAFLD (11) | 82% (9) | 45% (5) | Not significant, p = 0.4545 | [54] |
| Taiwan | 63% HBV+, 40.2% HCV+ (195) | 29.2% (57) | 16.5% (31/188) | Not significant, p = 0.2055 | [34] |
| Korea | 74.3% HBV+, 5.7% HCV+; (105) | 39% (41) | 14.6% (15) | Not significant, p = 0.568 | [46] |
| China | 83% HBV+ (190) | 30% (57) | 24.3% (17/70) | Not significant, p = 0.535 | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotiyal, S.; Evason, K.J. Exploring the Interplay of Telomerase Reverse Transcriptase and β-Catenin in Hepatocellular Carcinoma. Cancers 2021, 13, 4202. https://doi.org/10.3390/cancers13164202
Kotiyal S, Evason KJ. Exploring the Interplay of Telomerase Reverse Transcriptase and β-Catenin in Hepatocellular Carcinoma. Cancers. 2021; 13(16):4202. https://doi.org/10.3390/cancers13164202
Chicago/Turabian StyleKotiyal, Srishti, and Kimberley Jane Evason. 2021. "Exploring the Interplay of Telomerase Reverse Transcriptase and β-Catenin in Hepatocellular Carcinoma" Cancers 13, no. 16: 4202. https://doi.org/10.3390/cancers13164202
APA StyleKotiyal, S., & Evason, K. J. (2021). Exploring the Interplay of Telomerase Reverse Transcriptase and β-Catenin in Hepatocellular Carcinoma. Cancers, 13(16), 4202. https://doi.org/10.3390/cancers13164202