1. Introduction
MicroRNAs (miRNAs) are endogenous small non-coding RNAs of 19–21 nt in length that control the expression of many genes at the post-transcriptional level, mostly through base pairing between a short “seed” region 5′ of the miRNA sequence (typically bases 28) and the 3′UTR of target genes [
1,
2], but imperfect centered sites, as well as coding region targeting, have also been reported [
3,
4]. This interaction leads to translational inhibition and transcript destabilization [
2]. A single miRNA is able to target hundreds of coding genes, and therefore miRNAs have wide effects on cell phenotype [
5]. They are involved in all biological processes, under normal and pathological conditions, including in cancer where they can act as oncogenes or as tumor suppressor genes [
6,
7].
Because of their frequent over- or under-expression in cancer, miRNAs are often considered possible therapeutic tools or targets [
6,
8]. But despite numerous still ongoing clinical trials, the current limitations for the clinical use of nucleic acids—such as siRNAs, miRNAs or anti-miRNAs—have undermined such strategies to date [
8]. However, understanding the determinants of the cytotoxic phenotype triggered by a given miRNA might enable the identification of druggable targets and thus, the design of innovative therapeutic strategies aiming to mimic its effects [
9,
10]. Ovarian cancer constitutes the leading cause of death from gynecological malignancies in developed countries, with a low 5-year survival rate of less than 40% [
11]. The main cause of therapeutic failure is the resistance to existing treatments, therefore innovative strategies are urgently needed for this disease [
12].
A large number of studies reporting cytotoxic effects of a given miRNA in cancer cells attribute the main part of such effect to the downregulation of a single direct target. Although quite useful for the description of miRNA targets, such study design might miss the most relevant determinants of miRNAs’ effects for two reasons. First, miRNAs are characterized by acting on hundreds of target genes. Second, owing to this high number of deregulated targets, the identification of indirect targets of miRNAs, e.g., downstream of directly targeted genes, should also be highly relevant to understand how a miRNA can trigger a given phenotype.
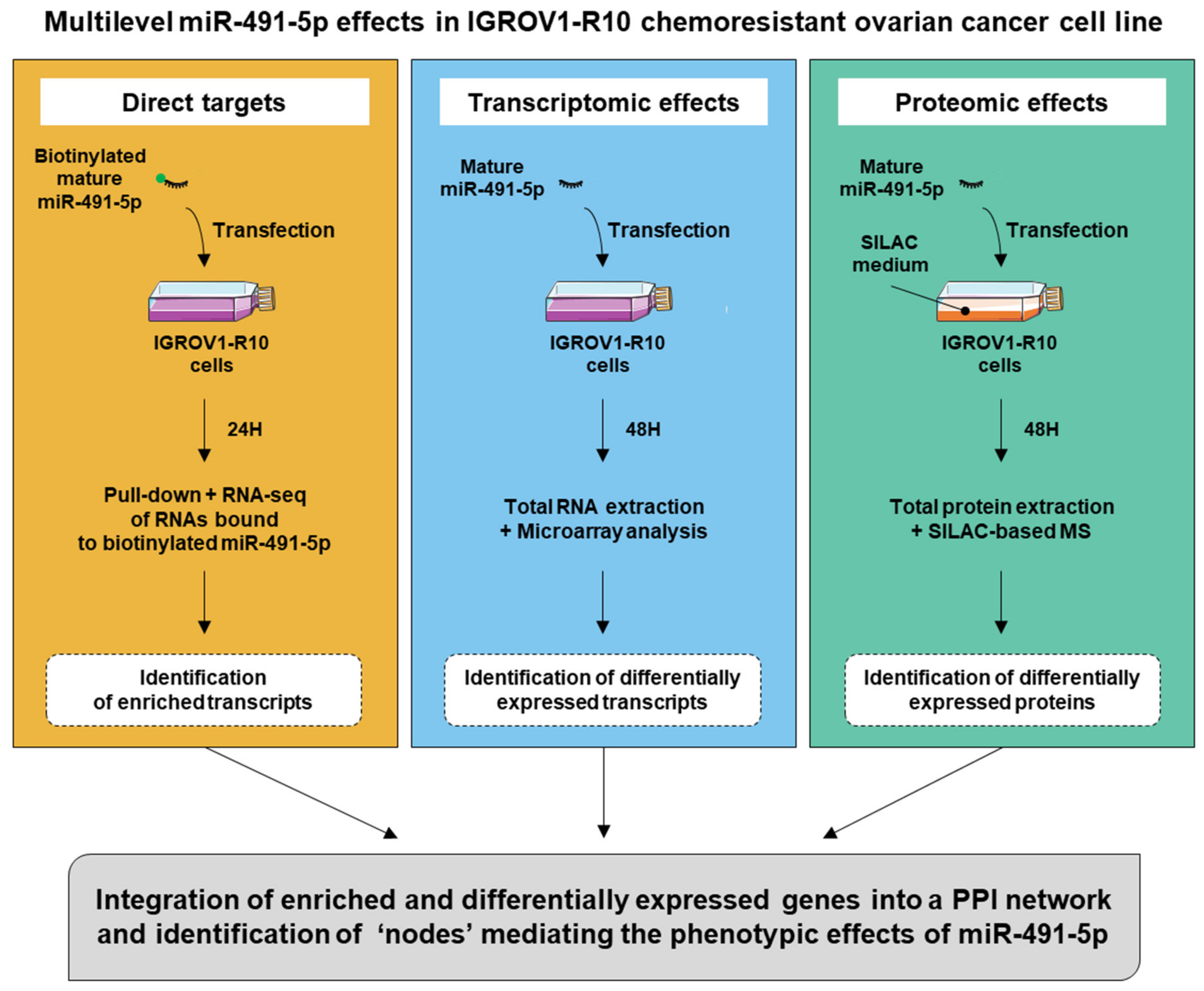
In order to obtain a comprehensive view of the cell-wide effects of miR-491-5p beyond a single—or a few—direct targets, we chose to study its effects at different molecular levels: direct interaction with targeted transcripts, transcriptomic effects and proteomic effects. By integrating this multi-omics data through a network-based approach, we aimed to identify the most critical determinants of its phenotypic action.
We chose to use miR-491-5p as a model system because it appeared among the top hits for its cytotoxic effects, in the IGROV1-R10 chemoresistant cell line in a screen of a library of 1233 miRNAs [
10]. Moreover, some of the most significant targets mediating its cytotoxic effects in ovarian cancer cells are already identified [
9]. This model allowed us to confirm that our strategy identifies some already known critical targets and pathways involved in the effects of miR-491-5p on ovarian cancer cells. We could also point to targets that are important mediators of miR-491-5p cytotoxic activities in ovarian cancer cells and propose new pharmacological combinations.
We then show that, by combining several high-throughput approaches to explore the effects of a given miRNA, we could identify some critical determinants of its phenotypic effects. Our strategy did imply easily available technologies and bioinformatic tools for the analysis of such data. Our results did also point to several new potential targets that might prove themselves very interesting for the treatment of ovarian cancer.
3. Discussion
It is now well acknowledged that miRNAs are deeply involved in cancer biology, wherein their deregulated expression displays oncogenic or tumor suppressive functions [
6,
7,
21]. The re-introduction of specific miRNAs, or their inhibition, is able to trigger cell death in cancer cells from various origins [
8,
22,
23]. This observation therefore led to the development of several strategies aiming to deliver miRNAs in vivo as a therapeutic option against cancer. However, despite numerous clinical trials, the safe and efficient delivery of miRNAs to tumors in human is yet to be achieved [
8].
We postulated that mimicking the effects of a miRNA using drugs already available or easily amenable to clinical practice would constitute a valuable proxy for the clinical use of miRNAs. We previously showed that miR-491-5p [
9] and miR-3622b-5p [
10] directly target Bcl-xL and EGFR in ovarian cancer cells, and that the combined use of Bcl-xL and EGFR inhibitors, respectively, in clinical trials (ABT-263), which are FDA-approved for the treatment of non-small-cell lung cancer (gefitinib), could efficiently recapitulate the cytotoxicity induced by both of these miRNAs. Although positive, the results of this previous study did bear two drawbacks. First, the step-by-step approach we followed for the identification of these two direct targets was a time-consuming process, and such approaches rely heavily on the accuracy of target-prediction algorithms, which remain imperfect. Second, any approach focusing on a single or a few direct targets of a miRNA of interest is limited at least to some extent because of miRNAs’ multitarget mode of action.
The strategy we chose for this study gathered information at multiple stages of miRNA action: physical association with RNAs directly targeted and the transcriptomic and proteomic consequences of miRNA action. We chose to use miR-491-5p because our knowledge of two important targets of this miRNA, Bcl-xL and EGFR [
9], could benchmark the results we would obtain and quality-control our pipeline of analysis. Moreover, we expected that the use of datasets generated with different methodological approaches would compensate for false positive and false negative results, which would most likely originate with each methodology.
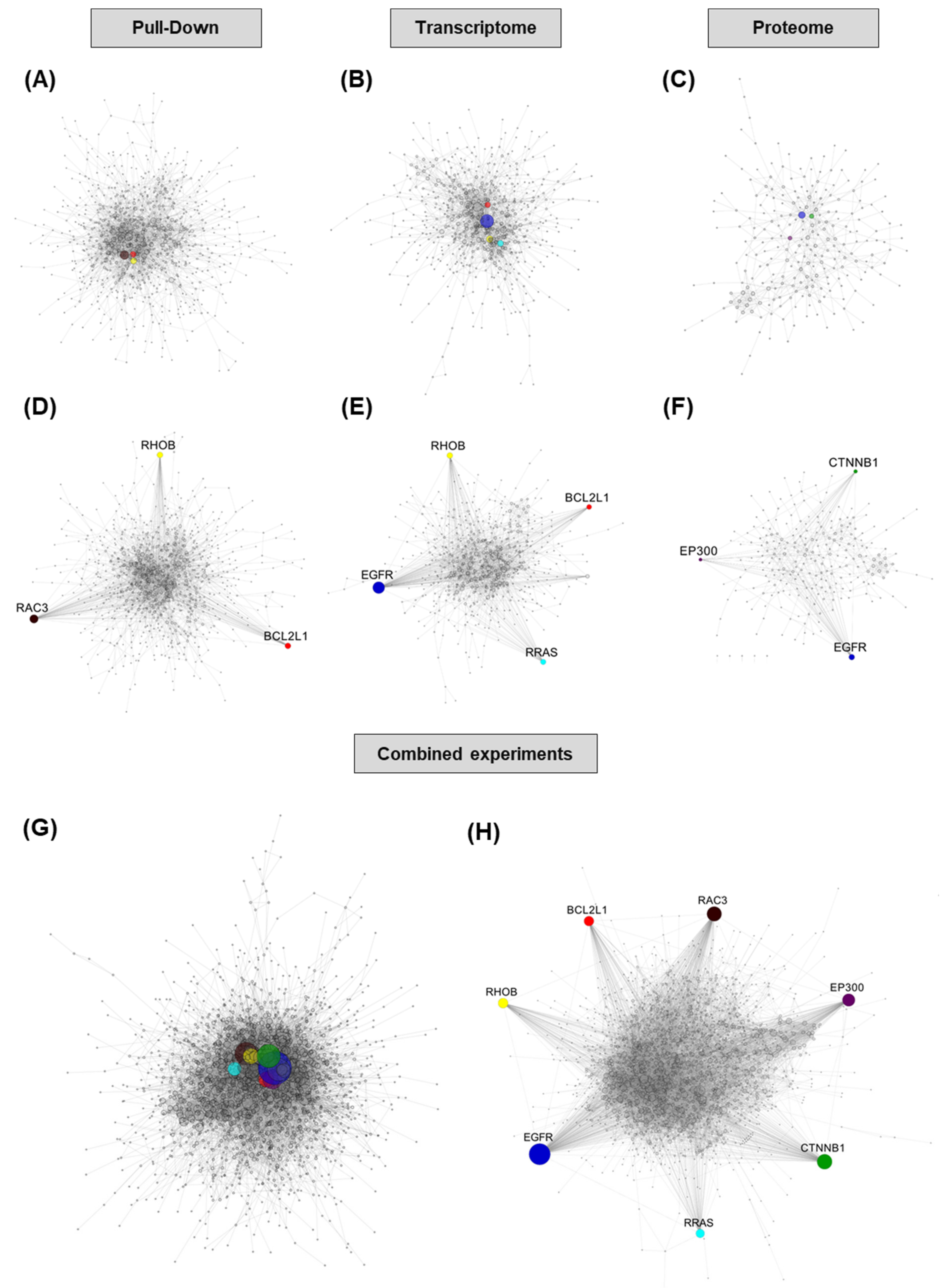
According to the literature, 12 genes have been demonstrated to date to be direct targets of miR-491-5p in different models. Eight of them are highlighted in at least one of our experiments (five with the transcriptomic experiment, six with the pull-down experiment and three with the proteomic experiment,
Table S3). Among the four remaining validated targets, MMP9 and NOTCH3 are expressed at background levels in IGROV1-R10 cells and therefore, cannot be taken into account. IGF2BP1 down-regulation is close to the cut-off for transcriptomic and proteomic data and is likely a moderately affected target. The last validated target we failed to identify is TP53, which is relatively highly expressed in our transcriptomic data. A reported mutation for TP53 in the IGROV1 cell line (parental to IGROV1-R10) [
24] is heterozygous and a SNP and therefore, should not impair miRNA targeting, although we did not test the integrity of miR-491-5p target sites in TP53 3′UTR in the cisplatin-resistant IGROV1-R10 cells. Altogether, the identification of eight out of ten already validated possible direct targets of miR-491-5p underlines the robustness of our approach. This is especially valid because of the cell-line- and cell-type-specific spectra of the targets of miRNAs [
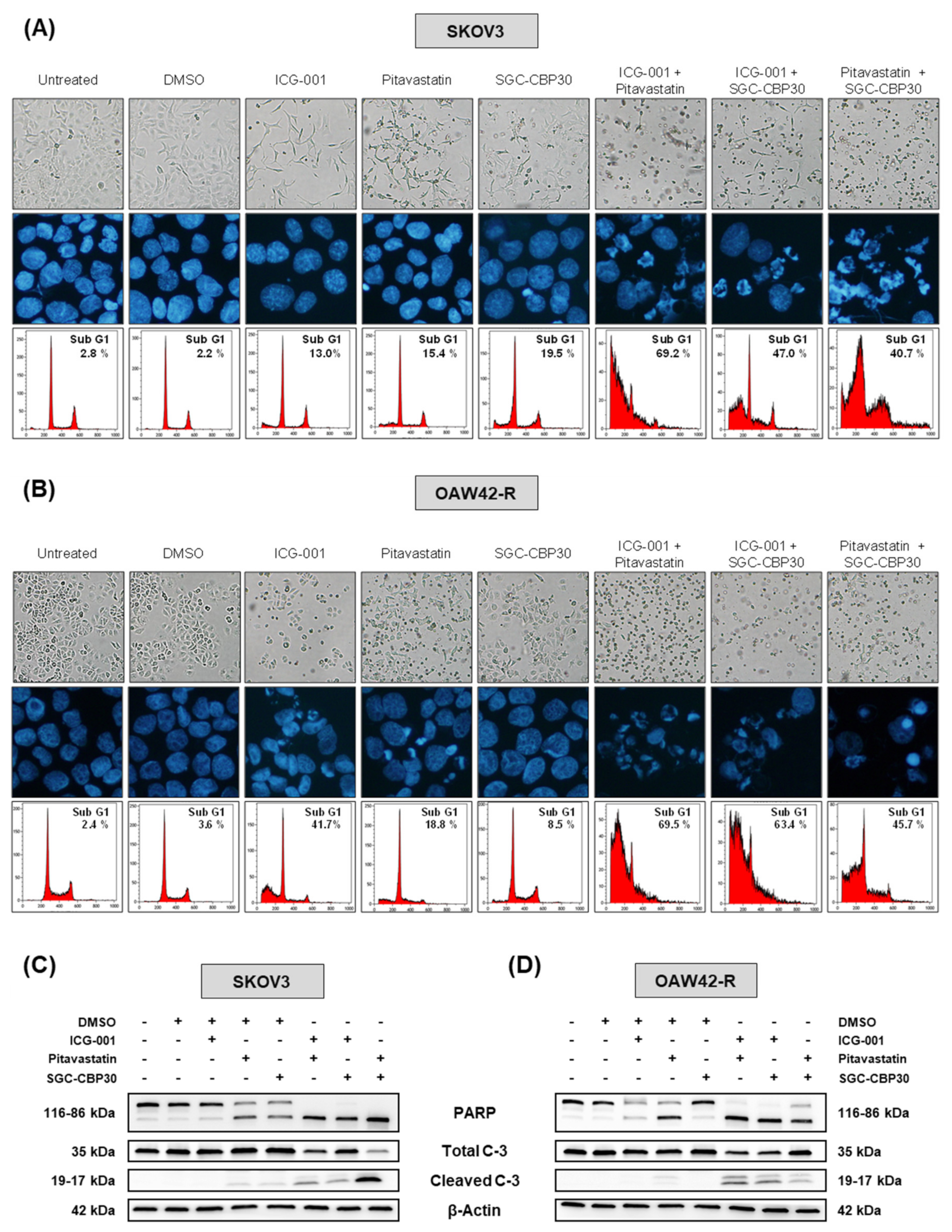
25]. This context specificity of their broad-range effects is likely to explain why miR-491-5p is not as cytotoxic in SKOV3 and OAW42-R cells as it is in IGROV1-R10. We postulate that, while these two cell lines are vulnerable to the specific inhibition of our selected nodes, the direct and indirect effects of miR-491-5p in these cells would be different enough from the ones occurring in IGROV1-R10 to preclude the same phenotype to be triggered upon miR-491-5p transfection.
Among the hubs we selected, only two have been previously demonstrated to be direct targets of miR-491-5p, EGFR and Bcl-xL. Based on our data, CTNNB1 and EP300 are likely to be indirect rather than direct targets. They are both down-regulated in proteomic data, but their transcriptomic levels are steady, and their transcripts are not enriched in pull-down. EP300 is however predicted to be a target of miR-491-5p by the MirTar website (
http://mirtar.mbc.nctu.edu.tw/human/ (accessed on 16 November 2020)) through an 8-mer site located in the coding region of its transcript. RAC3 is enriched in pull-down, but its transcript levels are steady and it is not detected in proteomics. It also has a predicted miR-491-5p binding site in its coding region according to MirTar. RRAS and RHOB are predicted to be direct targets of miR-491-5p through 3′UTR sites by TargetScan [
26] (
http://www.targetscan.org (accessed on 16 November 2020)). RRAS transcript is down-regulated in our data, but protein down-regulation and transcript enrichment in pull-down experiments do not reach cut-offs. RHOB protein is not detected in our proteomic experiment, but its transcript levels and pull-down enrichment values strongly suggest that it could be a genuine direct target of miR-491-5p. Further studies will be needed to assess which of these hubs do constitute direct targets of miR-491-5p.
Since our goal was to identify proteins or pathways modules that could be readily targeted with inhibitor molecules in order to emulate the effects of miR-491-5p, we chose to use a network-based approach to integrate data from our three experiments, without taking into consideration the direct or indirect nature of miR-491-5p effects on differentially expressed genes.
In this regard, pathway analysis of our data pointed to MAPK (a consequence of EGFR inhibition) and RHO-GTPases, which reflect the involvement of some interesting targets. Pathway analysis also underlined cell motion, and we could indeed validate miR-491-5p effects on cell invasion and wound healing in the SKOV3 cell line, in which no cell toxicity was induced with this miRNA. Overall, although informative and apparently reliable, pathway analysis with the most commonly used databases did not help us to identify candidate targets for the emulation of miR-491-5p effects. The construction of PPI networks proved itself to be much more efficient to point clearly towards potential targets. We could therefore successfully identify hubs whose combined inhibition recapitulates the cytotoxic phenotype of miR-491-5p. No single dataset was able to identify all five targets of interest, so focusing on a single method of analysis would miss some potentially highly interesting targets. The combination of the data from different methods to build PPI networks has led, as expected, to greater network complexity and, most importantly, has allowed us to pinpoint the most interesting targets. However, the use of all our three methods of analysis might not be essential. The combination of transcriptome and proteome data or pull-down and proteome data enabled the identification of our targets of interest. It could therefore be sufficient to perform only two experiments to get valuable results. One potential drawback of our approach at this point is that highly studied proteins tend to be the nodes with the most potential connections in a network. Although this could have led us to underestimate the role of some poorly studied factors, it is less likely that some specific inhibitors under clinical or pre-clinical consideration would have been available for such factors. We studied the effects of miR-491-5p in ovarian cancer cells, but our approach is indeed applicable to virtually any cellular context, in miRNA or to identify the determinants of any phenotype induced by the down-regulation of long non-coding RNAs (lncRNAs) for instance, which also have the ability to regulate the expression of several hundred genes and are deeply involved in cancer biology.
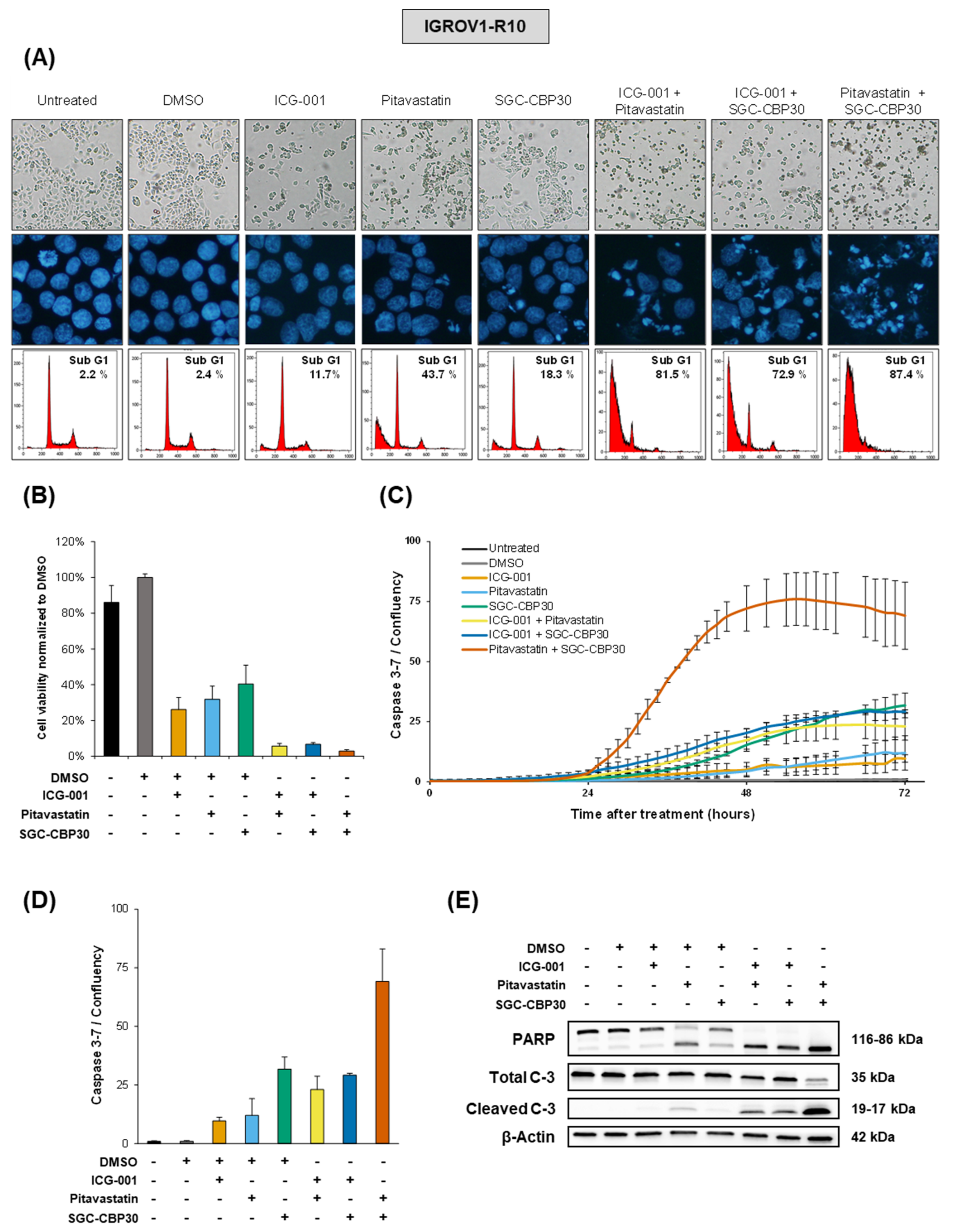
While miR-491-5p affects all the selected hubs together, our data show that targeting only two at a time efficiently mimicked its cytotoxic effects. This is of particular interest since the use of combination strategies using more than two molecules would not be realistically adaptable to a clinical setting. We postulate that pharmacological inhibitors would have a much stronger effect on their targets than the miRNA itself, whose effect relies on the moderate inhibition of a large number of targets. Moreover, our drug combination approach allowed the use of lower dose of each inhibitor, therefore lowering potential off-target and side effects. The use of a combination of pharmacological inhibitors appears therefore as a balanced strategy between synergistic effects and the strength of single-target inhibition.
Out of the five hubs we identified, Bcl-xL and EGFR already have pharmacological inhibitors available in clinical practice, ABT-263 [
27] and gefitinib [
28] respectively. Either of those used in combination with pitavastatin, ICG-001 or SGC-CBP30 is cytotoxic to ovarian cancer cells to an extent comparable to a combination of pitavastatin, ICG-001 and SGC-CBP30 (
Figure S7). Out of the three inhibitors we used, pitavastatin is the most advanced in clinics, since its use is already FDA-approved for lowering blood cholesterol, and it has been shown that it is an interesting option for the treatment of ovarian carcinoma [
14]. Interestingly, it has been suggested that the use of statins alone might induce side effects, and that its use at a lower concentration in combination with other drugs might reduce these [
14]. In addition, it was shown that small GTPase inhibition is able to sensitize ovarian cancer cells to cisplatin [
29]. ICG-001’s clinically useable form, PRI-724, has also been shown to sensitize ovarian cancer cells to cisplatin [
16]. Interestingly, PRI-724 has been shown to be safe and tolerable for patients with cirrhosis [
30], and the results of several early phase clinical trials with this molecule in pancreatic adenocarcinoma (NCT01764477) and myeloid malignancies (NCT01606579) have not been published yet. P300 has been described either as a tumor suppressor or an oncogene in tumors, depending on the tissue of origin [
31]. The use of SGC-CBP30 or other P300 inhibitor in ovarian cancer cells had not been reported yet. However, CBP/P300 bromodomain inhibitors have been proposed to be potentially interesting therapeutic tools, most likely through synergistic effects to improve the response to existing treatments [
16]. CCS1477 compound, a selective and orally bioavailable inhibitor of the bromodomain of p300 and CBP is being evaluated for its safety and efficacy in two clinical trials in hematological malignancies (NCT04068597) and in castration-resistant prostate cancer and other solid tumors (NCT03568656), both still in the recruitment phase. Interestingly, it has also been reported that P300 bromodomain is involved in IL6 signaling, which also appears as one of the most-connected hubs in our combined experiment (
Table 1).
More specifically in the context of ovarian cancer, the recently FDA-approved PARP inhibitors, increasing the amount of double strand DNA breaks, hold great promise for the treatment of this disease. Indeed, ovarian cancers represent the first cause of death from gynecological malignancies in developed countries, and the 5-year overall survival has stagnated over the past decades. Interestingly, it has been shown that the FDA-approved Wnt inhibitor pyrvinium pamoate, which down-regulates CTNNB1, synergizes with the PARP inhibitor olaparib in ovarian cancer cells and PDX models [
32]. In addition, P300 is also a co-activator BRCA1 [
33], and it mediates histone acetylation at the sites of double strand DNA breaks [
34] thus facilitating DNA repair; its inhibition might therefore constitute another interesting strategy to sensitize ovarian cancer to the action of PARP inhibitors.
4. Materials and Methods
4.1. Cell Culture and Treatments
IGROV1-R10 and SKOV3 cells were grown in RPMI Medium 1640, supplemented with 2 mM Glutamax, 25 µM HEPES, 10% fetal calf serum and 33 mM sodium bicarbonate (ThermoFisher Scientific, Illkirch, France). OAW42-R cells were grown in DMEM (Gibco, Fisher Scientific Bioblock, Illkirch, France), supplemented with 10% insulin (Novo Nordisk, Bagsvaerd, Denmark). The SKOV3 cell line was purchased from ATCC (LGS Standards, Molsheim, France). The IGROV1 cell line was kindly provided by Dr J. Bénard (Institute G. Roussy, Villejuif, France). The OAW42 cell line was purchased from ECACC (Sigma Aldrich, St Quentin-Fallavier, France). IGROV1-R10 and OAW42-R cells were obtained by mimicking a clinical protocol of the administration of cisplatin in vitro on IGROV1 and OAW42 cells, as detailed previously [
35].All cells lines were maintained in a 5% CO2 humidified atmosphere at 37 °C. Ovarian cancer cell lines were certified mycoplasma-free. For drug treatments, cells were treated the day after plating with growth media supplemented with an appropriate volume of drug solubilized in DMSO to ensure the desired drug concentration. When appropriate (i.e., for conditions where a single drug was used), extra DMSO was added to ensure that all conditions (except the untreated one) were exposed to the same DMSO concentration. At the endpoint of the experiments, the cell layer was trypsinized and centrifuged. Cell pellets were washed in cold PBS and processed accordingly for further analysis. ICG-001 (catalog number: 4505), pitavastatin (catalog number: 4942) and SGC-CBP30 (catalog number: 4889) were purchased from Tocris/bio-techne (Noyal Châtillon sur Seiche, France).
4.2. Transfection of miRNA
Hsa-miR-491-5p and miRNA negative control #1, CN-001000-01, were purchased from Dharmacon (ThermoFisher Scientific). Additionally, 3′ biotinylated miR-491-5p (5′ AGUGGGGAACCCUUCCAUGAGG 3′) and 3′ biotinylated cel-miR-65-5p (5′ CGCUCAUUCUGCCGGUUGUUAUG 3′) were purchased from Eurogentec (Liege, Belgium). Exponentially growing cells were seeded at 250,000 cells per 25 cm2 flask. Twenty-four hours after seeding, miRNA duplexes were diluted in OptiMEM (Life Technologies, Saint Aubin, France), and cells were transfected using INTERFERin (Polyplus-Transfection, Strasbourg, France) according to the manufacturer’s protocol with the indicated miRNA to a final concentration of 20 nM for unmodified miRNAs or 60 nM for biotinylated miRNAs.
4.3. Western Blotting
Pelleted cells were rinsed with ice-cold PBS 1X and lysed in RIPA 30 min on ice (50 mM Tris-HCl (pH 8), 150 mM NaCl, 1% Nonidet P-40, 5 mM EDTA, 10 mM NaF, 4 mM PMSF, 2 mM aprotinin, 10 mM NaPPi, 1 mM Na3VO4 and a complete mini mixture of protease inhibitors (Roche Applied Science)). After centrifugation (13,000× g, 4 °C, 10 min) to remove non-soluble cell debris, protein concentrations were measured using the Bradford assay. Then, 30 μg of protein were separated by SDS–PAGE on 4–15% gradient polyacrylamide gel (Biorad) and transferred to PVDF-membranes (Millipore) using the Trans-Blot Turbo Transfer system (Bio-Rad, Marnes-la-Coquette, France). After blocking for 1 h at RT with 5% (v/v) non-fat dry milk in TBS with 0.05% (v/v) Tween20 (T-TBS), membranes were incubated overnight at 4 °C in blocking buffer with the following primary antibodies at the indicated dilutions: PARP (1:1000) (9542), total and cleaved caspase-3 (1:500) (9662) (Cell Signaling Technology, Ozyme, Saint Quentin en Yvelines, France) and appropriate horseradish-peroxidase-conjugated secondary antibodies (CST or GE HealthCare Europe GmbH, Velizy-Villacoublay, France) were used, and signals were detected using enhanced chemiluminescence (GE HealthCare Europe GmbH, Velizy-Villacoublay, France). Blots were also hybridized with β-actin (1:5000) (Eurobio, Courtaboeuf, France), monoclonal antibodies to control protein loading. Each immunoblot is representative of three distinct experiments.
4.4. Flow Cytometry
Cells were detached by trypsinization, washed with PBS, fixed in 70% ethanol and stored at −20 °C until analysis. Fixed cells were centrifuged (2000 rpm, 5 min) and incubated for 30 min at 37 °C in PBS. After centrifugation, cells were resuspended and stained with propidium iodide using the DNA-Prep Coulter Reagent Kit (Beckman Coulter, Villepinte, France) and were analyzed using an EPICS XL flow cytometer (Beckman Coulter). Computerized gating was applied on the side and forward scattering to exclude small debris and on a pulse width and integral peak of red fluorescence to eliminate aggregates. The data were analyzed by Expo32 acquisition software (Beckman Coulter).
4.5. RNA Extraction
RNA was extracted from cell pellets (except for biotin pull-down experiments, see below) using TRIzol Reagent (ThermoFisher Scientific). RNA was resuspended in 43 µL nuclease-free water, 2 µL RQ1 DNAseI and 5 µL 10X DNase buffer (Promega, Charbonnières-les-Bains, France) and incubated 1 h at 37 °C. After DNA digestion, RNA was purified with TRIzol, resuspended in nuclease-free water and dosed and quality-controlled on a NanoDrop 2000 spectrophotometer (ThermoFisher Scientific).
4.6. Biotin Pull-Down of miR-491-5p-Linked RNA
Our protocol was adapted from the one published by Lal et al. [
36]. IGROV1-R10 cells were seeded in 25 cm
2 flasks the day before transfection with biotinylated miRNAs and harvested by trypsinization 24 h after transfection. Cells were pelleted and washed once in cold PBS. Cells were then resuspended in 700 µL lysis buffer (20 mM Tris-HCl pH 7.5, 5 mM MgCl2, 100 mM KCl, 0.3% NP-40, protease inhibitor and RNase inhibitor 60 U/mL (RNase OUT, ThermoFisher Scientific)), then incubated on ice for 5 min. Samples were then centrifuged (4 °C, 130,00×
g) to remove cell debris and nuclei. Then, 10% of the lysate was sampled and submitted to RNA extraction with Trizol reagent, to be later used as “input”, while the rest was incubated on 50 µL magnetic streptavidin beads (Dynabeads MyOne Streptavidin T1, ThermoFisher Scientific) at 4 °C for 4 h under agitation. Before this incubation step, beads were washed twice and blocked by incubation in lysis buffer supplemented with BSA (1 mg/mL) (Promega) and yeast tRNA (1 mg/mL) (Sigma-Aldrich) at 4 °C for 2 h under agitation. At the end of the 4 h incubation period with lysate, beads were washed 5 times in 500 µL lysis buffer. After the final wash, they were resuspended in Trizol reagent to proceed with RNA purification. After RT and qPCR, transcript enrichment was calculated as follows after normalization: (biotin-miR-491-5p Pull-Down/biotin-cel-miR-65 Pull-Down)/(biotin-miR-491-5p Input/biotin-cel-miR-Input). This method of enrichment calculation was aimed at normalizing the decrease in miR-491-5p-targeted RNA levels in biotin-miR-491-5p-transfected samples relative to biotin-cel-miR-65. The pull-down experiment was run 3 times independently. The RNA from the 3 experiments was pulled together to constitute the samples submitted to NGS. NGS was run once for each of the 4 samples: biotin-miR-491-5p Pull-Down, biotin-cel-miR-65 Pull-Down, biotin-miR-491-5p Input, biotin-cel-miR Input.
4.7. Pulled-Down Data Sequencing and Processing
Total RNA was sent to IntegraGen SA (Evry, France) for library construction with TruSeq Stranded Total RNA Sample Prep’ Illumina, and paired-end 2 × 75 nt sequencing was performed on HiSeq 2500 (Illumina). Reads were mapped with reference genome (GRCh38) using STAR mapper tool version 2.5.2a. To count reads that were aligned in each gene, the featureCounts tool (version 1.5.0) [
37] were used with the GENCODE annotation (release 24, in GTF format) downloaded from gencodegens.org. Only read pairs that had both ends successfully aligned were considered. Chimeric reads as well as duplicated reads were not counted. All genes having 0 counts in all samples were filtered out before subsequent analysis. Raw counts were normalized using the size factor-based approach implemented in DESeq2 package (version 1.10.1) [
38]. Normalized counts were then used to compute the enrichment ratio following the formula described above.
4.8. Microarray Analysis
IGROV1-R10 cells were seeded in 25 cm2 flasks the day before transfection with miRNAs and harvested by trypsinization 48 h after transfection. RNA was extracted and the concentration of purified RNA from one control miRNA and one miR-491-5p-transfected sample was assessed using the Qubit reagent assay RNA quantification kit (Thermo Fisher Scientific). Sample RNAs (100 ng) were converted to biotin-labeled single-strand cDNAs using the Affymetrix Genechip WT Plus. Labeled and fragmented ss-cDNA (5.5 µg) was hybridized to Affymetrix arrays (Genechip HTA human array). The data obtained for the whole set of samples were normalized by the RMA process (Affymetrix Expression Consol). Probe-set annotation, quantitative expressions of all the transcripts and comparisons between the different groups of samples were analyzed using the Affymetrix software TAC.v3.
4.9. Proteomics and SILAC-Based Mass Spectrometry Analysis
IGROV1-R10 were grown in SILAC RPMI Medium 1640 supplemented with 2 mM Glutamax, 25 µM HEPES, 10% dialyzed fetal bovine serum and 33 mM sodium bicarbonate (ThermoFisher Scientific, Illkirch, France). Medium was also supplemented with standard L-lysine and L-arginine (referred to as the “light” medium) or with 13C615N2 L-lysine and 13C615N4 L-arginine (referred to as the “heavy” medium). Cells were grown in either light or heavy medium for at least 7 doubling times to ensure >95% labeling of cellular proteins, according to the manufacturer’s protocol. Cells grown in both light and heavy media were seeded in 25 cm
2 flasks the day before transfection with control miRNA or with miR-491-5p, for a total of 4 experimental conditions. Cells were harvested by trypsinization 48 h after transfection, and proteins were purified. Equal amounts of protein extracts from heavy/miRNA control and light/miR-491-5p were mixed together, as well as light/miRNA control and heavy/miR-491-5p. Both protein mixes, each from one experiment, were subjected to mass spectrometry analysis. Protein extracts were separated on SDS–PAGE gels (10%, Invitrogen at 30mA for 1 h) and stained with colloidal blue staining (LabSafe GEL BlueTM GBiosciences). Gel slices were excised (7 bands), and proteins were reduced with 10 mM DTT prior to alkylation with 55 mM iodoacetamide. After washing and shrinking the gel pieces with 100% MeCN, in-gel digestion was performed using trypsin (Promega) overnight in 25 mM NH4HCO3 at 30 °C. Peptides were analyzed by LC-MS/MS using an RSLCnano system (Ultimate 3000, ThermoFisher Scientific) coupled to an Orbitrap Fusion Tribrid mass spectrometer (ThermoFisher Scientific). Peptides were loaded onto a C18-reversed phase column (300-μm inner diameter × 5 mm; ThermoFisher Scientific), separated and MS data acquired using Xcalibur software. Peptide separation was performed over a multistep gradient of 95 min from 1% to 32% (vol/vol) acetonitrile (75-μm inner diameter × 50 cm; nanoViper C18, 3 μm, 100Å, Acclaim PepMapTM, ThermoFisher Scientific). Full-scan MS was acquired in the Orbitrap analyzer with a resolution set to 120,000, and ions from each full scan were HCD fragmented and analyzed in the linear ion trap. Data were acquired using the Xcalibur software (v 3.0), and the resulting spectra were interrogated by Sequest HT through Proteome Discoverer (v 1.4, ThermoFisher Scientific) with the SwissProt Homo Sapiens database (032015). We set carbamidomethyl cysteine, the oxidation of methionine, N-terminal acetylation, heavy 13C615N2-lysine (Lys8) and 13C615N4-arginine (Arg10) and medium 2H4-lysine (Lys4) and 13C6-arginine (Arg6) as variable modifications. We set the specificity of trypsin digestion and allowed 2 missed cleavage sites, and we set the mass tolerances in MS and MS/MS to 10 ppm and 0.6 Da, respectively. The resulting files were further processed by using myProMS (v 3.5) [
39]. The Sequest HT target and decoy search result were validated at 1% false discovery rate (FDR) with Percolator. For SILAC-based protein quantification, peptides XICs (extracted ion chromatograms) were retrieved from Proteome DiscovererTM. Scale normalization was applied to compensate for mixing errors of the different SILAC cultures. Protein ratios were computed as the geometrical mean of related peptides. To estimate ratio significance, a t test was performed with a Benjamini–Hochberg FDR control threshold set to 0.05. (All quantified proteins have at least 3 peptides quantified (all peptides selected)).
4.10. Sylamer Analysis
The enrichment of all possible words of different length (from 5 to >8) at the 3′ untranslated regions (3′UTR) were systematically assessed using the approach based on the hypergeometric probability distribution implemented in the Sylamer tool (version 08-281) [
40]. Furthermore, 3′UTR sequences were previously sorted according to the fold-enrichment (for the pull-down experiment result, from top enriched to top depleted genes) and the fold-change (for transcriptomics and proteomics experiment, from top up-regulated to top down-regulated genes). The 3′UTR sequence corresponding to each annotated transcript was downloaded from the Ensembl web site (via biomart). As we performed a gene level analysis, we ensured that, for any gene having multiple annotated transcripts in the database, the longest 3′UTR sequence were retained. Using Sylamer, we computed then the
p-value of over-representation of each word in a set of sequences (window) defined from the top of the ranked 3′UTR list compared to the rest of the sequences in the list (leading window). This
p-value corresponded to the
p-value of the under-representation of the same word in the leading window compared to the current window due to the symmetric property of the hypergeometric distribution. The size of the window was constantly increased until the current window included all the sequences in the list. Sylamer reported the
p-value result of each word (with a specific length) and each window size in a text format table that we used to plot the result with an R function inspired from an R script available in Sylamer itself. In this plot, for a given word, the (inverse) peak of
p-values indicates a cut-off that separate the ranked sequence list in two parts: the first part includes 3′UTR sequences wherein the given word is under-represented, and the second part includes 3′UTR sequences wherein the word is over-represented with the lowest
p-value. For more details about the algorithm and the statistical model implemented in Sylamer, please refer to van Dongen et al. 2008 [
40] (especially the Supplementary Methods part). For our study, for the pull-down and transcriptomics experiments, we set the starting window size and the increment size to 500 sequences. For proteomics experiment, due to the small number of detected proteins (compared to the two previous experiments), we decided to set the same parameter to 100 sequences in order to have more clear resolution.
4.11. Network Analysis and Visualization
Human protein–protein interaction (PPI) information was downloaded from the STRING (version 10). The “combine_score” column was used to filter out low confident interactions. Interactions having a “combine_score” lower than 400 were filtered out. The filtered table of PPI interactions was merged with the rest of the datasets. The biomaRt bioconductor R package was used to map gene identifiers. Cytoscape (version 3) [
41] software was used to build, visualize and characterize the resulting networks.
4.12. Statistical Analysis
Data processing, data analysis and data integration were performed inside an R (version 3.2.3) environment. Network analysis were performed inside Cytoscape (version 3) [
41].
4.13. Live-Cell Wound Healing Assay
SKOV3 cells (3.5 × 103 per well) were seeded in 96-well plates. The following day, cells were transfected with miR-491-5p or miRNA negative control and then were incubated in the IncuCyte S3 system (Sartorius, Goettingen, Germany) for real-time imaging, with three fields imaged per well under 10× magnification every hour. Data were analyzed using the IncuCyte Confluence software, which quantified cell surface area coverage as confluence values. IncuCyte experiments were performed in triplicate. Cell confluence was graphed over time to evaluate the characteristics of proliferation in the presence of molecules.
4.14. Kinetic Quantification of Caspase-3/7 Mediated Apoptosis Using Live-Cell Time-Lapse Imaging
Caspase-3/7 activity was assessed using the IncuCyte Caspase-3/7 Green Apoptosis Assay Reagent (Sartorius) as described previously [
42]. Briefly, this assay consists of an inert peptide, a caspase -3/7 recognition site and the peptide NucViewTM 488 (Sartorius). The full-length caspase-3/7 reagent is non-fluorescent and is confined to the cytoplasm. Upon the induction of apoptosis, caspase-3/7 cleaves the bond between the inert peptide and NucViewTM 488. Liberated NucViewTM 488 has a high affinity for nuclear DNA and is fluorescent in the green spectrum; thus, caspase-3/7 activation correlates with an increase in fluorescent green nuclei. To assess apoptosis, a total of 6 × 10
3 IGROV1-R10 cells were cultured in 96-well plates and monitored in the IncuCyte S3 acquiring images (objective × 10) every 1 h in 2 separate regions per well after transfection or treatment with the indicated molecules. The live-cell phase contrast images were used to calculate confluence using the IncuCyte software, and to provide morphology information. Each experiment was done in triplicate and the accumulation of caspase-3/7 over time was normalized to cell confluence.
4.15. CellTiter-Glo Assay
ATP levels were quantified using CellTiter-Glo 3D cell viability assay (Promega) according to the manufacturer’s instruction, and luminescence was measured using Centro XS3 LB 960 (Berthold Technologies, Bad Wildbad, Germany) with Miko Win 2000 software. All viability results were normalized to DMSO.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}