
The Impact of Induction Regimes on Immune Responses in Patients with Multiple Myeloma



Abstract
Simple Summary
Abstract
1. Introduction
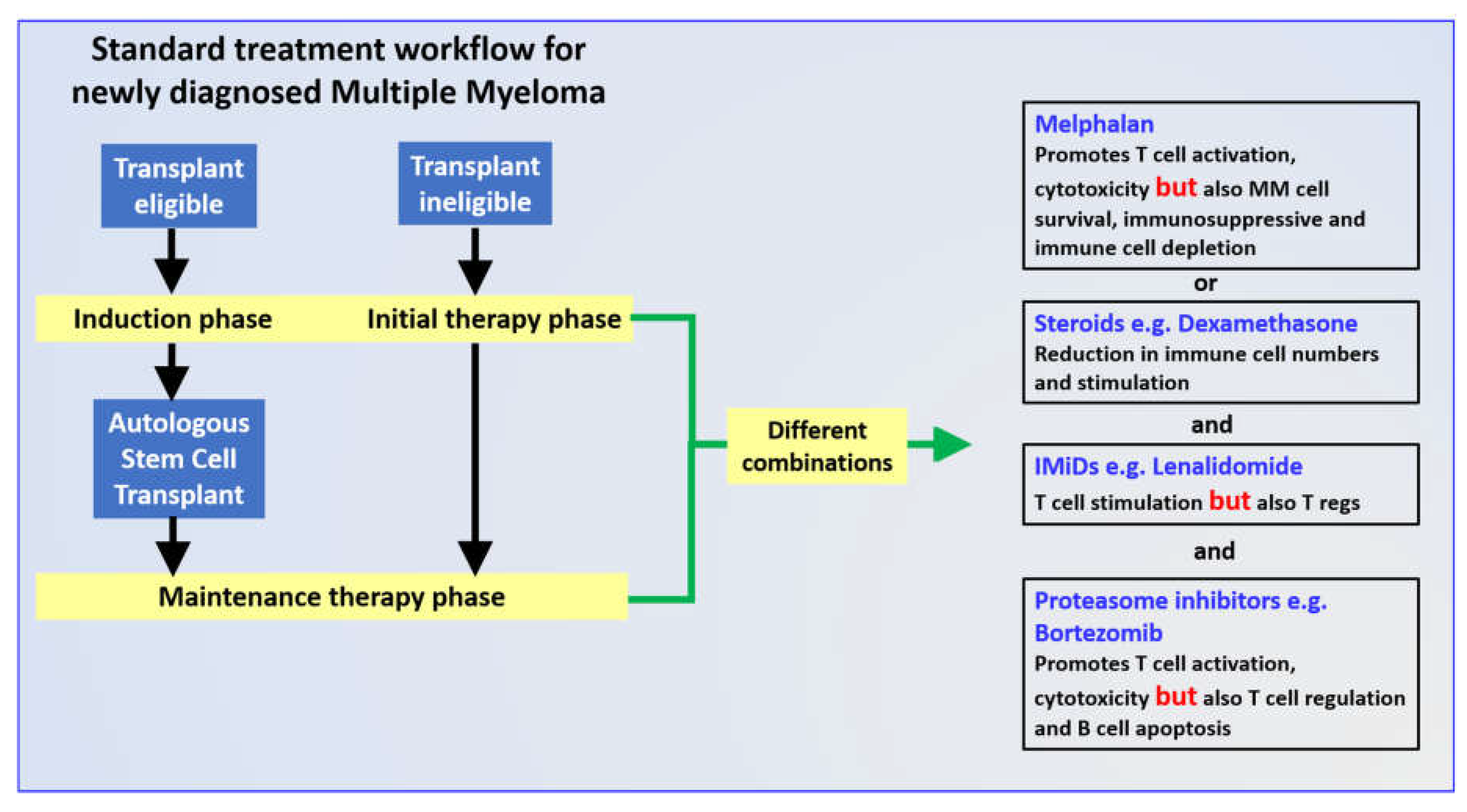
2. Immune Efforts of Individual Induction Regime Drugs
2.1. Melphalan
2.2. Dexamethasone
2.3. Lenalidomide
2.4. Bortezomib
3. Combination Induction Regimes
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shen, Y.J.; Mishima, Y.; Shi, J.; Sklavenitis-Pistofidis, R.; Redd, R.A.; Moschetta, M.; Manier, S.; Roccaro, A.M.; Sacco, A.; Tai, Y.-T.; et al. Progression signature underlies clonal evolution and dissemination of multiple myeloma. Blood 2021, 137, 2360–2372. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Landgren, O. MGUS and Smoldering Multiple Myeloma: Diagnosis and Epidemiology. In Plasma Cell Dyscrasias; Macmillan Education: London, UK, 2016; Volume 169, pp. 3–12. [Google Scholar]
- Lomas, O.; Tahri, S.; Ghobrial, I.M. The microenvironment in myeloma. Curr. Opin. Oncol. 2020, 32, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat. Rev. Cancer 2020, 1, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar] [CrossRef]
- Gay, F.; Engelhardt, M.; Terpos, E.; Wäsch, R.; Giaccone, L.; Auner, H.; Caers, J.; Gramatzki, M.; Van De Donk, N.; Oliva, S.; et al. From transplant to novel cellular therapies in multiple myeloma: European Myeloma Network guidelines and future perspectives. Haematologica 2017, 103, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Vuckovic, S.; Minnie, S.; Smith, D.; Gartlan, K.H.; Watkins, T.S.; Markey, K.A.; Mukhopadhyay, P.; Guillerey, C.; Kuns, R.D.; Locke, K.R.; et al. Bone marrow transplantation generates T cell–dependent control of myeloma in mice. J. Clin. Investig. 2018, 129, 106–121. [Google Scholar] [CrossRef]
- Minnie, S.A.; Hill, G.R. Autologous Stem Cell Transplantation for Myeloma: Cytoreduction or an Immunotherapy? Front. Immunol. 2021, 12, 1288. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef]
- Ma, T.; Shi, J.; Liu, H. Chimeric antigen receptor T cell targeting B cell maturation antigen immunotherapy is promising for multiple myeloma. Ann. Hematol. 2019, 98, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Arteche-López, A.; Kreutzman, A.; Alegre, A.; Martín, P.S.; Aguado, B.; Pardo, M.G.; Espiño, M.; Villar, L.M.; Belmonte, D.G.; de la Camara, R.; et al. Multiple myeloma patients in long-term complete response after autologous stem cell transplantation express a particular immune signature with potential prognostic implication. Bone Marrow Transplant. 2017, 52, 832–838. [Google Scholar] [CrossRef]
- Ho, C.M.; McCarthy, P.L.; Wallace, P.; Zhang, Y.; Fora, A.; Mellors, P.; Tario, J.D.; McCarthy, B.L.S.; Chen, G.L.; Holstein, S.A.; et al. Immune signatures associated with improved progression-free and overall survival for myeloma patients treated with AHSCT. Blood Adv. 2017, 1, 1056–1066. [Google Scholar] [CrossRef]
- Goicoechea, I.; Puig, N.; Cedena, M.-T.; Burgos, L.; Cordón, L.; Vidriales, M.-B.; Flores-Montero, J.; Gutierrez, N.C.; Calasanz, M.-J.; Ramos, M.-L.M.; et al. Deep MRD profiling defines outcome and unveils different modes of treatment resistance in standard- and high-risk myeloma. Blood 2021, 137, 49–60. [Google Scholar] [CrossRef]
- Paiva, B.; Van Dongen, J.J.M.; Orfao, A. New criteria for response assessment: Role of minimal residual disease in multiple myeloma. Blood 2015, 125, 3059–3068. [Google Scholar] [CrossRef]
- Munshi, N.C.; Avet-Loiseau, H.; Anderson, K.C.; Neri, P.; Paiva, B.; Samur, M.; Dimopoulos, M.; Kulakova, M.; Lam, A.; Hashim, M.; et al. A large meta-analysis establishes the role of MRD negativity in long-term survival outcomes in patients with multiple myeloma. Blood Adv. 2020, 4, 5988–5999. [Google Scholar] [CrossRef]
- Fairfield, H.; Falank, C.; Avery, L.; Reagan, M.R. Multiple myeloma in the marrow: Pathogenesis and treatments. Ann. N. Y. Acad. Sci. 2016, 1364, 32–51. [Google Scholar] [CrossRef]
- Hou, J.; Wei, R.; Qian, J.; Wang, R.; Fan, Z.; Gu, C.; Yang, Y. The impact of the bone marrow microenvironment on mul-tiple myeloma (Review). Oncol. Rep. 2019, 42, 1272–1282. [Google Scholar] [PubMed]
- Falco, P.; Bringhen, S.; Avonto, I.; Gay, F.; Morabito, F.; Boccadoro, M.; Palumbo, A. Melphalan and its role in the management of patients with multiple myeloma. Expert Rev. Anticancer. Ther. 2007, 7, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Poczta, A.; Rogalska, A.; Marczak, A. Treatment of Multiple Myeloma and the Role of Melphalan in the Era of Modern Therapies—Current Research and Clinical Approaches. J. Clin. Med. 2021, 10, 1841. [Google Scholar] [CrossRef]
- Shaw, P.; E Nath, C.; Lazarus, H.M. Not too little, not too much—Just right! (Better ways to give high dose melphalan). Bone Marrow Transplant. 2014, 49, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Ding, Z.-C.; Cao, Y.; Liu, C.; Habtetsion, T.; Yu, M.; Lemos, H.D.P.; Salman, H.; Xu, H.; Mellor, A.L.; et al. Alkylating Agent Melphalan Augments the Efficacy of Adoptive Immunotherapy Using Tumor-Specific CD4+ T Cells. J. Immunol. 2015, 194, 2011–2021. [Google Scholar] [CrossRef]
- Krysko, D.; Garg, A.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Robillard, N.; Jego, J.; Pellat-Deceunynck, C.; Pineau, D.; Puthier, D.; Mellerin, M.; Barille, S.; Rapp, M.; Harousseau, J.-L.; Amiot, M.; et al. CD28, A marker associated with tumoral expansion in multiple myeloma. Clin. Cancer Res. 1998, 4, 1521–1526. [Google Scholar] [PubMed]
- Bahlis, N.J.; King, A.M.; Kolonias, D.; Carlson, L.M.; Liu, H.Y.; Hussein, M.A.; Terebelo, H.R.; Byrne, G.E.; Levine, B.L.; Boise, L.; et al. CD28-mediated regulation of multiple myeloma cell proliferation and survival. Blood 2007, 109, 5002–5010. [Google Scholar] [CrossRef] [PubMed]
- Giles, A.J.; Hutchinson, M.-K.; Sonnemann, H.M.; Jung, J.; Fecci, P.E.; Ratnam, N.M.; Zhang, W.; Song, H.; Bailey, R.; Davis, D.; et al. Dexamethasone-induced immunosuppression: Mechanisms and implications for immunotherapy. J. Immunother. Cancer 2018, 6, 51. [Google Scholar] [CrossRef]
- Presti, M.; Westergaard, M.C.W.; Draghi, A.; Chamberlain, C.A.; Gokuldass, A.; Svane, I.M.; Donia, M. The effects of targeted immune-regulatory strategies on tumor-specific T-cell responses in vitro. Cancer Immunol. Immunother. 2021, 70, 1771–1776. [Google Scholar] [CrossRef]
- Nucci, M.; Anaissie, E. Infections in Patients with Multiple Myeloma in the Era of High-Dose Therapy and Novel Agents. Clin. Infect. Dis. 2009, 49, 1211–1225. [Google Scholar] [CrossRef]
- Holstein, S.A.; McCarthy, P.L. Immunomodulatory Drugs in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Drugs 2017, 77, 505–520. [Google Scholar] [CrossRef]
- Muller, G.W.; Chen, R.; Huang, S.-Y.; Corral, L.G.; Wong, L.M.; Patterson, R.T.; Chen, Y.; Kaplan, G.; Stirling, D.I. Amino-substituted thalidomide analogs: Potent inhibitors of TNF-α production. Bioorg. Med. Chem. Lett. 1999, 9, 1625–1630. [Google Scholar] [CrossRef]
- Chung, D.J.; Pronschinske, K.B.; Shyer, J.A.; Sharma, S.; Leung, S.; Curran, S.A.; Lesokhin, A.M.; Devlin, S.M.; Giralt, S.A.; Young, J. T-cell Exhaustion in Multiple Myeloma Relapse after Autotransplant: Optimal Timing of Immunotherapy. Cancer Immunol. Res. 2016, 4, 61–71. [Google Scholar] [CrossRef]
- Galustian, C.; Meyer, B.; Labarthe, M.-C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D.; et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2008, 58, 1033–1045. [Google Scholar] [CrossRef]
- Shi, J.; Tricot, G.J.; Garg, T.K.; Malaviarachchi, P.A.; Szmania, S.M.; Kellum, R.E.; Storrie, B.; Mulder, A.; Shaughnessy, J.J.D.; Barlogie, B.; et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell–mediated lysis of myeloma. Blood 2008, 111, 1309–1317. [Google Scholar] [CrossRef]
- Feng, X.; Yan, J.; Wang, Y.; Zierath, J.R.; Nordenskjöld, M.; Henter, J.-I.; Fadeel, B.; Zheng, C. The proteasome inhibitor bortezomib disrupts tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression and natural killer (NK) cell killing of TRAIL receptor-positive multiple myeloma cells. Mol. Immunol. 2010, 47, 2388–2396. [Google Scholar] [CrossRef]
- Pellom, S.T.; Dudimah, D.F.; Thounaojam, M.; Sayers, T.J.; Shanker, A. Modulatory effects of bortezomib on host immune cell functions. Immunotherapy 2015, 7, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Thounaojam, M.C.; Dudimah, D.F.; Pellom, S.T., Jr.; Uzhachenko, R.V.; Carbone, D.P.; Dikov, M.M.; Shanker, A. Bortezomib enhances expression of effector molecules in anti-tumor CD8+ T lymphocytes by promoting Notch-nuclear factor-κB crosstalk. Oncotarget 2015, 6, 32439–32455. [Google Scholar] [CrossRef] [PubMed]
- Neeson, P.J.; Hsu, A.K.; Chen, Y.R.; Halse, H.M.; Loh, J.; Cordy, R.; Fielding, K.; E Davis, J.; Noske, J.; Davenport, A.J.; et al. Induction of potent NK cell-dependent anti-myeloma cytotoxic T cells in response to combined mapatumumab and bortezomib. OncoImmunology 2015, 4, e1038011. [Google Scholar] [CrossRef]
- Cascio, P.; Oliva, L.; Cerruti, F.; Mariani, E.; Pasqualetto, E.; Cenci, S.; Sitia, R. Dampening Ab responses using proteasome inhibitors followingin vivo B cell activation. Eur. J. Immunol. 2008, 38, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Mulder, A.; Heidt, S.; Vergunst, M.; Roelen, D.L.; Claas, F.H. Proteasome Inhibition Profoundly Affects Activated Human B Cells. Transplant. 2013, 95, 1331–1337. [Google Scholar] [CrossRef]
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The paradox of Th17 cell functions in tumor immunity. Cell. Immunol. 2017, 322, 15–25. [Google Scholar] [CrossRef]
- Ercetin, A.P.; Ozcan, M.A.; Aktas, S.; Yuksel, F.; Solmaz, S.M.; Sevindik, G.O.; Katgi, A.; Piskin, O.; Undar, B. Ex vivo evaluation of the effect of regulatory T cells on the anti-tumor activity of bortezomib in multiple myeloma. Exp. Hematol. 2016, 44, 223–230. [Google Scholar] [CrossRef]
- Nencioni, A.; Schwarzenberg, K.; Brauer, K.M.; Schmidt, S.M.; Ballestrero, A.; Grünebach, F.; Brossart, P. Proteasome inhibitor bortezomib modulates TLR4-induced dendritic cell activation. Blood 2006, 108, 551–558. [Google Scholar] [CrossRef]
- Spisek, R.; Charalambous, A.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Bortezomib enhances dendritic cell (DC)–mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: Therapeutic implications. Blood 2007, 109, 4839–4845. [Google Scholar] [CrossRef]
- Hadjiaggelidou, C.; Mandala, E.; Terpos, E.; Yiannaki, E.; Markala, D.; Triantafyllou, T.; Papatheodorou, A.; Gkastari, V.; Verrou, E.; Papanikolaou, A.; et al. Evaluation of regulatory T cells (Tregs) alterations in patients with multiple myeloma treated with bortezomib or lenalidomide plus dexamethasone: Correlations with treatment outcome. Ann. Hematol. 2019, 98, 1457–1466. [Google Scholar] [CrossRef]
- Nair, J.R.; Carlson, L.M.; Koorella, C.; Rozanski, C.H.; Byrne, G.E.; Bergsagel, P.L.; Shaughnessy, J.P.; Boise, L.; Chanan-Khan, A.; Lee, K.P. CD28 Expressed on Malignant Plasma Cells Induces a Prosurvival and Immunosuppressive Microenvironment. J. Immunol. 2011, 187, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Aldea, M.; Orillard, E.; Mansi, L.; Marabelle, A.; Scotte, F.; Lambotte, O.; Michot, J.-M. How to manage patients with corticosteroids in oncology in the era of immunotherapy? Eur. J. Cancer 2020, 141, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Lossignol, D. A little help from steroids in oncology. J. Transl. Intern. Med. 2016, 4, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Aston, W.J.; Hope, D.E.; Cook, A.M.; Boon, L.; Dick, I.; Nowak, A.K.; Lake, R.A.; Lesterhuis, W.J. Dexamethasone differentially depletes tumour and peripheral blood lymphocytes and can impact the efficacy of chemotherapy/checkpoint blockade combination treatment. OncoImmunology 2019, 8, e1641390. [Google Scholar] [CrossRef]
- Wandler, A.M.; Huang, B.; Craig, J.W.; Hayes, K.; Yan, H.; Meyer, L.K.; Scacchetti, A.; Monsalve, G.; Dail, M.; Li, Q.; et al. Loss of glucocorticoid receptor expression mediates in vivo dexamethasone resistance in T-cell acute lymphoblastic leukemia. Leukemia 2020, 34, 2025–2037. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Wang, Y.; Fu, C.-C.; Diao, F.; Song, L.-N.; Li, Z.-B.; Yang, R.; Lu, J. Dexamethasone enhances cell resistance to chemotherapy by increasing adhesion to extracellular matrix in human ovarian cancer cells. Endocr. Relat. Cancer 2010, 17, 39–50. [Google Scholar] [CrossRef]
- Weber, J.S.; Kähler, K.C.; Hauschild, A. Management of Immune-Related Adverse Events and Kinetics of Response With Ipilimumab. J. Clin. Oncol. 2012, 30, 2691–2697. [Google Scholar] [CrossRef] [PubMed]
- Okoye, I.S.; Xu, L.; Walker, J.; Elahi, S. The glucocorticoids prednisone and dexamethasone differentially modulate T cell function in response to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade. Cancer Immunol. Immunother. 2020, 69, 1423–1436. [Google Scholar] [CrossRef]
- Vormehr, M.; Lehar, S.; Kranz, L.M.; Tahtinen, S.; Oei, Y.; Javinal, V.; Delamarre, L.; Walzer, K.C.; Diken, M.; Kreiter, S.; et al. Dexamethasone premedication suppresses vaccine-induced immune responses against cancer. OncoImmunology 2020, 9, 1758004. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Bringhen, S.; Anttila, P.; Capra, M.; Cavo, M.; Cole, C.; Gasparetto, C.; Hungria, V.; Jenner, M.; Vorobyev, V.; et al. Isatuximab as monotherapy and combined with dexamethasone in patients with relapsed/refractory multiple myeloma. Blood 2021, 137, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- A Hussein, M. Thalidomide: Present and future in multiple myeloma. Expert Rev. Anticancer. Ther. 2005, 5, 25–31. [Google Scholar] [CrossRef]
- Botting, J. The history of thalidomide. Drug News Perspect. 2002, 15, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Ridings, J.E. The Thalidomide Disaster, Lessons from the Past. Methods Mol. Biol. 2013, 947, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.B.; Dredge, K.; Dalgleish, A.G. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat. Rev. Cancer 2004, 4, 314–322. [Google Scholar] [CrossRef]
- Vallet, S.; Palumbo, A.; Raje, N.; Boccadoro, M.; Anderson, K.C. Thalidomide and lenalidomide: Mechanism-based potential drug combinations. Leuk. Lymphoma 2008, 49, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Kim, Y.; Gast, S.-M.; Endo, T.; Lu, D.; Carson, D.; Schmidt-Wolf, I.G.H. Increased in vivo efficacy of lenalidomide and thalidomide by addition of ethacrynic acid. In Vivo 2011, 25, 325–333. [Google Scholar]
- Lopez-Girona, A.; Mendy, D.; Ito, T.A.; Miller, K.H.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.E.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, G. Cereblon attenuates DNA damage-induced apoptosis by regulating the transcription-independent function of p53. Cell Death Dis. 2019, 10, 69. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G., Jr. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Guillerey, C.; Nakamura, K.; Vuckovic, S.; Hill, G.; Smyth, M.J. Immune responses in multiple myeloma: Role of the natural immune surveillance and potential of immunotherapies. Cell. Mol. Life Sci. 2016, 73, 1569–1589. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Ma, D.; Wang, P.; Cao, L.; Lu, T.; Fang, Q.; Zhao, J.; Wang, J. Potential crosstalk of the interleukin-6-heme oxygenase-1-dependent mechanism involved in resistance to lenalidomide in multiple myeloma cells. FEBS J. 2015, 283, 834–849. [Google Scholar] [CrossRef]
- Neuber, B.; Dai, J.; Waraich, W.A.; Awwad, M.H.; Engelhardt, M.; Schmitt, M.; Medenhoff, S.; Witzens-Harig, M.; Ho, A.D.; Goldschmidt, H.; et al. Lenalidomide overcomes the immunosuppression of regulatory CD8+CD28− T-cells. Oncotarget 2017, 8, 98200–98214. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; E Witzig, T.; Dispenzieri, A.; Lacy, M.Q.; E Wellik, L.; Fonseca, R.; A Lust, J.; A Gertz, M.; A Kyle, R.; Greipp, P.R.; et al. Effect of thalidomide therapy on bone marrow angiogenesis in multiple myeloma. Leukemia 2004, 18, 624–627. [Google Scholar] [CrossRef][Green Version]
- Bolzoni, M.; Storti, P.; Bonomini, S.; Todoerti, K.; Guasco, D.; Toscani, D.; Agnelli, L.; Neri, A.; Rizzoli, V.; Giuliani, N. Immunomodulatory drugs lenalidomide and pomalidomide inhibit multiple myeloma-induced osteoclast formation and the RANKL/OPG ratio in the myeloma microenvironment targeting the expression of adhesion molecules. Exp. Hematol. 2013, 41, 387–397.e1. [Google Scholar] [CrossRef] [PubMed]
- Görgün, G.T.; Samur, M.K.; Cowens, K.B.; Paula, S.; Bianchi, G.; Anderson, J.E.; White, R.E.; Singh, A.; Ohguchi, H.; Suzuki, R.; et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4607–4618. [Google Scholar] [CrossRef]
- Luptakova, K.; Rosenblatt, J.; Glotzbecker, B.; Mills, H.; Stroopinsky, D.; Kufe, T.; Vasir, B.; Arnason, J.; Tzachanis, D.; Zwicker, J.; et al. Lenalidomide enhances anti-myeloma cellular immunity. Cancer Immunol. Immunother. 2013, 62, 39–49. [Google Scholar] [CrossRef]
- Jungkunz-Stier, I.; Zekl, M.; Stühmer, T.; Einsele, H.; Seggewiss-Bernhardt, R. Modulation of natural killer cell effector functions through lenalidomide/dasatinib and their combined effects against multiple myeloma cells. Leuk. Lymphoma 2013, 55, 168–176. [Google Scholar] [CrossRef]
- De Keersmaecker, B.; Fostier, K.; Corthals, J.; Wilgenhof, S.; Heirman, C.; Aerts, J.L.; Thielemans, K.; Schots, R. Immunomodulatory drugs improve the immune environment for dendritic cell-based immunotherapy in multiple myeloma patients after autologous stem cell transplantation. Cancer Immunol. Immunother. 2014, 63, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Brissot, E.; Clavert, A.; Blin, N.; Roland, V.; Guillaume, T.; Dubruille, V.; Mahé, B.; Gastinne, T.; Le Gouill, S.; Gaugler, B.; et al. Impact of lenalidomide on immune functions in the setting of maintenance therapy for multiple myeloma. Leukemia 2015, 29, 2098–2100. [Google Scholar] [CrossRef]
- Di Lullo, G.; Marcatti, M.; Heltai, S.; Tresoldi, C.; Paganoni, A.M.; Bordignon, C.; Ciceri, F.; Protti, M.P. Immunomodulatory Drugs in the Context of Autologous Hematopoietic Stem Cell Transplantation Associate with Reduced Pro-tumor T Cell Subsets in Multiple Myeloma. Front. Immunol. 2019, 9, 3171. [Google Scholar] [CrossRef]
- Fostier, K.; Caers, J.; Meuleman, N.; Broos, K.; Corthals, J.; Thielemans, K.; Schots, R.; De Keersmaecker, B. Impact of lenalidomide maintenance on the immune environment of multiple myeloma patients with low tumor burden after autologous stem cell transplantation. Oncotarget 2018, 9, 20476–20489. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, M.; Chiu, M.I.; Pagano, M. The ubiquitin-mediated proteolytic pathway as a therapeutic area. J. Mol. Med. 1997, 75, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. Regulation of NF-κB by ubiquitination. Curr. Opin. Immunol. 2013, 25, 4–12. [Google Scholar] [CrossRef]
- Traenckner, E.; Wilk, S.; Baeuerle, P. A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-alpha that is still bound to NF-kappa B. EMBO J. 1994, 13, 5433–5441. [Google Scholar] [CrossRef]
- Schenkein, D. Proteasome Inhibitors in the Treatment of B-Cell Malignancies. Clin. Lymphoma 2002, 3, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Matta, H.; Chaudhary, P.M. The proteasome inhibitor bortezomib (PS-341) inhibits growth and induces apoptosis in primary effusion lymphoma cells. Cancer Biol. Ther. 2005, 4, 84–89. [Google Scholar] [CrossRef]
- Bruning, A.; Vogel, M.; Mylonas, I.; Friese, K.; Burges, A. Bortezomib targets the caspase-like proteasome activity in cervical cancer cells, triggering apoptosis that can be enhanced by nelfinavir. Curr. Cancer Drug Targets 2011, 11, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Selimovic, D.; Porzig, B.B.; El-Khattouti, A.; Badura, H.E.; Ahmad, M.; Ghanjati, F.; Santourlidis, S.; Haikel, Y.; Hassan, M. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell. Signal. 2013, 25, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Łuczkowska, K.; Rogińska, D.; Ulańczyk, Z.; Paczkowska, E.; Schmidt, C.A.; Machaliński, B. Molecular Mechanisms of Bortezomib Action: Novel Evidence for the miRNA–mRNA Interaction Involvement. Int. J. Mol. Sci. 2020, 21, 350. [Google Scholar] [CrossRef]
- Stuhler, G.; Nekova, T.S. Myeloma cell sensitivity to bortezomib is associated with Dicer1 expression. Blood 2014, 124, 657–658. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade ®: U.S. FDA Approval for the Treatment of Multiple Myeloma Progressing on Prior Therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Duan, J.; Zhou, L. The paradoxical pharmacological mechanisms of lenalidomide and bortezomib in the treatment of multiple myeloma. Anti-Cancer Drugs 2021, 32, 227–232. [Google Scholar] [CrossRef]
- Straube, C.; Wehner, R.; Wendisch, M.; Bornhauser, M.; Bachmann, M.; Rieber, E.P.; Schmitz, M. Bortezomib significantly impairs the immunostimulatory capacity of human myeloid blood dendritic cells. Leukemia 2007, 21, 1464–1471. [Google Scholar] [CrossRef]
- Gullà, A.; Morelli, E.; Samur, M.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Rao, P.; Talluri, S.; Tai, Y.-T.; Chauhan, D.; et al. Mechanisms, biologic sequelae and clinical benefits of bortezomib-induced immunogenic cell death in multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, e162. [Google Scholar] [CrossRef]
- Gulla, A.; Morelli, E.; Samur, M.K.; Botta, C.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Malvestiti, S.; Prabhala, R.H.; Talluri, S.; et al. Bortezomib Induces Anti–Multiple Myeloma Immune Response Mediated by cGAS/STING Pathway Activation. Blood Cancer Discov. 2021, 2, 1–16. [Google Scholar] [CrossRef]
- Terlizzi, M.; Colarusso, C.; Pinto, A.; Sorrentino, R. Role of Plasmacytoid Dendritic Cells in Cancer. In Cancer Immunology; Springer: Cham, Switzerland, 2020; pp. 147–169. [Google Scholar]
- Shabaneh, T.B.; Downey, S.L.; Goddard, A.L.; Screen, M.; Lucas, M.M.; Eastman, A.; Kisselev, A.F. Molecular Basis of Differential Sensitivity of Myeloma Cells to Clinically Relevant Bolus Treatment with Bortezomib. PLoS ONE 2013, 8, e56132. [Google Scholar] [CrossRef] [PubMed]
- Škrott, Z.; Cvek, B. Linking the activity of bortezomib in multiple myeloma and autoimmune diseases. Crit. Rev. Oncol. 2014, 92, 61–70. [Google Scholar] [CrossRef]
- Bennett, M.K.; Kirk, C.J. Development of Proteasome Inhibitors in Oncology and Autoimmune Diseases—Web of Science Core Collection. Available online: https://www.webofscience.com/wos/woscc/full-record/WOS:000258642700003 (accessed on 11 July 2021).
- Hofmann, K.; Clauder, A.-K.; Manz, R.A. Targeting B Cells and Plasma Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 835. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, B.S.; Park, Y.; Lee, J.G.; Lim, B.J.; Jeong, H.J.; Kim, Y.S.; Huh, K.H. The Effect of Bortezomib on Antibody-Mediated Rejection after Kidney Transplantation. Yonsei Med. J. 2015, 56, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Everly, M.J.; Terasaki, P.I.; Hopfield, J.; Trivedi, H.L.; Kaneku, H. Protective Immunity Remains Intact After Antibody Removal by Means of Proteasome Inhibition. Transplantation 2010, 90, 1493–1498. [Google Scholar] [CrossRef]
- Celotto, K.; Nair, J.; Lee, K. The Effect of Bortezomib Treatment on Antibody Titers Against Common Viral And Vaccine Antigens. Clin. Lymphoma Myeloma Leuk. 2015, 15, e179. [Google Scholar] [CrossRef]
- Shanker, A.; Pellom, S.T.; Dudimah, D.F.; Thounaojam, M.C.; De Kluyver, R.L.; Brooks, A.D.; Yagita, H.; McVicar, D.W.; Murphy, W.J.; Longo, D.L.; et al. Bortezomib Improves Adoptive T-cell Therapy by Sensitizing Cancer Cells to FasL Cytotoxicity. Cancer Res. 2015, 75, 5260–5272. [Google Scholar] [CrossRef]
- Pellom, S.T., Jr.; Dudimah, D.F.; Thounaojam, M.; Uzhachenko, R.V.; Singhal, A.; Richmond, A.; Shanker, A. Bortezomib augments lymphocyte stimulatory cytokine signaling in the tumor microenvironment to sustain CD8+T cell antitumor function. Oncotarget 2016, 8, 8604–8621. [Google Scholar] [CrossRef] [PubMed]
- Renrick, A.N.; Thounaojam, M.C.; de Aquino, M.T.P.; Chaudhuri, E.; Pandhare, J.; Dash, C.; Shanker, A. Bortezomib Sustains T Cell Function by Inducing miR-155-Mediated Downregulation of SOCS1 and SHIP1. Front. Immunol. 2021, 12, 69. [Google Scholar] [CrossRef]
- Xiaohui, W.; Lingyun, S.; Tian, J.; Wang, X.; Chen, Q.; Rui, K.; Ma, J.; Wang, S.; Wang, Q.; Wang, X.; et al. Proteasome inhibition suppresses Th17 cell generation and ameliorates autoimmune development in experimental Sjögren’s syndrome. Cell. Mol. Immunol. 2017, 14, 924–934. [Google Scholar] [CrossRef]
- Plitas, G.; Rudensky, A.Y. Regulatory T Cells in Cancer. Annu. Rev. Cancer Biol. 2020, 4, 459–477. [Google Scholar] [CrossRef]
- Blanco, B.; Pérez-Simón, J.A.; Sánchez-Abarca, L.I.; Caballero-Velazquez, T.; Gutierrez-Cossío, S.; Hernández-Campo, P.; Campelo, M.D.; Herrero-Sanchez, C.; Rodriguez-Serrano, C.; Santamaría, C.; et al. Treatment with bortezomib of human CD4+ T cells preserves natural regulatory T cells and allows the emergence of a distinct suppressor T-cell population. Haematologica 2009, 94, 975–983. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Moreau, P.; Terpos, E.; Mateos, M.; Zweegman, S.; Cook, G.; Delforge, M.; Hájek, R.; Schjesvold, F.; Cavo, M.; et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2021, 32, 309–322. [Google Scholar] [CrossRef]
- Paul, B.; Lipe, B.; Ocio, E.M.; Usmani, S.Z. Induction Therapy for Newly Diagnosed Multiple Myeloma. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, e176–e186. [Google Scholar] [CrossRef]
- Rosiñol, L.; Oriol, A.; Rios, R.; Sureda, A.; Blanchard, M.J.; Hernández, M.T.; Martínez-Martínez, R.; Moraleda, J.M.; Jarque, I.; Bargay, J.; et al. Bortezomib, lenalidomide, and dexamethasone as induction therapy prior to autologous transplant in multiple myeloma. Blood 2019, 134, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Cook, J.; Johnson, I.; Higgins, A.; Sidana, S.; Warsame, R.; Gonsalves, W.; Gertz, M.A.; Buadi, F.; Lacy, M.; Kapoor, P.; et al. Outcomes with different administration schedules of bortezomib in bortezomib, lenalidomide and dexamethasone (VRd) as first-line therapy in multiple myeloma. Am. J. Hematol. 2021, 96, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Arcuri, L.J.; Americo, A.D. Treatment of relapsed/refractory multiple myeloma in the bortezomib and lenalidomide era: A systematic review and network meta-analysis. Ann. Hematol. 2021, 100, 725–734. [Google Scholar] [CrossRef]
- Hsu, A.K.; Quach, H.; Tai, T.; Prince, H.M.; Harrison, S.J.; Trapani, J.A.; Smyth, M.J.; Neeson, P.; Ritchie, D.S. The immunostimulatory effect of lenalidomide on NK-cell function is profoundly inhibited by concurrent dexamethasone therapy. Blood 2011, 117, 1605–1613. [Google Scholar] [CrossRef]
- Gandhi, A.K.; Kang, J.; Capone, L.; Parton, A.; Wu, L.; Zhang, L.H.; Mendy, D.; Lopez-Girona, A.; Tran, T.; Sapinoso, L.; et al. Dexamethasone Synergizes with Lenalidomide to Inhibit Multiple Myeloma Tumor Growth, But Reduces Lenalidomide-Induced Immunomodulation of T and NK Cell Function. Curr. Cancer Drug Targets 2010, 10, 155–167. [Google Scholar] [CrossRef]
- Paiva, B.D.L.; Mateos, M.V.; Sanchez-Abarca, L.I.; Puig, N.; Vidriales, M.-B.; Corral, L.L.; Sánchez, L.A.C.; Hernandez, M.T.; Bargay, J.; De Arriba, F.; et al. Immune status of high-risk smoldering multiple myeloma patients and its therapeutic modulation under LenDex: A longitudinal analysis. Blood 2016, 127, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Casneuf, T.; Adams, H.C.; van de Donk, N.W.; Abraham, Y.; Bald, J.; Vanhoof, G.; Van der Borght, K.; Smets, T.; Foulk, B.; Nielsen, K.C.; et al. Deep immune profiling of patients treated with lenalidomide and dexamethasone with or without daratumumab. Leukemia 2021, 35, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, K.; Tsakirakis, N.; Malandrakis, P.; Vitsos, P.; Metousis, A.; Orologas-Stavrou, N.; Ntanasis-Stathopoulos, I.; Kanellias, N.; Eleutherakis-Papaiakovou, E.; Pothos, P.; et al. Deep Phenotyping Reveals Distinct Immune Signatures Correlating with Prognostication, Treatment Responses, and MRD Status in Multiple Myeloma. Cancers 2020, 12, 3245. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-E.; Lim, J.-Y.; Ryu, D.-B.; Kim, T.W.; Yoon, J.-H.; Cho, B.-S.; Eom, K.-S.; Kim, Y.-J.; Kim, H.-J.; Lee, S.; et al. Circulating immune cell phenotype can predict the outcome of lenalidomide plus low-dose dexamethasone treatment in patients with refractory/relapsed multiple myeloma. Cancer Immunol. Immunother. 2016, 65, 983–994. [Google Scholar] [CrossRef]
- Martino, M.; Recchia, A.G.; Console, G.; Gentile, M.; Cimminiello, M.; Gallo, G.A.; Ferreri, A.; Naso, V.; Irrera, G.; Mes-sina, G.; et al. Can we improve the conditioning regimen before autologous stem cell transplantation in multiple myeloma? Expert Opin. Orphan Drugs 2017, 5, 875–887. [Google Scholar] [CrossRef]
- Lee, S.J.; Borrello, I. Role of the Immune Response in Disease Progression and Therapy in Multiple Myeloma. In Cancer Treatment and Research; Springer Science and Business Media LLC: Berlin, Germany, 2016; Volume 169, pp. 207–225. [Google Scholar]
- Yamamoto, L.; Amodio, N.; Gulla, A.; Anderson, K.C. Harnessing the Immune System Against Multiple Myeloma: Challenges and Opportunities. Front. Oncol. 2021, 10, 3160. [Google Scholar] [CrossRef]
- Papadimitriou, K.; Kostopoulos, I.V.; Tsopanidou, A.; Orologas-Stavrou, N.; Kastritis, E.; Tsitsilonis, O.; Dimopoulos, M.A.; Terpos, E. Ex vivo models simulating the bone marrow environment and predicting response to therapy in multiple myeloma. Cancers 2020, 12, 2006. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C. An Overview of Organoid and 3-Dimensional Models in Multiple Myeloma. Cancer J. 2021, 27, 239–246. [Google Scholar] [CrossRef]
- Rangel-Pozzo, A.; Yu, P.; LaL, S.; Asbaghi, Y.; Sisdelli, L.; Tammur, P.; Tamm, A.; Punab, M.; Klewes, L.; Louis, S.; et al. Telomere Architecture Correlates with Aggressiveness in Multiple Myeloma. Cancers 2021, 13, 1969. [Google Scholar] [CrossRef]
- Fakhari, S.; Stefanucci, A.; Mollica, A.; Nikkhoo, B.; Tafsiri, E.; Jalili, A.; Mirzaie, S. Designing new generation of potent inhibitors against membrane-type matrix metalloproteinase-2: A computational effort against multiple myeloma. J. Biomol. Struct. Dyn. 2019, 38, 3879–3891. [Google Scholar] [CrossRef]
- Sun, J.; Muz, B.; Alhallak, K.; Markovic, M.; Gurley, S.; Wang, Z.; Guenthner, N.; Wasden, K.; Fiala, M.; King, J.; et al. Targeting CD47 as a Novel Immunotherapy for Multiple Myeloma. Cancers 2020, 12, 305. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug | Essential MOA | Effects on Innate Immune System | System | Ref | Effects on Adaptive Immune System | System | Ref |
|---|---|---|---|---|---|---|---|
| Melphalan (Mel) | Induction of DNA–DNA, DNA–protein cross-links leading to disruption of DNA replication and transcription; tumor cell death | Negative -Transient depletion of monocytes, granulocytes, and conventional DCs | AM | [21] | Positive -Transient burst of inflammatory cytokines; | AM | [21] |
| -Priming of tumor-infiltrating activated, CD8+ TEM cells | AM | [21] | |||||
| -Induction of ICD | AM | [21] | |||||
| -Transient burst of inflammatory cytokines | AM | [21] | -Proliferation of antigen-specific CD4+ T cells | AM | [21] | ||
| -Enhances B7 expression of tumor cells and DCs | EV | [23] | |||||
| Negative CD28+MM/B7+DC interaction enhances MM cell survival | EV | [23,24] | |||||
| Dexamethasone (Dex) | -Immunosuppression, particularly T cells | Negative -Reduction in T cell stimulation by upregulation of CTLA-4/PD-1 expression | IV | [25] | |||
| -Reduction in cytokine production | IV | [25] | |||||
| -Reduction in peripheral blood CD4+, CD8+ T cells, T-regs, B cells, and NK cells | IV | [25] | |||||
| -Reduction in TIL activity vs. autologous tumor cells | IV | [26] | |||||
| -Risk factor for infection in MM patients | Cl | [27] | |||||
| Lenalidomide (Revlamid®) (Rev) | -Immunomodulation, antiangiogenic | Positive -Enhances production of IFN-γ and TNF-α in NK cells | EV, Cl | [28] | Positive -Inhibition of pro-inflammatory cytokines TNF-α, IL-1b, -6 -12 | IV | [29] |
| -Increased production of IL-2, IFN-γ | EV, Cl | [28] | |||||
| -Increased IFN-γ production in CD8+ T cells | EV, Cl | [28] | |||||
| - Tregs decline as CD8+ T cells expand | EV, Cl | [30] | |||||
| -Binds cereblon leading to reduction in growth of MM cells | Negative May enhance production of Tregs | IV, EV | [31] | ||||
| -Inhibits production of IL-6 by BM stromal cells | |||||||
| Bortezomib (Velcade®) (Vel) | -Inhibitor of proteasomal enzymes | Conflicting -Effect of NK activity | IV, EV | [32,33] | Positive Low-dose -Increase in CD8+T cell activation and production of IL-2,-12,-15 | AM, IV | [34,35] |
| -Increase in CD8+ T cell and NK cytotoxicity | IV | [36] | |||||
| Negative -Decreased polyclonal antibody production; increased B cell apoptosis | AM, IV | [37,38] | |||||
| Conflicting -Effect on Th17+ T cells | AM | [39,40] | |||||
| -blocks activation of NFκB | -Effect on DC activity | IV, EV | [41,42] | -Effect on Tregs | AM, Cl | [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Firer, M.A.; Shapira, M.Y.; Luboshits, G. The Impact of Induction Regimes on Immune Responses in Patients with Multiple Myeloma. Cancers 2021, 13, 4090. https://doi.org/10.3390/cancers13164090
Firer MA, Shapira MY, Luboshits G. The Impact of Induction Regimes on Immune Responses in Patients with Multiple Myeloma. Cancers. 2021; 13(16):4090. https://doi.org/10.3390/cancers13164090
Chicago/Turabian StyleFirer, Michael A., Michael Y. Shapira, and Galia Luboshits. 2021. "The Impact of Induction Regimes on Immune Responses in Patients with Multiple Myeloma" Cancers 13, no. 16: 4090. https://doi.org/10.3390/cancers13164090
APA StyleFirer, M. A., Shapira, M. Y., & Luboshits, G. (2021). The Impact of Induction Regimes on Immune Responses in Patients with Multiple Myeloma. Cancers, 13(16), 4090. https://doi.org/10.3390/cancers13164090