
Diving into the Pleural Fluid: Liquid Biopsy for Metastatic Malignant Pleural Effusions

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
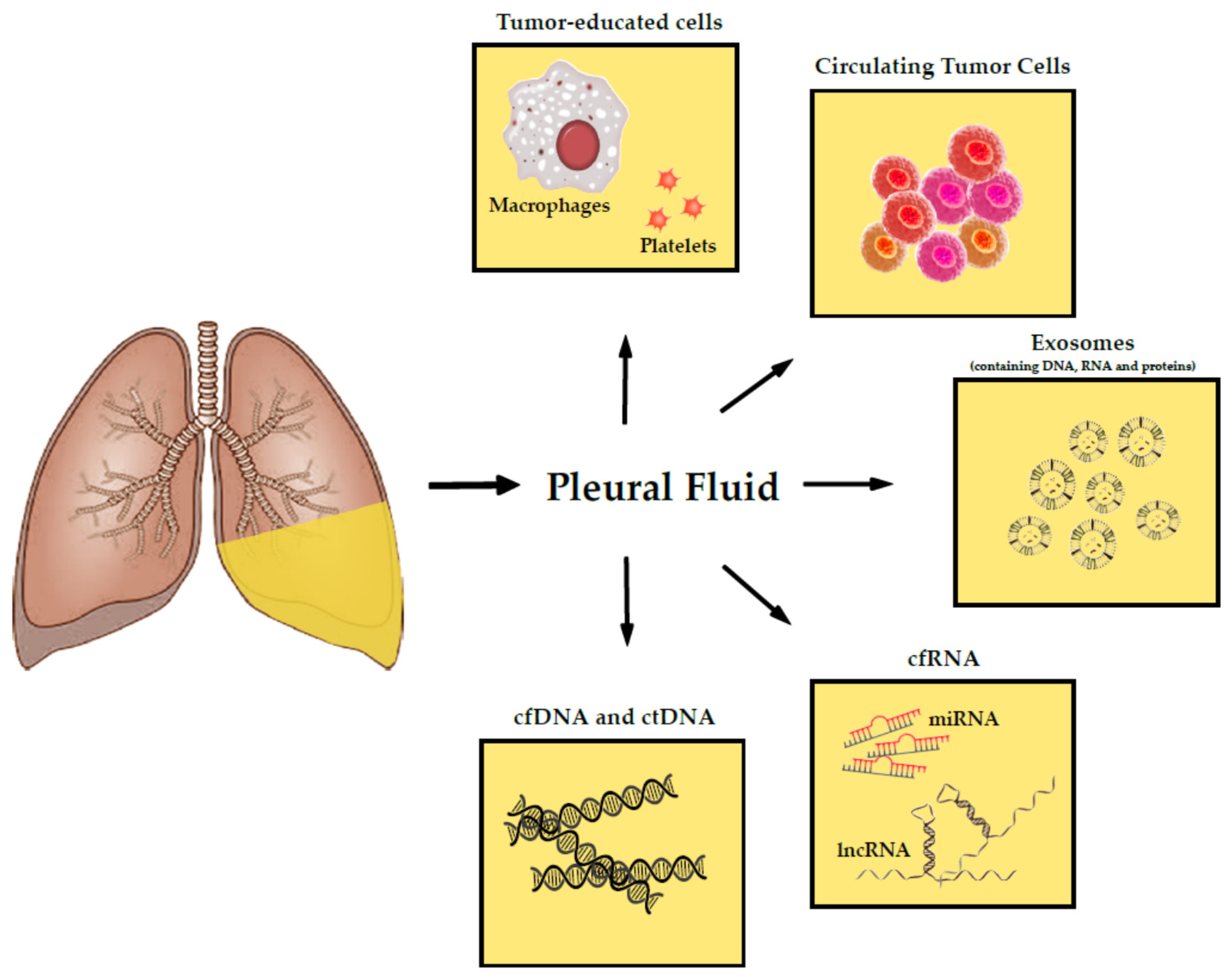
2. Tumor-Derived Products in the Pleural Fluid
2.1. Nucleic Acids
2.1.1. Cell-Free DNA and Circulating Tumor DNA
Genomic Strategies for Mutation Profiling in Pleural Fluid
Approved Clinical Tests for Routine Testing
Limitations of the Detection of Mutations on ctDNA
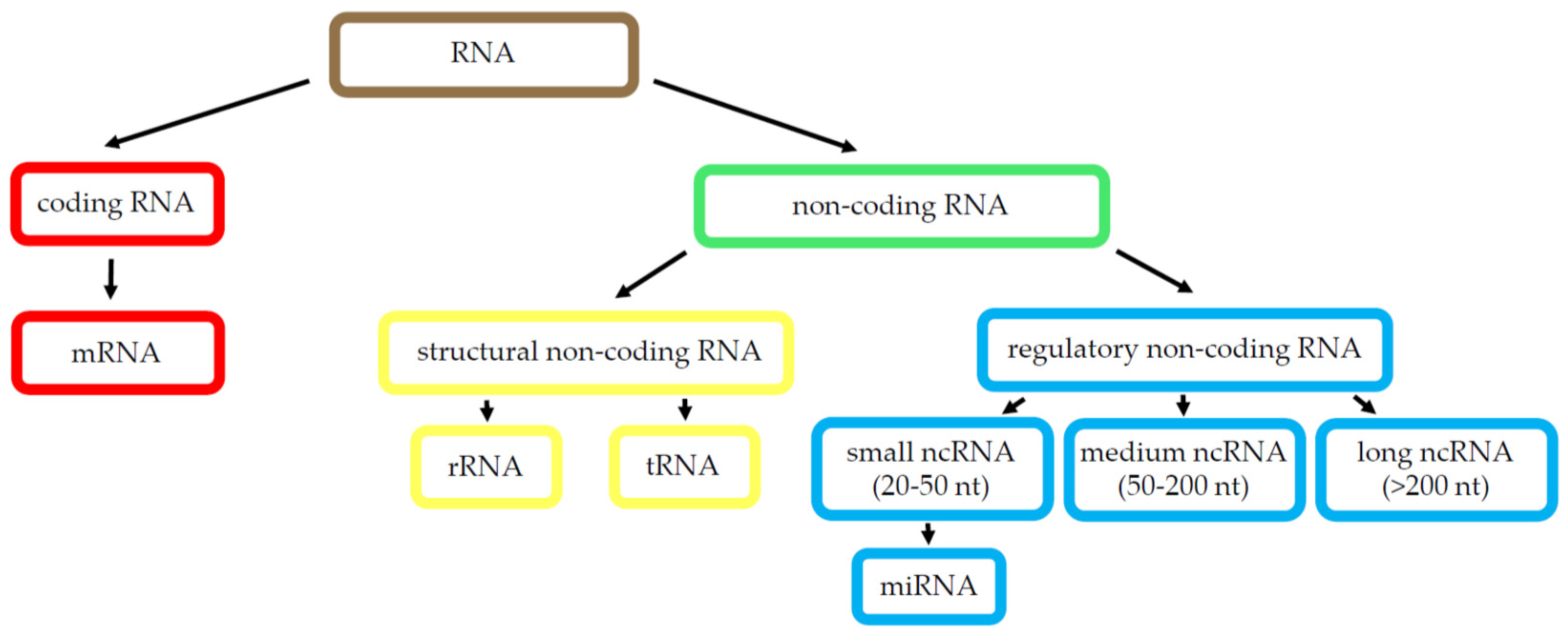
2.1.2. RNA Species
MicroRNA
Long Non-Coding RNA
Limitations of the ncRNA Implementation
2.2. Exosomes
2.3. Cell Entities
2.4. Circulating Tumor Cells
3. Future Perspectives: Tumor-Educated Cells
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Porcel, J.M.; Light, R.W. Pleural effusions. Dis. Mon. 2013, 59, 29–57. [Google Scholar] [CrossRef]
- Porcel, J.M.; Esquerda, A.; Vives, M.; Bielsa, S. Etiology of Pleural Effusions: Analysis of More Than 3000 Consecutive Thoracenteses. Arch. Bronconeumol. 2014, 50, 161–165. [Google Scholar] [CrossRef]
- Porcel, J.M.; Quirós, M.; Gatius, S.; Bielsa, S. Examination of cytological smears and cell blocks of pleural fluid: Complementary diagnostic value for malignant effusions. Revista Clínica Española 2017, 217, 144–148. [Google Scholar] [CrossRef]
- Porcel, J.M. Diagnosis and characterization of malignant effusions through pleural fluid cytological examination. Curr. Opin. Pulm. Med. 2019, 25, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Mandel, P.; Metais, P. Nuclear Acids in Human Blood Plasma. Available online: https://pubmed.ncbi.nlm.nih.gov/18875018/ (accessed on 23 January 2021).
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the Serum of Cancer Patients and the Effect of Therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Benlloch, S.; Martí-Ciriquián, J.L.; Galbis-Caravajal, J.M.; Martín, C.; Sánchez-Payá, J.; Rodríguez-Paniagua, J.M.; Romero, S.; Massutí, B. Cell-free DNA concentration in pleural fluid and serum: Quantitative approach and potential prognostic factor in patients with cancer and pleural effusions. Clin. Lung Cancer 2006, 8, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Martignano, F.; Molinari, C.; Gurioli, G.; Calistri, D.; De Giorgi, U.; Conteduca, V.; Casadio, V. The potential use of urine cell free DNA as a marker for cancer. Expert Rev. Mol. Diagn. 2016, 16, 1283–1290. [Google Scholar] [CrossRef]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709. [Google Scholar] [CrossRef]
- Hyun, K.A.; Gwak, H.; Lee, J.; Kwak, B.; Jung, H. Il Salivary exosome and cell-free DNA for cancer detection. Micromachines 2018, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Umu, S.U.; Langseth, H.; Bucher-Johannessen, C.; Fromm, B.; Keller, A.; Meese, E.; Lauritzen, M.; Leithaug, M.; Lyle, R.; Rounge, T.B. A comprehensive profile of circulating RNAs in human serum. RNA Biol. 2018, 15, 242–250. [Google Scholar] [CrossRef]
- de Souza, A.G.; Bastos, V.A.F.; Fujimura, P.T.; Ferreira, I.C.C.; Leal, L.F.; da Silva, L.S.; Laus, A.C.; Reis, R.M.; Martins, M.M.; Santos, P.S.; et al. Cell-free DNA promotes malignant transformation in non-tumor cells. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.H.M.; Chow, K.M.; Chan, A.T.C.; Leung, C.B.; Chan, L.Y.S.; Chow, K.C.K.; Lam, C.W.; Lo, Y.M.D. Quantitative analysis of pleural fluid cell-free DNA as a tool for the classification of pleural effusions. Clin. Chem. 2003, 49, 740–745. [Google Scholar] [CrossRef]
- Chetty, K. Transudative pleural effusions. Clin. Chest Med. 1985, 6, 49–54. [Google Scholar] [CrossRef]
- Porcel, J.M.; Light, R.W. Diagnostic approach to pleural effusion in adults. Am. Fam. Physician 2006, 73, 1211–1220. [Google Scholar]
- Santotoribio, J.D.; Cabrera-Alarcón, J.L.; Batalha-Caetano, P.; Macher, H.C.; Guerrero, J.M. Pleural fluid cell-free DNA in parapneumonic pleural effusion. Clin. Biochem. 2015, 48, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Yu, Y.; Qian, J.; Shen, L.; Ji, W.; Lu, S. Distinct profile of cell-free DNA in malignant pleural effusion of non-small cell lung cancer and its impact on clinical genetic testing. Int. J. Med. Sci. 2021, 18, 1510–1518. [Google Scholar] [CrossRef]
- Sriram, K.B.; Relan, V.; Clarke, B.E.; Duhig, E.E.; Windsor, M.N.; Matar, K.S.; Naidoo, R.; Passmore, L.; McCaul, E.; Courtney, D.; et al. Pleural fluid cell-free DNA integrity index to identify cytologically negative malignant pleural effusions including mesotheliomas. BMC Cancer 2012. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Vasioukhin, V.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Stroun, M. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br. J. Haematol. 1994, 86, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, G.D.; Pribish, D.M.; Valone, F.H.; Memoli, V.A.; Bzik, D.J.; Yao, S.L. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiol. Prev. Biomarkers 1994, 3, 67–71. [Google Scholar]
- Shi, Y.; Au, J.S.K.; Thongprasert, S.; Srinivasan, S.; Tsai, C.M.; Khoa, M.T.; Heeroma, K.; Itoh, Y.; Cornelio, G.; Yang, P.C. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J. Thorac. Oncol. 2014, 9, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Gahr, S.; Stoehr, R.; Geissinger, E.; Ficker, J.H.; Brueckl, W.M.; Gschwendtner, A.; Gattenloehner, S.; Fuchs, F.S.; Schulz, C.; Rieker, R.J.; et al. EGFR mutational status in a large series of Caucasian European NSCLC patients: Data from daily practice. Br. J. Cancer 2013, 109, 1821–1828. [Google Scholar] [CrossRef]
- Kerr, K.M.; Dafni, U.; Schulze, K.; Thunnissen, E.; Bubendorf, L.; Hager, H.; Finn, S.; Biernat, W.; Vliegen, L.; Losa, J.H.; et al. Prevalence and clinical association of gene mutations through multiplex mutation testing in patients with NSCLC: Results from the ETOP Lungscape Project. Ann. Oncol. 2018, 29, 200–208. [Google Scholar] [CrossRef]
- Huang, M.J.; Lim, K.H.; Tzen, C.Y.; Hsu, H.S.; Yen, Y.; Huang, B.S. EGFR mutations in malignant pleural effusion of non-small cell lung cancer: A case report. Lung Cancer 2005, 49, 413–415. [Google Scholar] [CrossRef]
- Lin, J.; Gu, Y.; Du, R.; Deng, M.; Lu, Y.; Ding, Y. Detection of EGFR mutation in supernatant, cell pellets of pleural effusion and tumor tissues from non-small cell lung cancer patients by high resolution melting analysis and sequencing. Int. J. Clin. Exp. Pathol. 2014, 7, 8813–8822. [Google Scholar]
- Tsai, T.H.; Wu, S.G.; Hsieh, M.S.; Yu, C.J.; Yang, J.C.H.; Shih, J.Y. Clinical and prognostic implications of RET rearrangements in metastatic lung adenocarcinoma patients with malignant pleural effusion. Lung Cancer 2015, 88, 208–214. [Google Scholar] [CrossRef]
- Han, H.S.; Eom, D.W.; Kim, J.H.; Kim, K.H.; Shin, H.M.; An, J.Y.; Lee, K.M.; Choe, K.H.; Lee, K.H.; Kim, S.T.; et al. EGFR mutation status in primary lung adenocarcinomas and corresponding metastatic lesions: Discordance in pleural metastases. Clin. Lung Cancer 2011, 12, 380–386. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, Y.; Wang, M.; Yap, W.S.; Chang, A.Y.C. Detection and comparison of epidermal growth factor receptor mutations in cells and fluid of malignant pleural effusion in non-small cell lung cancer. Lung Cancer 2008, 60, 175–182. [Google Scholar] [CrossRef]
- Kimura, H.; Fujiwara, Y.; Sone, T.; Kunitoh, H.; Tamura, T.; Kasahara, K.; Nishio, K. EGFR mutation status in tumour-derived DNA from pleural effusion fluid is a practical basis for predicting the response to gefitinib. Br. J. Cancer 2006, 95, 1390–1395. [Google Scholar] [CrossRef]
- Husain, H.; Nykin, D.; Bui, N.; Quan, D.; Gomez, G.; Woodward, B.; Venkatapathy, S.; Duttagupta, R.; Fung, E.; Lippman, S.M.; et al. Cell-Free DNA from Ascites and Pleural Effusions: Molecular Insights into Genomic Aberrations and Disease Biology. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef]
- Lee, J.S.; Hur, J.Y.; Kim, I.A.; Kim, H.J.; Choi, C.M.; Lee, J.C.; Kim, W.S.; Lee, K.Y. Liquid biopsy using the supernatant of a pleural effusion for EGFR genotyping in pulmonary adenocarcinoma patients: A comparison between cell-free DNA and extracellular vesicle-derived DNA. BMC Cancer 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, O.J.; Son, S.M.; Woo, C.G.; Jeong, Y.; Yang, Y.; Kwon, J.; Lee, K.H.; Han, H.S. EGFR mutation status in lung adenocarcinoma-associated malignant pleural effusion and efficacy of egfr tyrosine kinase inhibitors. Cancer Res. Treat. 2018, 50, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.D.; Kim, J.W.; Kim, K.H.; Ha, J.H.; Rhee, C.K.; Kim, S.J.; Kim, Y.K.; Park, C.K.; Lee, S.H.; Park, M.S.; et al. Detection and comparison of EGFR mutations in matched tumor tissues, cell blocks, pleural effusions, and sera from patients with NSCLC with malignant pleural effusion, by PNA clamping and direct sequencing. Lung Cancer 2013, 81, 207–212. [Google Scholar] [CrossRef]
- Han, H.S.; Lim, S.N.; An, J.Y.; Lee, K.M.; Choe, K.H.; Lee, K.H.; Kim, S.T.; Son, S.M.; Choi, S.Y.; Lee, H.C.; et al. Detection of EGFR mutation status in lung adenocarcinoma specimens with different proportions of tumor cells using two methods of differential sensitivity. J. Thorac. Oncol. 2012, 7, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Kim, J.; Kim, Y.; Cho, S.M.; Lee, K.A. Assessment of real-time PCR method for detection of EGFR mutation using both supernatant and cell pellet of malignant pleural effusion samples from non-small-cell lung cancer patients. Clin. Chem. Lab. Med. 2017, 55, 1962–1969. [Google Scholar] [CrossRef]
- Jian, G.; Songwen, Z.; Ling, Z.; Qinfang, D.; Jie, Z.; Liang, T.; Caicun, Z. Prediction of epidermal growth factor receptor mutations in the plasma/pleural effusion to efficacy of gefitinib treatment in advanced non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 1341–1347. [Google Scholar] [CrossRef]
- Wu, S.G.; Gow, C.H.; Yu, C.J.; Chang, Y.L.; Yang, C.H.; Hsu, Y.C.; Shih, J.Y.; Lee, Y.C.; Yang, P.C. Frequent epidermal growth factor receptor gene mutations in malignant pleural effusion of lung adenocarcinoma. Eur. Respir. J. 2008, 32, 924–930. [Google Scholar] [CrossRef]
- Akamatsu, H.; Koh, Y.; Kenmotsu, H.; Naito, T.; Serizawa, M.; Kimura, M.; Mori, K.; Imai, H.; Ono, A.; Shukuya, T.; et al. Multiplexed molecular profiling of lung cancer using pleural effusion. J. Thorac. Oncol. 2014, 9, 1048–1052. [Google Scholar] [CrossRef]
- Carter, J.; Miller, J.A.; Feller-Kopman, D.; Ettinger, D.; Sidransky, D.; Maleki, Z. Molecular profiling of malignant pleural effusion in metastatic non-small-cell lung carcinoma the effect of preanalytical factors. Ann. Am. Thorac. Soc. 2017, 14, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Shi, W.; Xu, H.; Hu, H.; Dong, Z.; Zhu, G.; Sun, Y.; Liu, B.; Gao, H.; et al. Droplet digital PCR improved the EGFR mutation diagnosis with pleural fluid samples in non-small-cell lung cancer patients. Clin. Chim. Acta 2017, 471, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Asaka, S.; Yoshizawa, A.; Saito, K.; Kobayashi, Y.; Yamamoto, H.; Negishi, T.; Nakata, R.; Matsuda, K.; Yamaguchi, A.; Honda, T. Rapid point-of-care testing for epidermal growth factor receptor gene mutations in patients with lung cancer using cell-free DNA from cytology specimen supernatants. Int. J. Oncol. 2018, 52, 2110–2118. [Google Scholar] [CrossRef]
- Buttitta, F.; Felicioni, L.; Del Grammastro, M.; Filice, G.; Di Lorito, A.; Malatesta, S.; Viola, P.; Centi, I.; D’Antuono, T.; Zappacosta, R.; et al. Effective assessment of egfr mutation status in bronchoalveolar lavage and pleural fluids by next-generation sequencing. Clin. Cancer Res. 2013, 19, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.; Hua, P.; Liu, N.; Li, Q.; Zhu, X.; Jiang, L.; Zheng, K.; Su, X. Targeted next-generation sequencing in cytology specimens for molecular profiling of lung adenocarcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 3647–3655. [Google Scholar]
- Zhang, P.; Wu, X.; Tang, M.; Nie, X.; Li, L. Detection of EGFR gene mutation status from pleural effusions and other body fluid specimens in patients with lung adenocarcinoma. Thorac. Cancer 2019, 10, 2218–2224. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Mooney, K.L.; Libiran, P.; Jones, C.D.; Joshi, R.; Lau, H.D.; Stehr, H.; Berry, G.J.; Zehnder, J.L.; Long, S.R.; et al. Targeted deep sequencing of cell-free DNA in serous body cavity fluids with malignant, suspicious, and benign cytology. Cancer Cytopathol. 2020, 128, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.; Sun, Y.; Wang, W.; Ye, J.; Zhang, D.; Gong, Z.; Yang, M. Multiplexed molecular profiling of lung cancer with malignant pleural effusion using next generation sequencing in Chinese patients. Oncol. Lett. 2020, 19, 3495–3505. [Google Scholar] [CrossRef]
- Liu, L.; Shao, D.; Deng, Q.; Tang, H.; Wang, J.; Liu, J.; Guo, F.; Lin, Y.; Peng, Z.; Mao, M.; et al. Next generation sequencing-based molecular profiling of lung adenocarcinoma using pleural effusion specimens. J. Thorac. Dis. 2018, 10, 2631–2637. [Google Scholar] [CrossRef]
- Vanni, I.; Tanda, E.T.; Spagnolo, F.; Andreotti, V.; Bruno, W.; Ghiorzo, P. The Current State of Molecular Testing in the BRAF-Mutated Melanoma Landscape. Front. Mol. Biosci. 2020, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Cai, P.; Xie, J.; Wei, Y. The diagnostic accuracy of digital PCR, ARMS and NGS for detecting KRAS mutation in cell-free DNA of patients with colorectal cancer: A systematic review and meta-analysis. PLoS ONE 2021, 16. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Xie, H.; Urrutia, R.; Mahipal, A. The promise of circulating tumor DNA (ctDNA) in the management of early-stage colon cancer: A critical review. Cancers 2020, 12, 2808. [Google Scholar] [CrossRef] [PubMed]
- Postel, M.; Roosen, A.; Laurent-Puig, P.; Taly, V.; Wang-Renault, S.F. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: A cancer diagnostic perspective. Expert Rev. Mol. Diagn. 2018, 18, 7–17. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.F. Precision medicine and testing for tumor biomarkers-are all tests born equal? JAMA Oncol. 2018, 4, 773–774. [Google Scholar] [CrossRef]
- Leighl, N.B.; Rekhtman, N.; Biermann, W.A.; Huang, J.; Mino-Kenudson, M.; Ramalingam, S.S.; West, H.; Whitlock, S.; Somerfield, M.R. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the college of American pathologists/international association for the study of lung cancer/association for molecular pathology guideline. J. Clin. Oncol. 2014, 32, 3673–3679. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhou, C.; Liam, C.K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR -Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR -Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Shen, L.; Ji, W.; Lu, S. Standardization of pleural effusion-based tumor mutation burden (TMB) estimation using capture-based targeted sequencing. Ann. Transl. Med. 2021, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Xu, X.; Chen, J.; Song, E.W. Non-coding RNAs: The new central dogma of cancer biology. Sci. China Life Sci. 2021, 64, 22–50. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Zen, K.; Zhang, C.Y. Circulating MicroRNAs: A novel class of biomarkers to diagnose and monitor human cancers. Med. Res. Rev. 2012, 32, 326–348. [Google Scholar] [CrossRef]
- Bianchi, F.; Nicassio, F.; Marzi, M.; Belloni, E.; Dall’Olio, V.; Bernard, L.; Pelosi, G.; Maisonneuve, P.; Veronesi, G.; Di Fiore, P.P. A serum circulating miRNA diagnostic test to identify asymptomatic high-risk individuals with early stage lung cancer. EMBO Mol. Med. 2011, 3, 495–503. [Google Scholar] [CrossRef]
- Boeri, M.; Verri, C.; Conte, D.; Roz, L.; Modena, P.; Facchinetti, F.; Calabrò, E.; Croce, C.M.; Pastorino, U.; Sozzi, G. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 3713–3718. [Google Scholar] [CrossRef]
- Shen, J.; Todd, N.W.; Zhang, H.; Yu, L.; Lingxiao, X.; Mei, Y.; Guarnera, M.; Liao, J.; Chou, A.; Lu, C.L.; et al. Plasma microRNAs as potential biomarkers for non-small-cell lung cancer. Lab. Investig. 2011, 91, 579–587. [Google Scholar] [CrossRef]
- Bao, Q.; Xu, Y.; Ding, M.; Chen, P. Identification of differentially expressed miRNAs in differentiating benign from malignant pleural effusion. Hereditas 2020, 157. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Jo, Y.N.; Lee, J.Y.; Choi, S.Y.; Jeong, Y.; Yun, J.; Lee, O.J. Identification of suitable reference genes for the relative quantification of microRNAs in pleural effusion. Oncol. Lett. 2014, 8, 1889–1895. [Google Scholar] [CrossRef]
- Wojczakowski, W.; Kobylarek, D.; Lindner, J.; Limphaibool, N.; Kaczmarek, M. MicroRNAs—Novel biomarkers for malignant pleural effusions. Wspolczesna Onkol. 2019, 23, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.M.; Yun, J.; Lee, O.J.; Han, H.S.; Lim, S.N.; An, J.Y.; Lee, K.H.; Lee, K.M.; Choe, K.H. Diagnostic Value of Circulating Extracellular miR-134, miR-185, and miR-22 Levels in Lung Adenocarcinoma-Associated Malignant Pleural Effusion. Cancer Res. Treat. 2014, 46, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Yun, J.; Lim, S.N.; Han, J.H.; Lee, K.H.; Kim, S.T.; Kang, M.H.; Son, S.M.; Lee, Y.M.; Choi, S.Y.; et al. Downregulation of cell-free miR-198 as a diagnostic biomarker for lung adenocarcinoma-associated malignant pleural effusion. Int. J. Cancer 2013, 133, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lv, M.; Shen, S.; Zhou, S.; Wang, P.; Chen, Y.; Liu, B.; Yu, L.; Hou, Y. Cell-free microRNA expression profiles in malignant effusion associated with patient survival in non-small cell lung cancer. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Peng, W.; Wang, J.; Shan, B.; Peng, Z.; Dong, Y.; Shi, W.; He, D.; Cheng, Y.; Zhao, W.; Zhang, C.; et al. Diagnostic and Prognostic Potential of Circulating Long Non-Coding RNAs in Non Small Cell Lung Cancer. Cell. Physiol. Biochem. 2018, 49, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hämmerle, M.; Eißmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Groß, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Zhou, X.L.; Song, Y.J.; Yu, C.H.; Zhu, W.G.; Tong, Y.S. Combination of long noncoding RNA MALAT 1 and carcinoembryonic antigen for the diagnosis of malignant pleural effusion caused by lung cancer. Onco. Targets. Ther. 2018, 11, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Greillier, L.; Roll, P.; Barlesi, F.; Robaglia-Schlupp, A.; Fraticelli, A.; Cau, P.; Astoul, P. Apport des puces à ADN dans le diagnostic étiologique des pleurésies: Étude de faisabilité. Rev. Mal. Respir. 2007, 24, 859–867. [Google Scholar] [CrossRef]
- Michael, C.W.; Davidson, B. Pre-analytical issues in effusion cytology. Pleura Peritoneum 2016, 1, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef]
- Liao, J.; Liu, R.; Shi, Y.J.; Yin, L.H.; Pu, Y.P. Exosome-shuttling microRNA-21 promotes cell migration and invasion-targeting PDCD4 in esophageal cancer. Int. J. Oncol. 2016, 48, 2567–2579. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Schartz, N.E.C.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; Le Chevalier, T.; et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef]
- Wada, J.; Onishi, H.; Suzuki, H.; Yamasaki, A.; Nagai, S.; Morisaki, T.; Katano, M. Surface-bound TGF-beta1 on effusion-derived exosomes participates in maintenance of number and suppressive function of regulatory T-cells in malignant effusions. Anticancer. Res. 2010, 30, 3747–3757. [Google Scholar]
- Alegre, E.; Rebmann, V.; Lemaoult, J.; Rodriguez, C.; Horn, P.A.; Díaz-Lagares, A.; Echeveste, J.I.; González, A. In vivo identification of an HLA-G complex as ubiquitinated protein circulating in exosomes. Eur. J. Immunol. 2013, 43, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- Bard, M.P.; Hegmans, J.P.; Hemmes, A.; Luider, T.M.; Willemsen, R.; Severijnen, L.A.A.; Van Meerbeeck, J.P.; Burgers, S.A.; Hoogsteden, H.C.; Lambrecht, B.N. Proteomic analysis of exosomes isolated from human malignant pleural effusions. Am. J. Respir. Cell Mol. Biol. 2004, 31, 114–121. [Google Scholar] [CrossRef]
- Park, J.O.; Choi, D.Y.; Choi, D.S.; Kim, H.J.; Kang, J.W.; Jung, J.H.; Lee, J.H.; Kim, J.; Freeman, M.R.; Lee, K.Y.; et al. Identification and characterization of proteins isolated from microvesicles derived from human lung cancer pleural effusions. Proteomics 2013, 13, 2125–2134. [Google Scholar] [CrossRef]
- Vaksman, O.; Tropé, C.; Davidson, B.; Reich, R. Exosome-derived miRNAs and ovarian carcinoma progression. Carcinogenesis 2014, 35, 2113–2120. [Google Scholar] [CrossRef]
- Watabe, S.; Kikuchi, Y.; Morita, S.; Komura, D.; Numakura, S.; Kumagai-Togashi, A.; Watanabe, M.; Matsutani, N.; Kawamura, M.; Yasuda, M.; et al. Clinicopathological significance of microRNA-21 in extracellular vesicles of pleural lavage fluid of lung adenocarcinoma and its functions inducing the mesothelial to mesenchymal transition. Cancer Med. 2020, 9, 2879–2890. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wang, Y.; Zou, Y.Q.; Chen, X.; Huang, B.; Liu, J.; Xu, Y.M.; Li, J.; Zhang, J.; Yang, W.M.; et al. Differential miRNA expression in pleural effusions derived from extracellular vesicles of patients with lung cancer, pulmonary tuberculosis, or pneumonia. Tumor Biol. 2016, 37, 15835–15845. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, H.; Mitani, A.; Saito, A.; Ishimori, T.; Saito, M.; Isago, H.; Jo, T.; Yamauchi, Y.; Tanaka, G.; Nagase, T. Exosomal MicroRNA expression profiling in patients with lung adenocarcinoma-associated malignant pleural effusion. Anticancer Res. 2018, 38, 6707–6714. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; De Petris, L.; Zhang, Y.; Brandén, E.; Koyi, H.; Novak, M.; Kanter, L.; Hååg, P.; Hurley, J.; Tadigotla, V.; et al. Exosomal RNA-profiling of pleural effusions identifies adenocarcinoma patients through elevated miR-200 and LCN2 expression. Lung Cancer 2018, 124, 45–52. [Google Scholar] [CrossRef]
- Roman-Canal, B.; Moiola, C.P.; Gatius, S.; Bonnin, S.; Ruiz-Miró, M.; González, E.; Ojanguren, A.; Recuero, J.L.; Gil-Moreno, A.; Falcón-Pérez, J.M.; et al. EV-associated miRNAs from pleural lavage as potential diagnostic biomarkers in lung cancer. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Song, Z.; Cai, Z.; Yan, J.; Shao, Y.W.; Zhang, Y. Liquid biopsies using pleural effusion-derived exosomal DNA in advanced lung adenocarcinoma. Transl. Lung Cancer Res. 2019, 8, 392–400. [Google Scholar] [CrossRef]
- Cully, M. Exosome-based candidates move into the clinic. Nat. Rev. Drug Discov. 2021, 20, 6–7. [Google Scholar] [CrossRef]
- Carneiro, F.P.; Muniz-Junqueira, M.I.; Pittella-Silva, F.; Vasconcelos Carneiro, M.D.; Takano, G.H.S.; De Sousa Vianna, L.M.; De Andrade, L.B.; De Castro, T.M.M.L.; Peres, I.; Dos Santos Borges, T.K.; et al. A panel of markers for identification of malignant and non-malignant cells in culture from effusions. Oncol. Rep. 2017, 38, 3538–3544. [Google Scholar] [CrossRef]
- Light, R.W. Clinical practice. Pleural effusion. N. Engl. J. Med. 2002, 346, 1971–1977. [Google Scholar] [CrossRef]
- Butler, T.P.; Gullino, P.M. Quantitation of Cell Shedding into Efferent Blood of Mammary Adenocarcinoma. Cancer Res. 1975, 35, 512–516. [Google Scholar]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, J.; Cadilha, B.L.; Markota, A.; Voigt, C.; Huang, Z.; Lin, P.P.; Wang, D.D.; Dai, J.; Kranz, G.; et al. Epithelial-type systemic breast carcinoma cells with a restricted mesenchymal transition are a major source of metastasis. Sci. Adv. 2019, 5. [Google Scholar] [CrossRef]
- Racila, E.; Euhus, D.; Weiss, A.J.; Rao, C.; McConnell, J.; Terstappen, L.W.M.M.; Uhr, J.W. Detection and characterization of carcinoma cells in the blood. Proc. Natl. Acad. Sci. USA 1998, 95, 4589–4594. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Stott, S.; Toner, M.; Maheswaran, S.; Haber, D.A. Circulating tumor cells: Approaches to isolation and characterization. J. Cell Biol. 2011, 192, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Fusi, A.; Klopocki, E.; Schmittel, A.; Tinhofer, I.; Nonnenmacher, A.; Keilholz, U. Negative enrichment by immunomagnetic nanobeads for unbiased characterization of circulating tumor cells from peripheral blood of cancer patients. J. Transl. Med. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Rath, B.; Klameth, L.; Hochmair, M. Receptor tyrosine kinase expression of circulating tumor cells in small cell lung cancer. Oncoscience 2015, 2, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Klameth, L.; Rath, B.; Thalhammer, T. Synergism of cyclin-Dependent kinase inhibitors with camptothecin derivatives in small cell lung cancer cell lines. Molecules 2014, 19, 2077–2088. [Google Scholar] [CrossRef]
- Hamilton, G.; Hochmair, M.; Rath, B.; Klameth, L.; Zeillinger, R. Small cell lung cancer: Circulating tumor cells of extended stage patients express a mesenchymal-epithelial transition phenotype. Cell Adhes. Migr. 2016, 10, 360–367. [Google Scholar] [CrossRef]
- Lustgarten, D.E.S.; Thompson, J.; Yu, G.; Vachani, A.; Vaidya, B.; Rao, C.; Connelly, M.; Udine, M.; Tan, K.S.; Heitjan, D.F.; et al. Use of circulating tumor cell technology (CELLSEARCH) for the diagnosis of malignant pleural effusions. Ann. Am. Thorac. Soc. 2013, 10, 582–589. [Google Scholar] [CrossRef]
- Beije, N.; Kraan, J.; den Bakker, M.A.; Maat, A.P.W.M.; van der Leest, C.; Cornelissen, R.; Van, N.M.; Martens, J.W.M.; Aerts, J.G.J.V.; Sleijfer, S. Improved diagnosis and prognostication of patients with pleural malignant mesothelioma using biomarkers in pleural effusions and peripheral blood samples—A short report. Cell. Oncol. 2017, 40, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Ge, F.; Zhang, H.; Wang, D.D.; Li, L.; Lin, P.P. Enhanced detection and comprehensive in situ phenotypic characterization of circulating and disseminated heteroploid epithelial and glioma tumor cells. Oncotarget 2015, 6, 27049–27064. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Z.; Li, Z.; Kim, J.; Deng, Y.; Li, Y.; Heath, J.R.; Wei, W.; Lu, S.; Shi, Q. High-throughput screening of rare metabolically active tumor cells in pleural effusion and peripheral blood of lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 2544–2549. [Google Scholar] [CrossRef]
- Situ, B.; Ye, X.; Zhao, Q.; Mai, L.; Huang, Y.; Wang, S.; Chen, J.; Li, B.; He, B.; Zhang, Y.; et al. Identification and Single-Cell Analysis of Viable Circulating Tumor Cells by a Mitochondrion-Specific AIE Bioprobe. Adv. Sci. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.P.; Gires, O.; Wang, D.D.; Li, L.; Wang, H. Comprehensive in situ co-detection of aneuploid circulating endothelial and tumor cells. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Peeters, D.J.; Fehm, T.; Nolé, F.; Gisbert-Criado, R.; Mavroudis, D.; Grisanti, S.; Generali, D.; Garcia-Saenz, J.A.; Stebbing, J.; et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol. 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Krebs, M.G.; Sloane, R.; Priest, L.; Lancashire, L.; Hou, J.M.; Greystoke, A.; Ward, T.H.; Ferraldeschi, R.; Hughes, A.; Clack, G.; et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 1556–1563. [Google Scholar] [CrossRef]
- Cohen, S.J.; Punt, C.J.A.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- Danila, D.C.; Heller, G.; Gignac, G.A.; Gonzalez-Espinoza, R.; Anand, A.; Tanaka, E.; Lilja, H.; Schwartz, L.; Larson, S.; Fleisher, M.; et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin. Cancer Res. 2007, 13, 7053–7058. [Google Scholar] [CrossRef]
- Psallidas, I.; Kalomenidis, I.; Porcel, J.M.; Robinson, B.W.; Stathopoulos, G.T. Malignant pleural effusion: From bench to bedside. Eur. Respir. Rev. 2016, 25, 189–198. [Google Scholar] [CrossRef]
- Lin, P. Aneuploid CTC and CEC. Diagnostics 2018, 8, 26. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Broglio, K.R.; Guarneri, V.; Jackson, S.; Fritsche, H.A.; Islam, R.; Dawood, S.; Reuben, J.M.; Kau, S.W.; Lara, J.M.; et al. Circulating tumor cells in metastatic breast cancer: Biologic staging beyond tumor burden. Clin. Breast Cancer 2007, 7, 34–42. [Google Scholar] [CrossRef]
- Fukuoka, K.; Tanaka, F.; Tsujimura, T.; Hashimoto-Tamaoki, T.; Hasegawa, S.; Nakano, T. [Exploratory study on the detection of markers for diagnosing early-stage malignant mesothelioma]. Nihon Eiseigaku Zasshi. 2011, 66, 553–557. [Google Scholar] [CrossRef][Green Version]
- Klement, G.L.; Yip, T.T.; Cassiola, F.; Kikuchi, L.; Cervi, D.; Podust, V.; Italiano, J.E.; Wheatley, E.; Abou-Slaybi, A.; Bender, E.; et al. Platelets actively sequester angiogenesis regulators. Blood 2009, 113, 2835–2842. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.M.; Tolley, N.D.; Bunting, M.; Schwertz, H.; Jiang, H.; Lindemann, S.; Yost, C.C.; Rubner, F.J.; Albertine, K.H.; Swoboda, K.J.; et al. Escaping the nuclear confines: Signal-dependent pre-mRNA splicing in anucleate platelets. Cell 2005, 122, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Wurdinger, T.; In’t Veld, S.G.J.G.; Best, M.G. Platelet RNA as pan-tumor biomarker for cancer detection. Cancer Res. 2020, 80, 1371–1373. [Google Scholar] [CrossRef] [PubMed]
- Calverley, D.C.; Phang, T.L.; Choudhury, Q.G.; Gao, B.; Oton, A.B.; Weyant, M.J.; Geraci, M.W. Significant downregulation of platelet gene expression in metastatic lung cancer. Clin. Transl. Sci. 2010, 3, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Best, M.G.; Sol, N.; Kooi, I.; Tannous, J.; Westerman, B.A.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Koster, J.; et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell 2015, 28, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef]
- Adams, D.L.; Martin, S.S.; Alpaugh, R.K.; Charpentier, M.; Tsai, S.; Bergan, R.C.; Ogden, I.M.; Catalona, W.; Chumsri, S.; Tang, C.M.; et al. Circulating giant macrophages as a potential biomarker of solid tumors. Proc. Natl. Acad. Sci. USA 2014, 111, 3514–3519. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Wu, Y.; Yang, Y.; Wang, J.; Niu, M.; Gao, S.; Qin, T.; Bao, D. Long noncoding RNA LINC00662 promotes M2 macrophage polarization and hepatocellular carcinoma progression via activating Wnt/β-catenin signaling. Mol. Oncol. 2020, 14, 462–483. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Ref | Year | Genomic Approach | Patients and Samples | Mutations Detected | Detection Rate | Concordance | Relevance |
|---|---|---|---|---|---|---|---|
| [29] | 2005 | PCR and direct DNA sequencing | 1 NSCLC patient: 1 PF | EGFR: exon 19 delE746-A750 | 100% | No EGFR status tested on tissue | Responsiveness to gefitinib therapy |
| [30] | 2014 | HRM and direct DNA sequencing | 36 NSCLC patients: 36 PFs and cell blocks, some of them paired with 22 tumor tissues | EGFR: exon 19 del; exon 21 L858R and exon 20 Ins773 | 50% in PFs 36.1% in cell blocks 59.1% in tissue | 90.8% PF-tissue 72.2% PF-cell block | PF cell-free supernatant is a better source for mutation detection than cell blocks |
| [31] | 2015 | Multiplex RTqPCR and DNA sequencing | 722 lung adenocarcinoma patients: 722 PF cell blocks | RET: KIF5B-RET and CCDC6-RET; EGFR: exon 19 del, exon 20, exon 21 L858R; KRAS: codons 12, 13, and 61; ALK: EML4-ALK | 2.4% RET 62.5% EGFR 1.8% KRAS 7.1% ALK | Not evaluated | Metastatic RET-rearranged lung adenocarcinoma patients have better prognosis than metastatic EGFR-mutant tumors |
| [32] | 2011 | Two-round PCR and DNA sequencing | 37 lung adenocarcinoma patients: 37 paired primary and metastatic tumors; 21 paired primary tumor and pleural metastases (PF cell blocks or pleural biopsy) | EGFR: exon 19 del; exon 21 L858R KRAS: codon 12–13 | EGFR: 48.6% in primary tumor; 43.2% in metastatic tumor and 38.1% in pleural metastasis KRAS: 2.7% in primary tumor and 5.4% in metastases | EGFR: 83.8% primary tumor-metastatic tumor; 85.7% primary tumor-pleural metastases KRAS: 50% primary tumor-metastatic tumor | Responsiveness to EGFR TKI is more likely to be correlated with EGFR status in metastatic lesions than in primary tumors |
| [33] | 2008 | PCR and PCR-RFLP followed by direct DNA sequencing | 26 NSCLC patients: 26 PFs paired with PF cell blocks | EGFR: exon 21 L858R; exon 19 delE746-A750 and E747–749delA750P; exon 20 S768I | PCR: 23.1% in both cell blocks and PFs PCR-RFLP: 50% in both cell blocks and PFs | PCR: 71.4% PF-cell blocks PCR-RFLP: 100% PF-cell blocks | PF cell-free supernatants and cell blocks are feasible clinical specimens for EGFR mutation detection |
| [34] | 2006 | PCR and direct DNA sequencing | 43 NSCLC patients: 43 PFs | EGFR: exon 19 E746_A750del, E746_T751del insA, and L747_T751del; exon 21 L858R | 25.6% in PFs | Not evaluated | EGFR status in PF is a predictor of response to gefitinib |
| [35] | 2017 | Microarray hybridation and NGS | 11 cancer patients with different primary tumors: lung, breast, pancreas, colon, rectal, and renal. Samples included ascites, PFs and plasma | Mutations on 9 gene-panel: BRAF, EGFR, IDH1, IDH2, KRAS, NRAS, PIK3CA, PTEN, and TP53 | 63% of patients had proper material to be analyzed 1 patient with CNV’s mutation gain in PF | Not evaluated | cfDNA from ascites and PFs provide additional information not detected in tumor and/or plasma |
| [36] | 2018 | PNA clamping | 50 lung adenocarcinoma patients: PF and EV PF | EGFR: exon 18 G719S, exon 19 del, exon 21 L858R | 59.4% in tissue 68.8% in EV PF 59.4% in PFs | 88% tissue-PF 91% tissue-EV PF 91% PF-EV PF | Liquid biopsy using EV-derived DNA is promising for EGFR genotyping, including the detection of the T790M resistance mutation |
| [37] | 2018 | PNA clamping | 40 lung adenocarcinoma patients: 40 PF cell blocks paired with 23 primary tissues | EGFR: exon 19 del, exon 21 L858R, exon 21 L861R | 72.5% in PFs 82.6% in tissue | 73.9% tissue-PF | EGFR mutation status in PF cell block is highly predictive of EGFR TKI efficacy |
| [38] | 2013 | PNA clamping and direct DNA sequencing | 37 NSCLC patients: tumor tissue, PF, PF cell blocks, and serum | EGFR: exon 18 G719X, exon 19 del, exon 20 S768I and L788L, exon 21 L858R, L861Q, and R832H | PNA: 35.7% in tissue, 33.3% in PF cell blocks, 27% in PFs, and 2.8% in serum Seq: 27.8% in tissue, 38.1% in PF cell blocks, 27% in PFs, and 2.8% in serum | 86% PNA-Seq in tissue 95% PNA-Seq in PF cell blocks 89% PNA-Seq in PFs 94% PNA-Seq in serum | PF has good diagnostic performance and PNA clamping method offers sensitive and accurate detection of EGFR allowing a better prediction of response to EGFR TKI |
| [39] | 2012 | PNA clamping and direct DNA sequencing | NSCLC patients: 41 PF cell blocks and 23 lung biopsies and/or resected tissues | EGFR: exon 19 del, exon 21 L858R, and L861Q | PNA: 39% in PF cell blocks and 69.6% in tissues Seq: 14.6% in PF cell blocks and 52.2 in tissues | PNA: 82.6% biopsy-resected tissues Seq: 100% biopsy-resected tissues | Both PNA clamping and DNA sequencing methods are complementary for the analysis of EGFR status in lung adenocarcinoma patients |
| [40] | 2017 | RTqPCR, PNA clamping, and DNA sequencing | 77 NSCLC patients: 77 PFs and cell blocks | EGFR: exon 18 G719X, exon 19 del, exon 20 T790M E20 insertion, exon 21 L858R | RTqPCR: 45.5% in PFs and/or PF cell blocks | 98.7% RTqPCR-PNA DNA seq | RTqPCR on follow-up PF samples is a promising approach when tissue is difficult to obtain |
| [41] | 2010 | RTqPCR | 88 NSCLC patients: 32 PFs and 56 plasma specimens | EGFR: exon 19 E746_A750del and L747–S752del, exon 21 L858R | 23.2% in plasma 28.1% in PF | Not evaluated | EGFR mutation detection by RTqPCR highly predicted the efficacy of gefitinib in advanced NSCLC |
| [42] | 2008 | RTqPCR and direct DNA sequencing | 136 lung adenocarcinoma patients: 136 cytologically-positive PF cell blocks; 91 lung adenocarcinoma resected patients: 91 tissue biopsies | EGFR: exon 21 L858R, L861Q and K861I; exon 19 del; exon 20 767–769 dupASV, 771_H773insYNP and H773Y | 50.5% in tumor tissue 68.4% in PF cell blocks | Not evaluated | Patients with MPE had a higher EGFR mutation rate than the surgically resected specimens suggesting EGFR TKI treatment for those patients having MPE |
| [43] | 2014 | Pyrosequencing and RTqPCR | 84 lung cancer patients: 102 PF cell blocks | EGFR, KRAS, BRAF, PIK3CA, NRAS, MEK1, AKT, PTEN, and ERBB2 | Global mutation rate: 42%. Per gene: EGFR: 29%, ALK: 5%, KRAS: 4% | 88% PF and FFPE tissue | Multiplexed molecular testing is suitable to monitor molecular profiles in PF |
| [44] | 2017 | FISH, Sanger seq, pyrosequencing, and NGS | 50 NSCLC patients: 27 PF cell blocks suitable for genetic analysis | EGFR: exon 21 L858R; exon 19 E746_A750del and L747_A750del, S768I and V769; exon 20 T790M; KRAS: codon 12 and 13 PI3KCA: E542K ALK: EML4-ALK fusion | Global mutation rate: 59% Per gene: EGFR: 33%, KRAS: 25.9%, PI3KCA: 3.7% and ALK: 3.7% | Not evaluated | Molecular profiling of PF is a viable alternative to testing solid tissue in advanced NSCLC |
| [45] | 2017 | Digital droplet PCR, ARMS, and direct PCR sequencing | 95 NSCLC patients: PF | EGFR: exon 19 A750-K757del and S752-L760del, exon 21 L858R | ddPCR: 75.4% ARMS: 61.3% Sanger seq: 43.8% | Not evaluated | ddPCR is feasible to detect EGFR mutations in PF |
| [46] | 2018 | Digital droplet PCR | 90 cancer patients (74 lung cancer): 80 bronchial lavages, 9 PFs, and 1 cerebrospinal fluid | EGFR: exon 19 E746-A750del, exon 20 T790M and exon 21 L858R | 13.3% in supernatants 16.7% in cell blocks | 96.7% supernatant-cell block | Cell-free supernatants are suitable for genetic analysis using ddPCR |
| [47] | 2013 | NGS | 830 lung adenocarcinoma patients: 33 bronchial lavages and 15 PF cell blocks | EGFR: exon 19del, exon 21 | 81% by NGS 16% by Sanger seq | 53.3% PF cell block-tissue 78.8% bronchial lavage cell block-tissue | NGS is a sensitive method for the detection of mutations in liquid specimens |
| [48] | 2018 | NGS, RTqPCR, and Sanger seq | 18 lung adenocarcinoma patients: 8 PF cell blocks and 10 tissues | EGFR: exon 19 E746-A750del, E746-T751del, S752-I759del, and T790M, exon 21 L858R; KRAS: G13C; PIK3CA: E545K | 46.7% by NGS 40.0% by RTqPCR 33.3% by Sanger seq | 75% PF cell block-tissue | High quality of DNA from FFPE PF cell blocks assures successful NGS |
| [49] | 2019 | NGS | 20 lung adenocarcinoma patients: 15 PFs, 2 pericardial fluids, 2 cerebrospinal fluids, 1 ascites, and 20 plasmas | EGFR: exon 19del, exon 21 L858R | Cell-free fluid: 100% PF, pericardial, ascites, and cerebrospinal; Cell block: 100% in PF, 50% in pericardial fluid and cerebrospinal fluid Plasma: 80% | 100% PF-PF cell block 50% pericardial fluid-pericardial fluid cell block 50% cerebrospinal fluid-cerebrospinal fluid cell block 86.7% PF-plasma | Cell-free body fluids have a higher detection rate and sensitivity for tumor-specific mutations than body fluid sediments or plasma |
| [50] | 2020 | NGS | 21 cancer patients (7 lung adenocarcinomas): 15 PFs, 5 peritoneal fluids, 1 pericardial fluid, 8 cell blocks, and 3 tissues | Mutations on 130 gene-panel of cancer related genes | 71.4% global mutation rate | 72.3% PF-tissue | cfDNA testing of effusion samples allows robust detection of clinically actionable genetic biomarkers |
| [51] | 2020 | NGS | 108 lung cancer patients: PF cell blocks | Mutations on 17 gene-panel containing lung cancer associated genes | 86% EGFR, 41.7% TP53, 9% BCL2, 21.3% BRAF, 19.4% PIK3CA, 21.3% PTEN, 18.5% FGFR1, 25% MET, 27.8% RET, 5.6% KRAS, and 5.6% ALK | Not evaluated | High capture efficiency and deep sequencing using NGS for molecular profiling of pleural effusions |
| [52] | 2018 | NGS and ARMS-PCR | 30 NSCLC patients: PF cell blocks and tissues | Mutations on 9 gene-panel containing lung cancer associated genes | Global mutation rate: 83.33% in both tissue and PF cell blocks | 86.7% PF cell blocks-tissue | High concordance rate on the detection of EGFR, KRAS, and ALK between tissue and PF cell blocks |
| Genomic Approach | Limit of Detection |
|---|---|
| Sanger sequencing | 20–25% |
| Pyrosequencing | 5–10% |
| High resolution melting | 5% |
| Peptide nucleic acid clamping | 5% |
| Real-time quantitative PCR | 0.5–5% |
| Amplification-refractory mutation system | 1% |
| ddPCR | 0.001–0.01% |
| NGS-Safe seq | 0.1% |
| NGS-CAPP seq | 0.01% |
| Name of the CTC Line | Primary Tumor | Technique for CTCs Isolation/Enrichment/Detection/Characterization | Major Findings/Basic Characterization | Reference |
|---|---|---|---|---|
| Not mentioned | Non-small cell lung cancer | CD45 depletion, and EpCAM and CD45 immunostaining | CD45− EpCAM− CK+ CTCs were identified in PEs with CTCs detected in matched peripheral blood | [108] |
| SCLC26A | Pancreatic cancer | Not mentioned | CTC had no expression of CHI3L1 and lower expression of tyrosine kinase receptors | [109] |
| SCLC26A | Pancreatic cancer | Not mentioned | CTCs were sensitive to camptothecin analogues | [110] |
| SCLC26A | Pancreatic cancer | Not mentioned | High expression of E-cadherin consistent with a mesenchymal-epithelial transition. Low expression of stem cell markers, which hamper the epithelial phenotype | [111] |
| Not mentioned | Benign, malignant epithelial, and non-epithelial cancers | Anti-EpCAM capture and CELLSEARCH® | More studies are needed to implement pleural CELLSEARCH. It could be useful in addition to traditional cytology | [112] |
| Not mentioned | Malignant mesothelioma | Variation of CELLSEARCH® for CTC enumeration based on the detection of MCAM | MCAM+ cells are malignant. Thus, they can help to identify malignancy in PE | [113] |
| Not mentioned | Pancreatic cancer | Anti-EpCAM capture and iFISH | CTCs were non-hematopoietic (CD45−) and polyploid | [114] |
| Not mentioned | Lung cancer | Identification of CTC by using a fluorescent glucose analog (2-NBDG) by high-throughput screening. Confirmation of CTC by single-cell sequencing | Most CTCs share the same oncogenic mutations as the primary tumors where they come from. Detection of emerging secondary mutations responsible for drug resistance before the manifestation of resistance. The reported method can complement traditional cytology for MPE diagnosis | [115] |
| Not mentioned | Lung and liver cancer | Mitochondria-targeting bioprobe with aggregation-induced emission activity. CTC characterization by single-cell sequencing | With this method of detection of CTCs there is less cell disruption; thus downstream single-cell sequencing gets less affected than using traditional cytokeratin markers | [116] |
| Not mentioned | Not mentioned | Antigen-independent subtraction enrichment and immunostaining-FISH (SE-iFISH) | Aneuploid circulating rare cells can be detected using the methodology described | [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorolla, M.A.; Sorolla, A.; Parisi, E.; Salud, A.; Porcel, J.M. Diving into the Pleural Fluid: Liquid Biopsy for Metastatic Malignant Pleural Effusions. Cancers 2021, 13, 2798. https://doi.org/10.3390/cancers13112798
Sorolla MA, Sorolla A, Parisi E, Salud A, Porcel JM. Diving into the Pleural Fluid: Liquid Biopsy for Metastatic Malignant Pleural Effusions. Cancers. 2021; 13(11):2798. https://doi.org/10.3390/cancers13112798
Chicago/Turabian StyleSorolla, Maria Alba, Anabel Sorolla, Eva Parisi, Antonieta Salud, and José M. Porcel. 2021. "Diving into the Pleural Fluid: Liquid Biopsy for Metastatic Malignant Pleural Effusions" Cancers 13, no. 11: 2798. https://doi.org/10.3390/cancers13112798
APA StyleSorolla, M. A., Sorolla, A., Parisi, E., Salud, A., & Porcel, J. M. (2021). Diving into the Pleural Fluid: Liquid Biopsy for Metastatic Malignant Pleural Effusions. Cancers, 13(11), 2798. https://doi.org/10.3390/cancers13112798