
Nature or Nurture? Role of the Bone Marrow Microenvironment in the Genesis and Maintenance of Myelodysplastic Syndromes



{kind=link}
{kind=link}
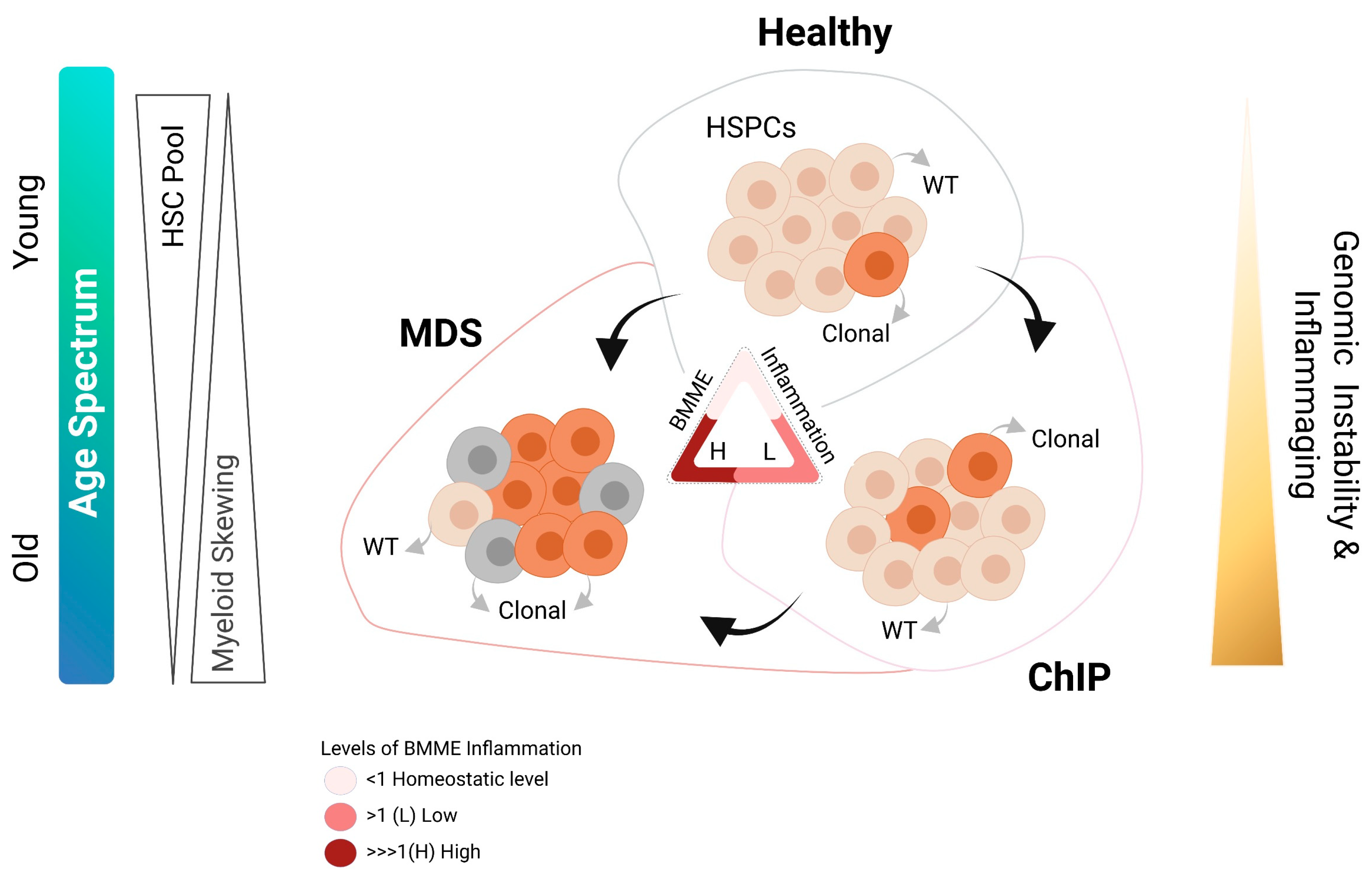
Abstract
Simple Summary
Abstract
1. Introduction
2. Bone Marrow Cellular Metropolis and the Hematopoietic Stem Cells
3. Initiators of MDS: HSPCs or BMME
3.1. Haematopoietic Stem Cells
3.2. Mesenchymal Stomal Cells
3.3. Endothelial Cells
3.4. Immune-BMME
4. BMME Inflammation: A Friend or a Foe
5. What Options Are There: Targeting the MDS HSPC-BMME Crosstalk
6. Future Direction
Author Contributions
Funding
Conflicts of Interest
References
- Mufti, G.J.; Bennett, J.M.; Goasguen, J.; Bain, B.J.; Baumann, I.; Brunning, R.; Cazzola, M.; Fenaux, P.; Germing, U.; Hellstrom-Lindberg, E.; et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica 2008, 93, 1712–1717. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P. Myelodysplastic syndromes current treatment algorithm 2018. Blood Cancer J. 2018, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef]
- Mian, S.A.; Smith, A.E.; Kulasekararaj, A.G.; Kizilors, A.; Mohamedali, A.M.; Lea, N.C.; Mitsopoulos, K.; Ford, K.; Nasser, E.; Seidl, T.; et al. Spliceosome mutations exhibit specific associations with epigenetic modifiers and proto-oncogenes mutated in myelodysplastic syndrome. Haematologica 2013, 98, 1058–1066. [Google Scholar] [CrossRef]
- Nazha, A.; Al-Issa, K.; Hamilton, B.K.; Radivoyevitch, T.; Gerds, A.T.; Mukherjee, S.; Adema, V.; Zarzour, A.; Abuhadra, N.; Patel, B.J.; et al. Adding molecular data to prognostic models can improve predictive power in treated patients with myelodysplastic syndromes. Leukemia 2017, 31, 2848–2850. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Shimamura, A. Genetic predisposition to MDS: Clinical features and clonal evolution. Blood 2019, 133, 1071–1085. [Google Scholar] [CrossRef]
- Crisa, E.; Boggione, P.; Nicolosi, M.; Mahmoud, A.M.; Al Essa, W.; Awikeh, B.; Aspesi, A.; Andorno, A.; Boldorini, R.; Dianzani, I.; et al. Genetic Predisposition to Myelodysplastic Syndromes: A Challenge for Adult Hematologists. Int. J. Mol. Sci. 2021, 22, 2525. [Google Scholar] [CrossRef]
- Palomo, L.; Acha, P.; Sole, F. Genetic Aspects of Myelodysplastic/Myeloproliferative Neoplasms. Cancers 2021, 13, 2120. [Google Scholar] [CrossRef]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Nazha, A.; Narkhede, M.; Radivoyevitch, T.; Seastone, D.J.; Patel, B.J.; Gerds, A.T.; Mukherjee, S.; Kalaycio, M.; Advani, A.; Przychodzen, B.; et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia 2016, 30, 2214–2220. [Google Scholar] [CrossRef]
- Bersanelli, M.; Travaglino, E.; Meggendorfer, M.; Matteuzzi, T.; Sala, C.; Mosca, E.; Chiereghin, C.; Di Nanni, N.; Gnocchi, M.; Zampini, M.; et al. Classification and Personalized Prognostic Assessment on the Basis of Clinical and Genomic Features in Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1223–1233. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lepine, G.; Mollica, L.; Szuber, N.; Dube, M.P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Hecker, J.S.; Hartmann, L.; Riviere, J.; Buck, M.C.; van der Garde, M.; Rothenberg-Thurley, M.; Fischer, L.; Winter, S.; Ksienzyk, B.; Ziemann, F.; et al. CHIP & HIPs: Clonal Hematopoiesis is Common in Hip Arthroplasty Patients and Associates with Autoimmune Disease. Blood 2021. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673. [Google Scholar] [CrossRef]
- Batsivari, A.; Grey, W.; Bonnet, D. Understanding of the crosstalk between normal residual hematopoietic stem cells and the leukemic niche in acute myeloid leukemia. Exp. Hematol. 2021, 95, 23–30. [Google Scholar] [CrossRef]
- Scadden, D.T. Nice neighborhood: Emerging concepts of the stem cell niche. Cell 2014, 157, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Goulard, M.; Dosquet, C.; Bonnet, D. Role of the microenvironment in myeloid malignancies. Cell Mol. Life Sci. 2018, 75, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Baccin, C.; Al-Sabah, J.; Velten, L.; Helbling, P.M.; Grunschlager, F.; Hernandez-Malmierca, P.; Nombela-Arrieta, C.; Steinmetz, L.M.; Trumpp, A.; Haas, S. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 2020, 22, 38–48. [Google Scholar] [CrossRef]
- Xu, C.; Gao, X.; Wei, Q.; Nakahara, F.; Zimmerman, S.E.; Mar, J.; Frenette, P.S. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat. Commun. 2018, 9, 2449. [Google Scholar] [CrossRef]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef]
- Kusumbe, A.P.; Ramasamy, S.K.; Itkin, T.; Mae, M.A.; Langen, U.H.; Betsholtz, C.; Lapidot, T.; Adams, R.H. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 2016, 532, 380–384. [Google Scholar] [CrossRef]
- Young, K.; Eudy, E.; Bell, R.; Loberg, M.A.; Stearns, T.; Sharma, D.; Velten, L.; Haas, S.; Filippi, M.D.; Trowbridge, J.J. Decline in IGF1 in the bone marrow microenvironment initiates hematopoietic stem cell aging. Cell Stem Cell 2021, 28, 1473–1482. [Google Scholar] [CrossRef]
- Feuerer, M.; Beckhove, P.; Mahnke, Y.; Hommel, M.; Kyewski, B.; Hamann, A.; Umansky, V.; Schirrmacher, V. Bone marrow microenvironment facilitating dendritic cell: CD4 T cell interactions and maintenance of CD4 memory. Int. J. Oncol. 2004, 25, 867–876. [Google Scholar]
- Schirrmacher, V.; Feuerer, M.; Fournier, P.; Ahlert, T.; Umansky, V.; Beckhove, P. T-cell priming in bone marrow: The potential for long-lasting protective anti-tumor immunity. Trends Mol. Med. 2003, 9, 526–534. [Google Scholar] [CrossRef]
- Kakiuchi, M.; Hirata, Y.; Robson, S.C.; Fujisaki, J. Transfer of stem cell niche-residential regulatory T cells prevents post-irradiation bone marrow injury. Haematologica 2021, 106, 891–893. [Google Scholar] [CrossRef]
- Hirata, Y.; Furuhashi, K.; Ishii, H.; Li, H.W.; Pinho, S.; Ding, L.; Robson, S.C.; Frenette, P.S.; Fujisaki, J. CD150(high) Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine. Cell Stem Cell 2018, 22, 445–453.e5. [Google Scholar] [CrossRef]
- Fujisaki, J.; Wu, J.; Carlson, A.L.; Silberstein, L.; Putheti, P.; Larocca, R.; Gao, W.; Saito, T.I.; Lo Celso, C.; Tsuyuzaki, H.; et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 2011, 474, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Kakiuchi, M.; Robson, S.C.; Fujisaki, J. CD150(high) CD4 T cells and CD150(high) regulatory T cells regulate hematopoietic stem cell quiescence via CD73. Haematologica 2019, 104, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Sims, N.A.; Pettit, A.R.; Barbier, V.; Nowlan, B.; Helwani, F.; Poulton, I.J.; van Rooijen, N.; Alexander, K.A.; Raggatt, L.J.; et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010, 116, 4815–4828. [Google Scholar] [CrossRef]
- Chow, A.; Lucas, D.; Hidalgo, A.; Mendez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef]
- Takizawa, H.; Fritsch, K.; Kovtonyuk, L.V.; Saito, Y.; Yakkala, C.; Jacobs, K.; Ahuja, A.K.; Lopes, M.; Hausmann, A.; Hardt, W.D.; et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 2017, 21, 225–240. [Google Scholar] [CrossRef]
- Fiorina, P.; Jurewicz, M.; Vergani, A.; Petrelli, A.; Carvello, M.; D’Addio, F.; Godwin, J.G.; Law, K.; Wu, E.; Tian, Z.; et al. Targeting the CXCR4-CXCL12 axis mobilizes autologous hematopoietic stem cells and prolongs islet allograft survival via programmed death ligand 1. J. Immunol. 2011, 186, 121–131. [Google Scholar] [CrossRef]
- Zheng, J.; Umikawa, M.; Zhang, S.; Huynh, H.; Silvany, R.; Chen, B.P.; Chen, L.; Zhang, C.C. Ex vivo expanded hematopoietic stem cells overcome the MHC barrier in allogeneic transplantation. Cell Stem Cell 2011, 9, 119–130. [Google Scholar] [CrossRef]
- Mian, S.A.; Rouault-Pierre, K.; Smith, A.E.; Seidl, T.; Pizzitola, I.; Kizilors, A.; Kulasekararaj, A.G.; Bonnet, D.; Mufti, G.J. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat. Commun. 2015, 6, 10004. [Google Scholar] [CrossRef] [PubMed]
- Woll, P.S.; Kjallquist, U.; Chowdhury, O.; Doolittle, H.; Wedge, D.C.; Thongjuea, S.; Erlandsson, R.; Ngara, M.; Anderson, K.; Deng, Q.; et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell 2014, 25, 794–808. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef]
- Rouault-Pierre, K.; Smith, A.E.; Mian, S.A.; Pizzitola, I.; Kulasekararaj, A.G.; Mufti, G.J.; Bonnet, D. Myelodysplastic syndrome can propagate from the multipotent progenitor compartment. Haematologica 2017, 102, e7–e10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Gong, Q.; Wang, Y.; Li, M.; Wang, L.; Ding, H.; Li, P. The biological function and clinical significance of SF3B1 mutations in cancer. Biomark. Res. 2020, 8, 38. [Google Scholar] [CrossRef]
- Wan, Y.; Wu, C.J. SF3B1 mutations in chronic lymphocytic leukemia. Blood 2013, 121, 4627–4634. [Google Scholar] [CrossRef]
- Chen, J.; Kao, Y.R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.R.; Montagna, C.; Will, B.; Verma, A.; Steidl, U. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med. 2019, 25, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef] [PubMed]
- van Galen, P.; Hovestadt, V.; Wadsworth Ii, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281. [Google Scholar] [CrossRef]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef]
- Heredia-Genestar, J.M.; Marques-Bonet, T.; Juan, D.; Navarro, A. Extreme differences between human germline and tumor mutation densities are driven by ancestral human-specific deviations. Nat. Commun. 2020, 11, 2512. [Google Scholar] [CrossRef] [PubMed]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef]
- Terao, C.; Suzuki, A.; Momozawa, Y.; Akiyama, M.; Ishigaki, K.; Yamamoto, K.; Matsuda, K.; Murakami, Y.; McCarroll, S.A.; Kubo, M.; et al. Chromosomal alterations among age-related haematopoietic clones in Japan. Nature 2020, 584, 130–135. [Google Scholar] [CrossRef]
- Loh, P.R.; Genovese, G.; McCarroll, S.A. Monogenic and polygenic inheritance become instruments for clonal selection. Nature 2020, 584, 136–141. [Google Scholar] [CrossRef]
- Taguchi, M.; Mishima, H.; Shiozawa, Y.; Hayashida, C.; Kinoshita, A.; Nannya, Y.; Makishima, H.; Horai, M.; Matsuo, M.; Sato, S.; et al. Genome analysis of myelodysplastic syndromes among atomic bomb survivors in Nagasaki. Haematologica 2020, 105, 358–365. [Google Scholar] [CrossRef]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260–264. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Kucab, J.E.; Zou, X.; Morganella, S.; Joel, M.; Nanda, A.S.; Nagy, E.; Gomez, C.; Degasperi, A.; Harris, R.; Jackson, S.P.; et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 2019, 177, 821–836. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Mangi, M.H.; Salisbury, J.R.; Mufti, G.J. Abnormal localization of immature precursors (ALIP) in the bone marrow of myelodysplastic syndromes: Current state of knowledge and future directions. Leuk. Res. 1991, 15, 627–639. [Google Scholar] [CrossRef]
- Bartl, R.; Frisch, B.; Baumgart, R. Morphologic classification of the myelodysplastic syndromes (MDS): Combined utilization of bone marrow aspirates and trephine biopsies. Leuk. Res. 1992, 16, 15–33. [Google Scholar] [CrossRef]
- Li, A.J.; Calvi, L.M. The microenvironment in myelodysplastic syndromes: Niche-mediated disease initiation and progression. Exp. Hematol. 2017, 55, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Geyh, S.; Oz, S.; Cadeddu, R.P.; Frobel, J.; Bruckner, B.; Kundgen, A.; Fenk, R.; Bruns, I.; Zilkens, C.; Hermsen, D.; et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia 2013, 27, 1841–1851. [Google Scholar] [CrossRef]
- Aanei, C.M.; Eloae, F.Z.; Flandrin-Gresta, P.; Tavernier, E.; Carasevici, E.; Guyotat, D.; Campos, L. Focal adhesion protein abnormalities in myelodysplastic mesenchymal stromal cells. Exp. Cell Res. 2011, 317, 2616–2629. [Google Scholar] [CrossRef] [PubMed]
- Aanei, C.M.; Flandrin, P.; Eloae, F.Z.; Carasevici, E.; Guyotat, D.; Wattel, E.; Campos, L. Intrinsic growth deficiencies of mesenchymal stromal cells in myelodysplastic syndromes. Stem Cells Dev. 2012, 21, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- Poon, Z.; Dighe, N.; Venkatesan, S.S.; Cheung, A.M.S.; Fan, X.; Bari, S.; Hota, M.; Ghosh, S.; Hwang, W.Y.K. Bone marrow MSCs in MDS: Contribution towards dysfunctional hematopoiesis and potential targets for disease response to hypomethylating therapy. Leukemia 2019, 33, 1487–1500. [Google Scholar] [CrossRef]
- Blau, O.; Baldus, C.D.; Hofmann, W.K.; Thiel, G.; Nolte, F.; Burmeister, T.; Turkmen, S.; Benlasfer, O.; Schumann, E.; Sindram, A.; et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011, 118, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- Blau, O.; Hofmann, W.K.; Baldus, C.D.; Thiel, G.; Serbent, V.; Schumann, E.; Thiel, E.; Blau, I.W. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp. Hematol. 2007, 35, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Corradi, G.; Baldazzi, C.; Ocadlikova, D.; Marconi, G.; Parisi, S.; Testoni, N.; Finelli, C.; Cavo, M.; Curti, A.; Ciciarello, M. Mesenchymal stromal cells from myelodysplastic and acute myeloid leukemia patients display in vitro reduced proliferative potential and similar capacity to support leukemia cell survival. Stem Cell Res. Ther. 2018, 9, 271. [Google Scholar] [CrossRef]
- Choi, H.; Kim, Y.; Kang, D.; Kwon, A.; Kim, J.; Min Kim, J.; Park, S.S.; Kim, Y.J.; Min, C.K.; Kim, M. Common and different alterations of bone marrow mesenchymal stromal cells in myelodysplastic syndrome and multiple myeloma. Cell Prolif. 2020, 53, e12819. [Google Scholar] [CrossRef]
- Fabiani, E.; Falconi, G.; Fianchi, L.; Guidi, F.; Bellesi, S.; Voso, M.T.; Leone, G.; D’Alo, F. Mutational analysis of bone marrow mesenchymal stromal cells in myeloid malignancies. Exp. Hematol. 2014, 42, 731–733. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, Z. Mesenchymal stem cells in pathogenesis of myelodysplastic syndromes. Stem Cell Investig. 2014, 1, 16. [Google Scholar]
- Falconi, G.; Fabiani, E.; Fianchi, L.; Criscuolo, M.; Raffaelli, C.S.; Bellesi, S.; Hohaus, S.; Voso, M.T.; D’Alo, F.; Leone, G. Impairment of PI3K/AKT and WNT/beta-catenin pathways in bone marrow mesenchymal stem cells isolated from patients with myelodysplastic syndromes. Exp. Hematol. 2016, 44, 75–83. [Google Scholar] [CrossRef]
- Weickert, M.T.; Hecker, J.S.; Buck, M.C.; Schreck, C.; Riviere, J.; Schiemann, M.; Schallmoser, K.; Bassermann, F.; Strunk, D.; Oostendorp, R.A.J.; et al. Bone marrow stromal cells from MDS and AML patients show increased adipogenic potential with reduced Delta-like-1 expression. Sci. Rep. 2021, 11, 5944. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Aanei, C.M.; Kesr, S.; Picot, T.; Guyotat, D.; Campos Catafal, L. Impaired Expression of Focal Adhesion Kinase in Mesenchymal Stromal Cells from Low-Risk Myelodysplastic Syndrome Patients. Front. Oncol. 2017, 7, 164. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Santamaria, C.; Muntion, S.; Roson, B.; Blanco, B.; Lopez-Villar, O.; Carrancio, S.; Sanchez-Guijo, F.M.; Diez-Campelo, M.; Alvarez-Fernandez, S.; Sarasquete, M.E.; et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica 2012, 97, 1218–1224. [Google Scholar] [CrossRef]
- Vasta, L.M.; Khan, N.E.; Higgs, C.P.; Harney, L.A.; Carr, A.G.; Harris, A.K.; Schultz, K.A.P.; McMaster, M.L.; Stewart, D.R. Hematologic indices in individuals with pathogenic germline DICER1 variants. Blood Adv. 2021, 5, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, D.; Fei, C.; Guo, J.; Gu, S.; Zhu, Y.; Xu, F.; Zhang, Z.; Wu, L.; Li, X.; et al. Down-regulation of Dicer1 promotes cellular senescence and decreases the differentiation and stem cell-supporting capacities of mesenchymal stromal cells in patients with myelodysplastic syndrome. Haematologica 2015, 100, 194–204. [Google Scholar] [CrossRef]
- Lin, Y.W.; Slape, C.; Zhang, Z.; Aplan, P.D. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood 2005, 106, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Weidner, H.; Rauner, M.; Trautmann, F.; Schmitt, J.; Balaian, E.; Mies, A.; Helas, S.; Baschant, U.; Khandanpour, C.; Bornhauser, M.; et al. Myelodysplastic syndromes and bone loss in mice and men. Leukemia 2017, 31, 1003–1007. [Google Scholar] [CrossRef]
- Weidner, H.; Baschant, U.; Lademann, F.; Ledesma Colunga, M.G.; Balaian, E.; Hofbauer, C.; Misof, B.M.; Roschger, P.; Blouin, S.; Richards, W.G.; et al. Increased FGF-23 levels are linked to ineffective erythropoiesis and impaired bone mineralization in myelodysplastic syndromes. JCI Insight 2020, 5, e137062. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef]
- Rouault-Pierre, K.; Mian, S.A.; Goulard, M.; Abarrategi, A.; Di Tulio, A.; Smith, A.E.; Mohamedali, A.; Best, S.; Nloga, A.M.; Kulasekararaj, A.G.; et al. Preclinical modeling of myelodysplastic syndromes. Leukemia 2017, 31, 2702–2708. [Google Scholar] [CrossRef] [PubMed]
- Krevvata, M.; Shan, X.; Zhou, C.; Dos Santos, C.; Habineza Ndikuyeze, G.; Secreto, A.; Glover, J.; Trotman, W.; Brake-Silla, G.; Nunez-Cruz, S.; et al. Cytokines increase engraftment of human acute myeloid leukemia cells in immunocompromised mice but not engraftment of human myelodysplastic syndrome cells. Haematologica 2018, 103, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Mian, S.A.; Abarrategi, A.; Kong, K.L.; Rouault-Pierre, K.; Wood, H.; Oedekoven, C.A.; Smith, A.E.; Batsivari, A.; Ariza-McNaughton, L.; Johnson, P.; et al. Ectopic humanized mesenchymal niche in mice enables robust engraftment of myelodysplastic stem cells. Blood Cancer Discov. 2021, 2, 135–145. [Google Scholar] [CrossRef]
- Antonelli, A.; Noort, W.A.; Jaques, J.; de Boer, B.; de Jong-Korlaar, R.; Brouwers-Vos, A.Z.; Lubbers-Aalders, L.; van Velzen, J.F.; Bloem, A.C.; Yuan, H.; et al. Establishing human leukemia xenograft mouse models by implanting human bone marrow-like scaffold-based niches. Blood 2016, 128, 2949–2959. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, A.; Thomas, D.; Corces, M.R.; Zhang, X.; Gratzinger, D.; Hong, W.J.; Schallmoser, K.; Strunk, D.; Majeti, R. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat. Med. 2016, 22, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.E.; Wagers, A.J.; Gulati, A.P.; Johnson, F.L.; Weissman, I.L. Physiological migration of hematopoietic stem and progenitor cells. Science 2001, 294, 1933–1936. [Google Scholar] [CrossRef]
- Bixel, M.G.; Kusumbe, A.P.; Ramasamy, S.K.; Sivaraj, K.K.; Butz, S.; Vestweber, D.; Adams, R.H. Flow Dynamics and HSPC Homing in Bone Marrow Microvessels. Cell Rep. 2017, 18, 1804–1816. [Google Scholar] [CrossRef]
- Flynn, C.M.; Kaufman, D.S. Donor cell leukemia: Insight into cancer stem cells and the stem cell niche. Blood 2007, 109, 2688–2692. [Google Scholar] [CrossRef][Green Version]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef]
- Frisch, B.J.; Ashton, J.M.; Xing, L.; Becker, M.W.; Jordan, C.T.; Calvi, L.M. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood 2012, 119, 540–550. [Google Scholar] [CrossRef]
- Wei, J.; Wunderlich, M.; Fox, C.; Alvarez, S.; Cigudosa, J.C.; Wilhelm, J.S.; Zheng, Y.; Cancelas, J.A.; Gu, Y.; Jansen, M.; et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell 2008, 13, 483–495. [Google Scholar] [CrossRef]
- Hawkins, E.D.; Duarte, D.; Akinduro, O.; Khorshed, R.A.; Passaro, D.; Nowicka, M.; Straszkowski, L.; Scott, M.K.; Rothery, S.; Ruivo, N.; et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016, 538, 518–522. [Google Scholar] [CrossRef]
- Wang, W.; Zimmerman, G.; Huang, X.; Yu, S.; Myers, J.; Wang, Y.; Moreton, S.; Nthale, J.; Awadallah, A.; Beck, R.; et al. Aberrant Notch Signaling in the Bone Marrow Microenvironment of Acute Lymphoid Leukemia Suppresses Osteoblast-Mediated Support of Hematopoietic Niche Function. Cancer Res. 2016, 76, 1641–1652. [Google Scholar] [CrossRef]
- Hameed, A.; Brady, J.J.; Dowling, P.; Clynes, M.; O’Gorman, P. Bone disease in multiple myeloma: Pathophysiology and management. Cancer Growth Metastasis 2014, 7, 33–42. [Google Scholar] [CrossRef]
- Hatfield, K.; Oyan, A.M.; Ersvaer, E.; Kalland, K.H.; Lassalle, P.; Gjertsen, B.T.; Bruserud, O. Primary human acute myeloid leukaemia cells increase the proliferation of microvascular endothelial cells through the release of soluble mediators. Br. J. Haematol. 2009, 144, 53–68. [Google Scholar] [CrossRef]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegue, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef]
- Krause, J.R. Bone Marrow Biopsy; Churchill Livingstone: New York, NY, USA, 1981; pp. 1–9. [Google Scholar]
- Tricot, G.; De Wolf-Peeters, C.; Hendrickx, B.; Verwilghen, R.L. Bone marrow histology in myelodysplastic syndromes. I. Histological findings in myelodysplastic syndromes and comparison with bone marrow smears. Br. J. Haematol. 1984, 57, 423–430. [Google Scholar] [CrossRef]
- Passaro, D.; Di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341. [Google Scholar] [CrossRef]
- Duarte, D.; Hawkins, E.D.; Akinduro, O.; Ang, H.; De Filippo, K.; Kong, I.Y.; Haltalli, M.; Ruivo, N.; Straszkowski, L.; Vervoort, S.J.; et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell 2018, 22, 64–77. [Google Scholar] [CrossRef]
- Kim, C.K.; Han, D.H.; Ji, Y.S.; Lee, M.S.; Min, C.W.; Park, S.K.; Kim, S.H.; Yun, J.; Kim, H.J.; Kim, K.H.; et al. Biomarkers of angiogenesis as prognostic factors in myelodysplastic syndrome patients treated with hypomethylating agents. Leuk. Res. 2016, 50, 21–28. [Google Scholar] [CrossRef]
- Korkolopoulou, P.; Apostolidou, E.; Pavlopoulos, P.M.; Kavantzas, N.; Vyniou, N.; Thymara, I.; Terpos, E.; Patsouris, E.; Yataganas, X.; Davaris, P. Prognostic evaluation of the microvascular network in myelodysplastic syndromes. Leukemia 2001, 15, 1369–1376. [Google Scholar] [CrossRef][Green Version]
- Pruneri, G.; Bertolini, F.; Soligo, D.; Carboni, N.; Cortelezzi, A.; Ferrucci, P.F.; Buffa, R.; Lambertenghi-Deliliers, G.; Pezzella, F. Angiogenesis in myelodysplastic syndromes. Br. J. Cancer 1999, 81, 1398–1401. [Google Scholar] [CrossRef]
- Brunner, B.; Gunsilius, E.; Schumacher, P.; Zwierzina, H.; Gastl, G.; Stauder, R. Blood levels of angiogenin and vascular endothelial growth factor are elevated in myelodysplastic syndromes and in acute myeloid leukemia. J. Hematother. Stem Cell Res. 2002, 11, 119–125. [Google Scholar] [CrossRef]
- Alexandrakis, M.G.; Passam, F.H.; Pappa, C.A.; Sfiridaki, K.; Tsirakis, G.; Damilakis, J.; Stathopoulos, E.N.; Kyriakou, D.S. Relation between bone marrow angiogenesis and serum levels of angiogenin in patients with myelodysplastic syndromes. Leuk. Res. 2005, 29, 41–46. [Google Scholar] [CrossRef]
- Alexandrakis, M.G.; Passam, F.H.; Pappa, C.A.; Damilakis, J.; Tsirakis, G.; Kandidaki, E.; Passam, A.M.; Stathopoulos, E.N.; Kyriakou, D.S. Serum evaluation of angiogenic cytokine basic fibroblast growth factor, hepatocyte growth factor and TNF-alpha in patients with myelodysplastic syndromes: Correlation with bone marrow microvascular density. Int. J. Immunopathol. Pharmacol. 2005, 18, 287–295. [Google Scholar] [CrossRef]
- Feng, X.; Scheinberg, P.; Wu, C.O.; Samsel, L.; Nunez, O.; Prince, C.; Ganetzky, R.D.; McCoy, J.P., Jr.; Maciejewski, J.P.; Young, N.S. Cytokine signature profiles in acquired aplastic anemia and myelodysplastic syndromes. Haematologica 2011, 96, 602–606. [Google Scholar] [CrossRef]
- Bellamy, W.T.; Richter, L.; Sirjani, D.; Roxas, C.; Glinsmann-Gibson, B.; Frutiger, Y.; Grogan, T.M.; List, A.F. Vascular endothelial cell growth factor is an autocrine promoter of abnormal localized immature myeloid precursors and leukemia progenitor formation in myelodysplastic syndromes. Blood 2001, 97, 1427–1434. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Teofili, L.; Martini, M.; Nuzzolo, E.R.; Capodimonti, S.; Iachininoto, M.G.; Cocomazzi, A.; Fabiani, E.; Voso, M.T.; Larocca, L.M. Endothelial progenitor cell dysfunction in myelodysplastic syndromes: Possible contribution of a defective vascular niche to myelodysplasia. Neoplasia 2015, 17, 401–409. [Google Scholar] [CrossRef]
- Houwerzijl, E.J.; van den Heuvel, F.A.; Blom, N.R.; van der Want, J.J.; Mulder, A.B.; Vellenga, E. Sinusoidal endothelial cells are damaged and display enhanced autophagy in myelodysplastic syndromes. Br. J. Haematol. 2013, 161, 443–446. [Google Scholar] [CrossRef]
- Della Porta, M.G.; Malcovati, L.; Rigolin, G.M.; Rosti, V.; Bonetti, E.; Travaglino, E.; Boveri, E.; Galli, A.; Boggi, S.; Ciccone, M.; et al. Immunophenotypic, cytogenetic and functional characterization of circulating endothelial cells in myelodysplastic syndromes. Leukemia 2008, 22, 530–537. [Google Scholar] [CrossRef]
- van Leeuwen-Kerkhoff, N.; Westers, T.M.; Poddighe, P.J.; de Gruijl, T.D.; Kordasti, S.; van de Loosdrecht, A.A. Thrombomodulin-expressing monocytes are associated with low-risk features in myelodysplastic syndromes and dampen excessive immune activation. Haematologica 2020, 105, 961–971. [Google Scholar] [CrossRef]
- Selimoglu-Buet, D.; Badaoui, B.; Benayoun, E.; Toma, A.; Fenaux, P.; Quesnel, B.; Etienne, G.; Braun, T.; Abermil, N.; Morabito, M.; et al. Accumulation of classical monocytes defines a subgroup of MDS that frequently evolves into CMML. Blood 2017, 130, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.A.; Galili, N.; Cerny, J.; Sechman, E.; Chen, S.S.; Loew, J.; Liu, Q.; Fadare, O.; Hasserjian, R.; Jones, D.; et al. Chronic myelomonocytic leukemia evolving from preexisting myelodysplasia shares many features with de novo disease. Am. J. Clin. Pathol. 2006, 126, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Gao, P.; Wu, N.; Shi, C.; Huang, Z.; Rong, C.; Sun, Y.; Sheng, L.; Ouyang, G.; Mu, Q. Elevated mature monocytes in bone marrow accompanied with a higher IPSS-R score predicts a poor prognosis in myelodysplastic syndromes. BMC Cancer 2021, 21, 546. [Google Scholar] [CrossRef]
- Sarhan, D.; Wang, J.; Sunil Arvindam, U.; Hallstrom, C.; Verneris, M.R.; Grzywacz, B.; Warlick, E.; Blazar, B.R.; Miller, J.S. Mesenchymal stromal cells shape the MDS microenvironment by inducing suppressive monocytes that dampen NK cell function. JCI Insight 2020, 5, e130155. [Google Scholar] [CrossRef]
- Hur, J.; Choi, J.I.; Lee, H.; Nham, P.; Kim, T.W.; Chae, C.W.; Yun, J.Y.; Kang, J.A.; Kang, J.; Lee, S.E.; et al. CD82/KAI1 Maintains the Dormancy of Long-Term Hematopoietic Stem Cells through Interaction with DARC-Expressing Macrophages. Cell Stem Cell 2016, 18, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xue, W.; Li, M.; Dong, M.; Wang, J.; Wang, X.; Li, X.; Chen, K.; Zhang, W.; Wu, S.; et al. VCAM-1(+) macrophages guide the homing of HSPCs to a vascular niche. Nature 2018, 564, 119–124. [Google Scholar] [CrossRef]
- Chow, A.; Huggins, M.; Ahmed, J.; Hashimoto, D.; Lucas, D.; Kunisaki, Y.; Pinho, S.; Leboeuf, M.; Noizat, C.; van Rooijen, N.; et al. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 2013, 19, 429–436. [Google Scholar] [CrossRef]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef]
- Petty, A.J.; Yang, Y. Tumor-Associated Macrophages in Hematologic Malignancies: New Insights and Targeted Therapies. Cells 2019, 8, 1526. [Google Scholar] [CrossRef]
- Pang, W.W.; Pluvinage, J.V.; Price, E.A.; Sridhar, K.; Arber, D.A.; Greenberg, P.L.; Schrier, S.L.; Park, C.Y.; Weissman, I.L. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc. Natl. Acad. Sci. USA 2013, 110, 3011–3016. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Y. The Clinical Significance of Tumor Associated Macrophages in Myelodysplastic Syndromes. Blood 2018, 132 (Suppl. S1), 5505. [Google Scholar] [CrossRef]
- Allampallam, K.; Shetty, V.; Mundle, S.; Dutt, D.; Kravitz, H.; Reddy, P.L.; Alvi, S.; Galili, N.; Saberwal, G.S.; Anthwal, S.; et al. Biological significance of proliferation, apoptosis, cytokines, and monocyte/macrophage cells in bone marrow biopsies of 145 patients with myelodysplastic syndrome. Int. J. Hematol. 2002, 75, 289–297. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Gabrilovich, D.I. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol. Immunother 2006, 55, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Invest. 2013, 123, 4595–4611. [Google Scholar] [CrossRef] [PubMed]
- Kittang, A.O.; Kordasti, S.; Sand, K.E.; Costantini, B.; Kramer, A.M.; Perezabellan, P.; Seidl, T.; Rye, K.P.; Hagen, K.M.; Kulasekararaj, A.; et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. Oncoimmunology 2016, 5, e1062208. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Zhao, B.; Basiorka, A.A.; Yang, J.; Cao, L.; Zhang, J.; List, A.; Ji, P. Age-related inflammatory bone marrow microenvironment induces ineffective erythropoiesis mimicking del(5q) MDS. Leukemia 2018, 32, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Younos, I.H.; Abe, F.; Talmadge, J.E. Myeloid-derived suppressor cells: Their role in the pathophysiology of hematologic malignancies and potential as therapeutic targets. Leuk. Lymphoma 2015, 56, 2251–2263. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Camacho, V.; Matkins, V.R.; Patel, S.B.; Lever, J.M.; Yang, Z.; Ying, L.; Landuyt, A.E.; Dean, E.C.; George, J.F.; Yang, H.; et al. Bone marrow Tregs mediate stromal cell function and support hematopoiesis via IL-10. JCI Insight 2020, 5, e135681. [Google Scholar] [CrossRef]
- Kordasti, S.Y.; Ingram, W.; Hayden, J.; Darling, D.; Barber, L.; Afzali, B.; Lombardi, G.; Wlodarski, M.W.; Maciejewski, J.P.; Farzaneh, F.; et al. CD4+CD25high Foxp3+ regulatory T cells in myelodysplastic syndrome (MDS). Blood 2007, 110, 847–850. [Google Scholar] [CrossRef]
- Costantini, B.; Kordasti, S.Y.; Kulasekararaj, A.G.; Jiang, J.; Seidl, T.; Abellan, P.P.; Mohamedali, A.; Thomas, N.S.; Farzaneh, F.; Mufti, G.J. The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica 2013, 98, 1196–1205. [Google Scholar] [CrossRef]
- Giovazzino, A.; Leone, S.; Rubino, V.; Palatucci, A.T.; Cerciello, G.; Alfinito, F.; Pane, F.; Ruggiero, G.; Terrazzano, G. Reduced regulatory T cells (Treg) in bone marrow preferentially associate with the expansion of cytotoxic T lymphocytes in low risk MDS patients. Br. J. Haematol. 2019, 185, 357–360. [Google Scholar] [CrossRef]
- Kotsianidis, I.; Bouchliou, I.; Nakou, E.; Spanoudakis, E.; Margaritis, D.; Christophoridou, A.V.; Anastasiades, A.; Tsigalou, C.; Bourikas, G.; Karadimitris, A.; et al. Kinetics, function and bone marrow trafficking of CD4+CD25+FOXP3+ regulatory T cells in myelodysplastic syndromes (MDS). Leukemia 2009, 23, 510–518. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Bjorkholm, M.; Hultcrantz, M.; Derolf, A.R.; Landgren, O.; Goldin, L.R. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 2897–2903. [Google Scholar] [CrossRef]
- Sato, T.; Onai, N.; Yoshihara, H.; Arai, F.; Suda, T.; Ohteki, T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat. Med. 2009, 15, 696–700. [Google Scholar] [CrossRef]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef]
- Matatall, K.A.; Shen, C.C.; Challen, G.A.; King, K.Y. Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cells 2014, 32, 3023–3030. [Google Scholar] [CrossRef]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 2010, 465, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, G.; Mills, T.S.; Rabe, J.L.; Chavez, J.S.; Kuldanek, S.; Kirkpatrick, G.; Noetzli, L.; Jubair, W.K.; Zanche, M.; Myers, J.R.; et al. Pro-inflammatory cytokine blockade attenuates myeloid expansion in a murine model of rheumatoid arthritis. Haematologica 2020, 105, 585–597. [Google Scholar] [CrossRef]
- Zhao, J.L.; Ma, C.; O’Connell, R.M.; Mehta, A.; DiLoreto, R.; Heath, J.R.; Baltimore, D. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell 2014, 14, 445–459. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef]
- Chen, Z.; Amro, E.M.; Becker, F.; Holzer, M.; Rasa, S.M.M.; Njeru, S.N.; Han, B.; Di Sanzo, S.; Chen, Y.; Tang, D.; et al. Cohesin-mediated NF-kappaB signaling limits hematopoietic stem cell self-renewal in aging and inflammation. J. Exp. Med. 2019, 216, 152–175. [Google Scholar] [CrossRef]
- Matatall, K.A.; Jeong, M.; Chen, S.; Sun, D.; Chen, F.; Mo, Q.; Kimmel, M.; King, K.Y. Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Rep. 2016, 17, 2584–2595. [Google Scholar] [CrossRef]
- Barreyro, L.; Will, B.; Bartholdy, B.; Zhou, L.; Todorova, T.I.; Stanley, R.F.; Ben-Neriah, S.; Montagna, C.; Parekh, S.; Pellagatti, A.; et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood 2012, 120, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, D.; Pietras, E.; Barry-Holson, K.; Mir, A.; Binnewies, M.; Jeanne, M.; Sala-Torra, O.; Radich, J.P.; Passegue, E. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell 2011, 20, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Stifter, G.; Heiss, S.; Gastl, G.; Tzankov, A.; Stauder, R. Over-expression of tumor necrosis factor-alpha in bone marrow biopsies from patients with myelodysplastic syndromes: Relationship to anemia and prognosis. Eur. J. Haematol. 2005, 75, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Rambaldi, A.; Torcia, M.; Dinarello, C.A.; Barbui, T.; Cozzolino, F. Modulation of cell proliferation and cytokine production in AML by recombinant interleukin-1 receptor antagonist. Leukemia 1993, 7 (Suppl. 2), S10–S12. [Google Scholar]
- Lambert, C.; Wu, Y.; Aanei, C. Bone Marrow Immunity and Myelodysplasia. Front. Oncol. 2016, 6, 172. [Google Scholar] [CrossRef]
- Navas, T.A.; Mohindru, M.; Estes, M.; Ma, J.Y.; Sokol, L.; Pahanish, P.; Parmar, S.; Haghnazari, E.; Zhou, L.; Collins, R.; et al. Inhibition of overactivated p38 MAPK can restore hematopoiesis in myelodysplastic syndrome progenitors. Blood 2006, 108, 4170–4177. [Google Scholar] [CrossRef]
- Kondo, A.; Yamashita, T.; Tamura, H.; Zhao, W.; Tsuji, T.; Shimizu, M.; Shinya, E.; Takahashi, H.; Tamada, K.; Chen, L.; et al. Interferon-gamma and tumor necrosis factor-alpha induce an immunoinhibitory molecule, B7-H1, via nuclear factor-kappaB activation in blasts in myelodysplastic syndromes. Blood 2010, 116, 1124–1131. [Google Scholar] [CrossRef]
- Ganan-Gomez, I.; Wei, Y.; Starczynowski, D.T.; Colla, S.; Yang, H.; Cabrero-Calvo, M.; Bohannan, Z.S.; Verma, A.; Steidl, U.; Garcia-Manero, G. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia 2015, 29, 1458–1469. [Google Scholar] [CrossRef]
- Wei, Y.; Dimicoli, S.; Bueso-Ramos, C.; Chen, R.; Yang, H.; Neuberg, D.; Pierce, S.; Jia, Y.; Zheng, H.; Wang, H.; et al. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia 2013, 27, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Ganan-Gomez, I.; Wei, Y.; Yang, H.; Pierce, S.; Bueso-Ramos, C.; Calin, G.; Boyano-Adanez Mdel, C.; Garcia-Manero, G. Overexpression of miR-125a in myelodysplastic syndrome CD34+ cells modulates NF-kappaB activation and enhances erythroid differentiation arrest. PLoS ONE 2014, 9, e93404. [Google Scholar] [CrossRef] [PubMed]
- Dimicoli, S.; Wei, Y.; Bueso-Ramos, C.; Yang, H.; Dinardo, C.; Jia, Y.; Zheng, H.; Fang, Z.; Nguyen, M.; Pierce, S.; et al. Overexpression of the toll-like receptor (TLR) signaling adaptor MYD88, but lack of genetic mutation, in myelodysplastic syndromes. PLoS ONE 2013, 8, e71120. [Google Scholar] [CrossRef] [PubMed]
- Briantais, A.; Seguier, J.; De Sainte Marie, B.; Mekinian, A.; Belizna, C.; Gondran, G.; Maurier, F.; Trouiller, S.; Willems, L.; Beyne-Rauzy, O.; et al. Inflammatory myopathies associated with myelodysplastic syndromes: A French multicenter case control study and literature review. Semin. Arthritis Rheum. 2021, 51, 845–852. [Google Scholar] [CrossRef]
- Seguier, J.; Gelsi-Boyer, V.; Ebbo, M.; Hamidou, Z.; Charbonnier, A.; Bernit, E.; Durand, J.M.; Harle, J.R.; Vey, N.; Schleinitz, N. Autoimmune diseases in myelodysplastic syndrome favors patients survival: A case control study and literature review. Autoimmun. Rev. 2019, 18, 36–42. [Google Scholar] [CrossRef]
- Wesner, N.; Drevon, L.; Guedon, A.; Fraison, J.B.; Trad, S.; Kahn, J.E.; Aouba, A.; Gillard, J.; Ponsoye, M.; Hanslik, T.; et al. Inflammatory disorders associated with trisomy 8-myelodysplastic syndromes: French retrospective case-control study. Eur. J. Haematol. 2019, 102, 63–69. [Google Scholar] [CrossRef]
- Geyh, S.; Rodriguez-Paredes, M.; Jager, P.; Koch, A.; Bormann, F.; Gutekunst, J.; Zilkens, C.; Germing, U.; Kobbe, G.; Lyko, F.; et al. Transforming growth factor beta1-mediated functional inhibition of mesenchymal stromal cells in myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2018, 103, 1462–1471. [Google Scholar] [CrossRef]
- Ward, G.A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; Meyer, B.S.; McLemore, A.F.; Vincelette, N.D.; Lam, N.B.; Aldrich, A.L.; Al Ali, N.H.; Padron, E.; et al. Oxidized mitochondrial DNA released after inflammasome activation is a disease biomarker for myelodysplastic syndromes. Blood Adv. 2021, 5, 2216–2228. [Google Scholar] [CrossRef]
- Schneider, R.K.; Schenone, M.; Ferreira, M.V.; Kramann, R.; Joyce, C.E.; Hartigan, C.; Beier, F.; Brummendorf, T.H.; Germing, U.; Platzbecker, U.; et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat. Med. 2016, 22, 288–297. [Google Scholar] [CrossRef]
- Hellstrom-Lindberg, E.; Tobiasson, M.; Greenberg, P. Myelodysplastic syndromes: Moving towards personalized management. Haematologica 2020, 105, 1765–1779. [Google Scholar] [CrossRef] [PubMed]
- Kubasch, A.S.; Fenaux, P.; Platzbecker, U. Development of luspatercept to treat ineffective erythropoiesis. Blood Adv. 2021, 5, 1565–1575. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Diez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Suragani, R.N.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.; Loveday, K.S.; et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Phase I First-in-Human Dose Escalation Study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 2021. [Google Scholar] [CrossRef]
- Henry, D.H.; Glaspy, J.; Harrup, R.A.; Mittelman, M.; Zhou, A.; Bradley, C.; Saha, G.; Bartels, P.; Robert, L.; Yu, K.-H.P. Roxadustat (FG4592; ASP1517; AZD9941) in the Treatment of Anemia in Patients with Lower Risk Myelodysplastic Syndrome (LR-MDS) and Low Red Blood Cell (RBC) Transfusion Burden (LTB). Blood 2019, 134 (Suppl. S1), 843. [Google Scholar] [CrossRef]
- Yan, Z.; Xu, G. A Novel Choice to Correct Inflammation-Induced Anemia in CKD: Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor Roxadustat. Front. Med. 2020, 7, 393. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Daver, N.G.; Xu, J.; Chao, M.; Chung, T.; Tan, A.; Wang, V.; Wei, A.; Vyas, P.; Sallman, D.A. Magrolimab + azacitidine versus azacitidine + placebo in untreated higher risk (HR) myelodysplastic syndrome (MDS): The phase 3, randomized, ENHANCE study. J. Clin. Oncol. 2021, 39 (Suppl. S15), TPS7055. [Google Scholar] [CrossRef]
- Ball, B.J.; Famulare, C.A.; Stein, E.M.; Tallman, M.S.; Derkach, A.; Roshal, M.; Gill, S.I.; Manning, B.M.; Koprivnikar, J.; McCloskey, J.; et al. Venetoclax and hypomethylating agents (HMAs) induce high response rates in MDS, including patients after HMA therapy failure. Blood Adv. 2020, 4, 2866–2870. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.S. Prospects for Venetoclax in Myelodysplastic Syndromes. Hematol. Oncol. Clin. N. Am. 2020, 34, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Stanchina, M.; Chaudhry, S.; Karr, M.; Taylor, J. Current State and Challenges in Development of Targeted Therapies in Myelodysplastic Syndromes (MDS). Hematology 2021, 2, 217–236. [Google Scholar]
- Jamieson, C.; Martinelli, G.; Papayannidis, C.; Cortes, J.E. Hedgehog Pathway Inhibitors: A New Therapeutic Class for the Treatment of Acute Myeloid Leukemia. Blood Cancer Discov. 2020, 1, 134–145. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mian, S.A.; Bonnet, D. Nature or Nurture? Role of the Bone Marrow Microenvironment in the Genesis and Maintenance of Myelodysplastic Syndromes. Cancers 2021, 13, 4116. https://doi.org/10.3390/cancers13164116
Mian SA, Bonnet D. Nature or Nurture? Role of the Bone Marrow Microenvironment in the Genesis and Maintenance of Myelodysplastic Syndromes. Cancers. 2021; 13(16):4116. https://doi.org/10.3390/cancers13164116
Chicago/Turabian StyleMian, Syed A., and Dominique Bonnet. 2021. "Nature or Nurture? Role of the Bone Marrow Microenvironment in the Genesis and Maintenance of Myelodysplastic Syndromes" Cancers 13, no. 16: 4116. https://doi.org/10.3390/cancers13164116
APA StyleMian, S. A., & Bonnet, D. (2021). Nature or Nurture? Role of the Bone Marrow Microenvironment in the Genesis and Maintenance of Myelodysplastic Syndromes. Cancers, 13(16), 4116. https://doi.org/10.3390/cancers13164116