
Is Emodin with Anticancer Effects Completely Innocent? Two Sides of the Coin

 ,
,  , ,
, ,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
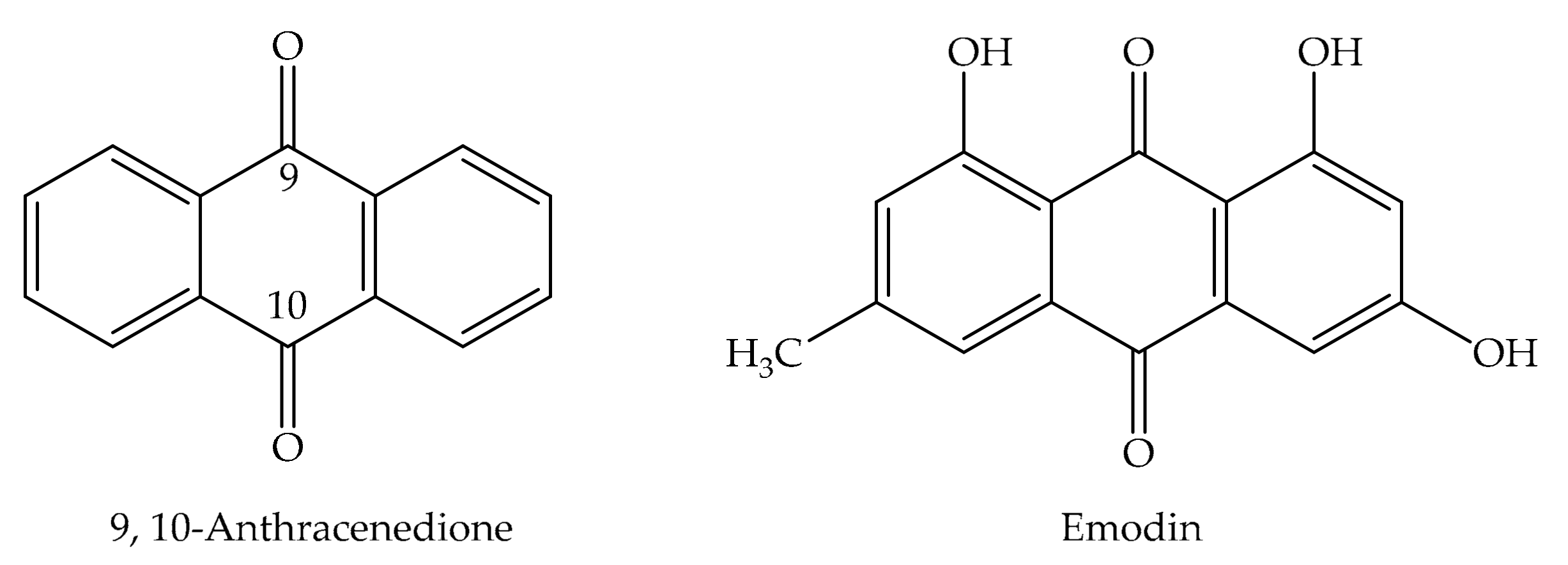
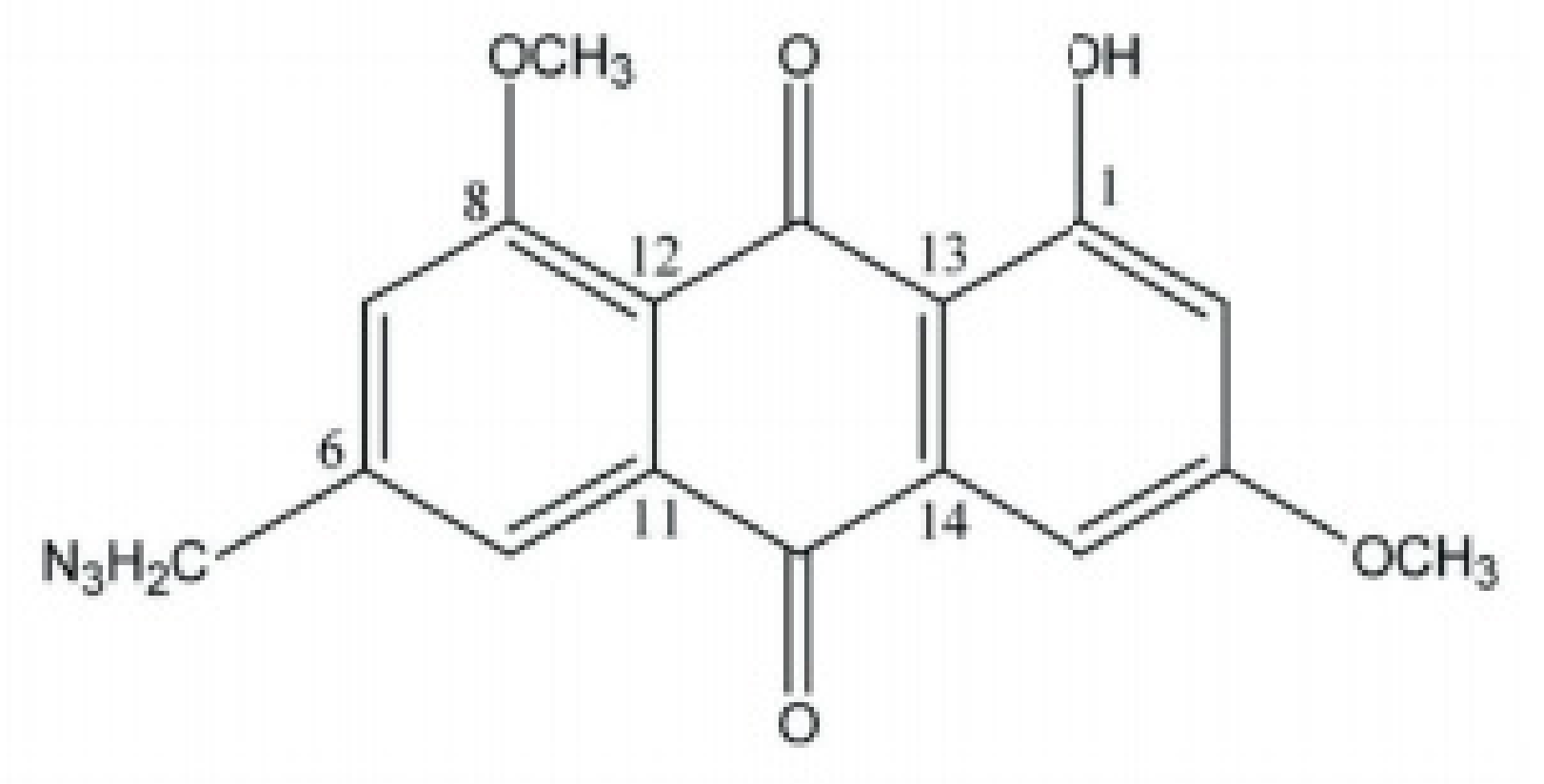
2. Chemical Properties of Emodin
2.1. Biosynthesis of Anthraquinones
2.2. Characterization of Anthraquinones
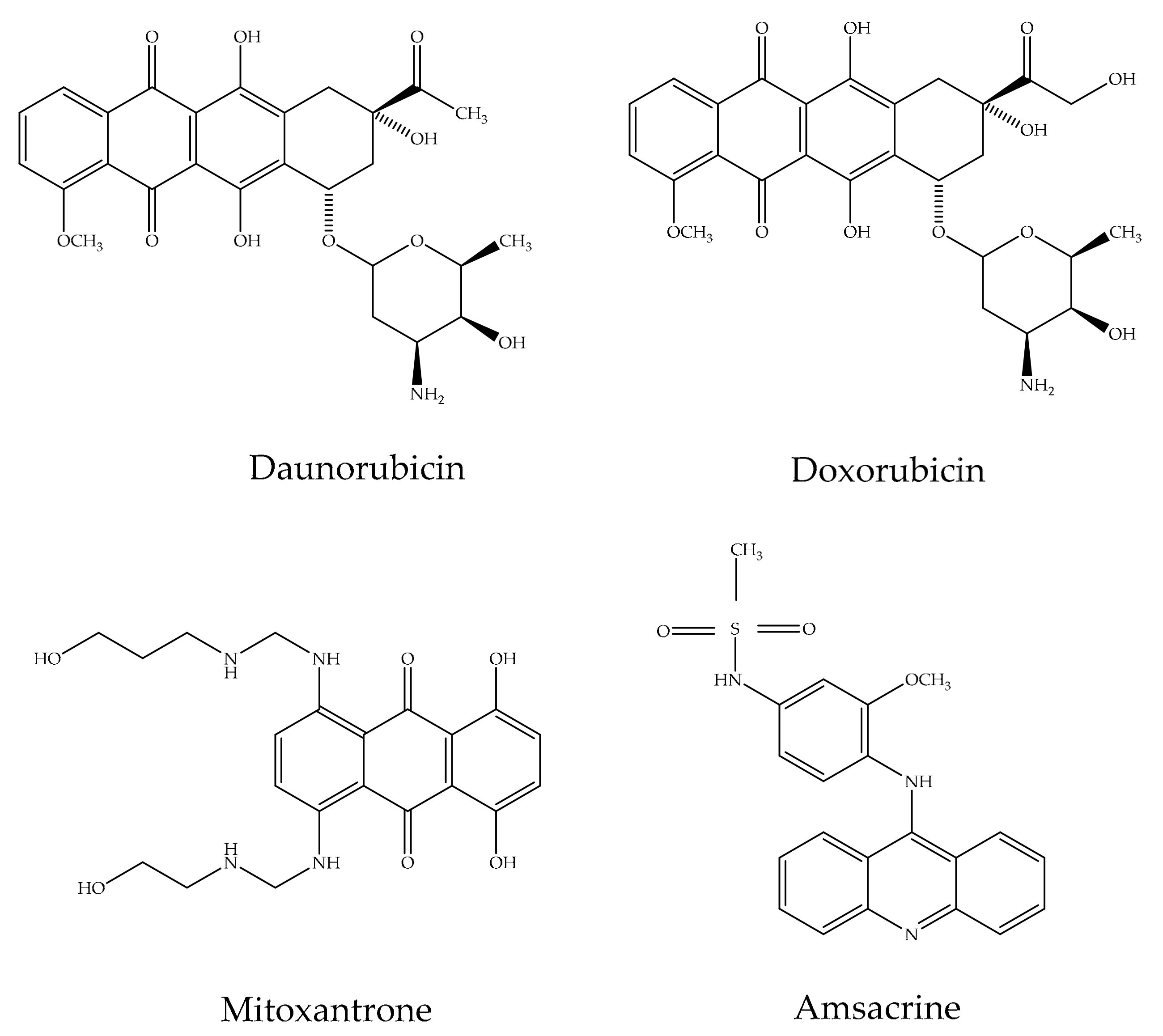
2.3. Pharmacological Applications of Anthraquinones
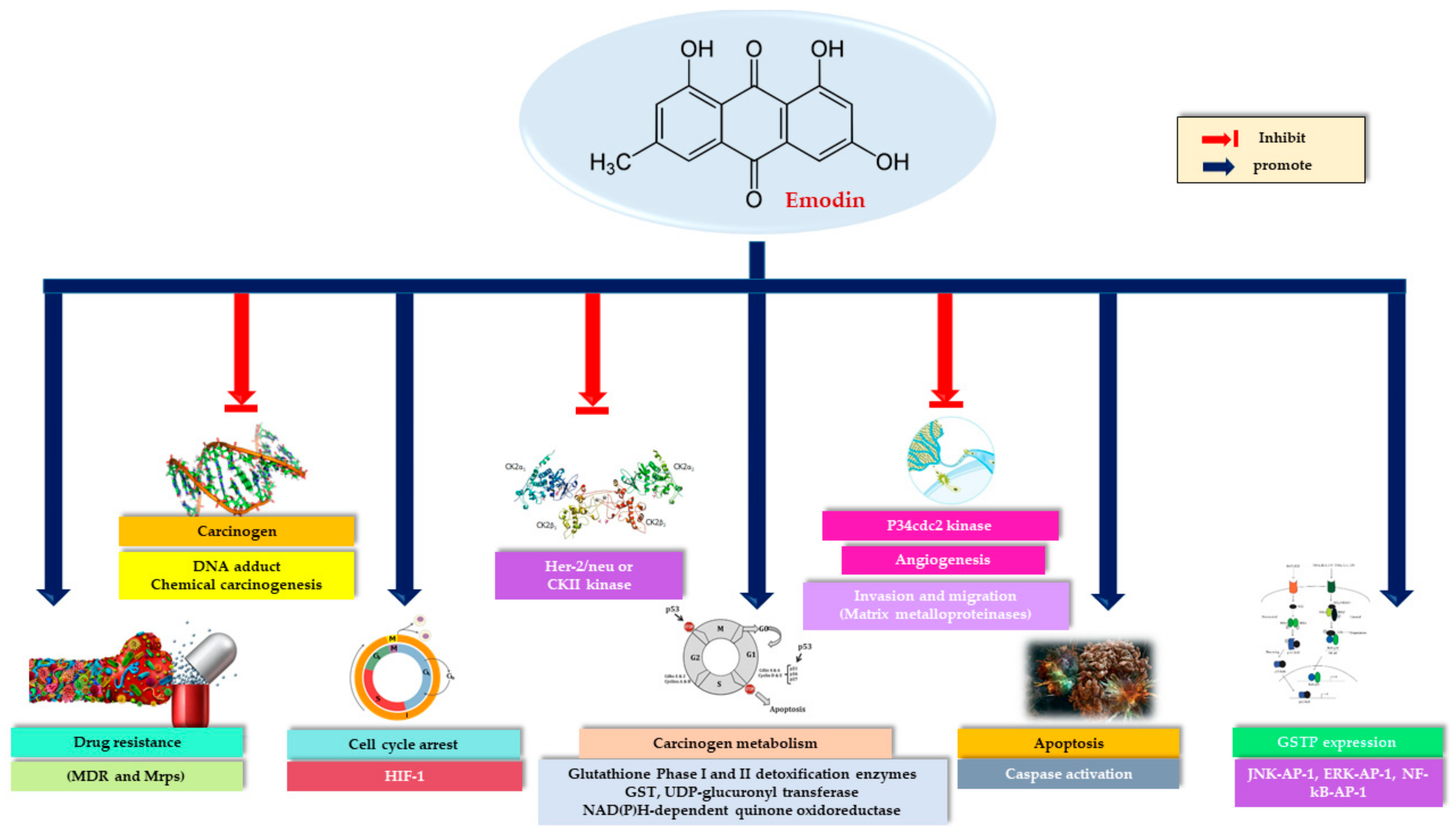
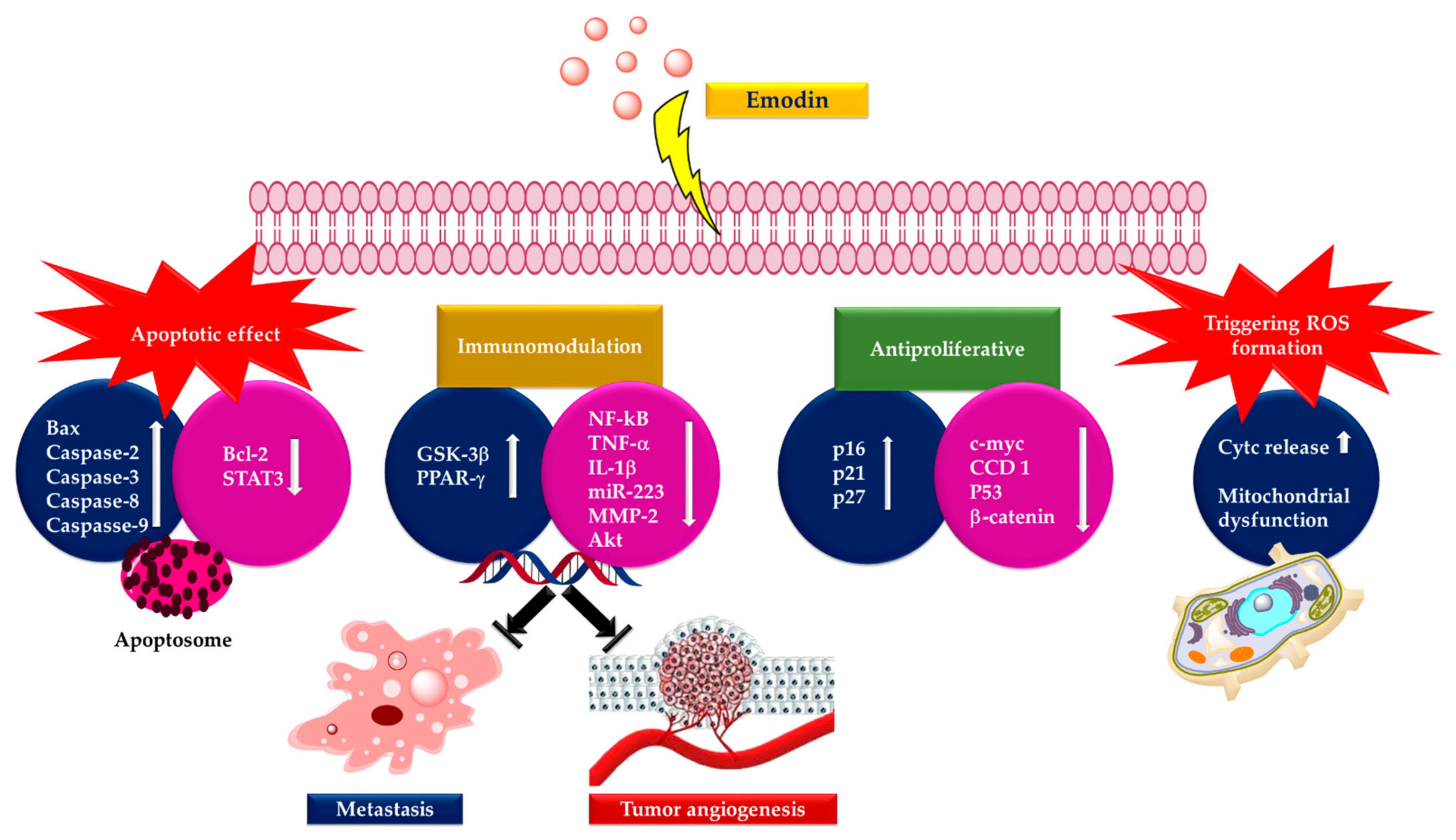
3. Anticancer Activity of Emodin
3.1. Lung Cancer
3.2. Breast Cancer
3.3. Gastric Cancer
3.4. Pancreatic Cancer
3.5. Hepatocellular Carcinoma
3.6. Gallbladder Cancer
3.7. Colon Cancer
3.8. Cervical Cancer
3.9. Ovarian Cancer
3.10. Prostate Cancer
3.11. Blood System Cancer
3.12. Tongue Squamous Cancer
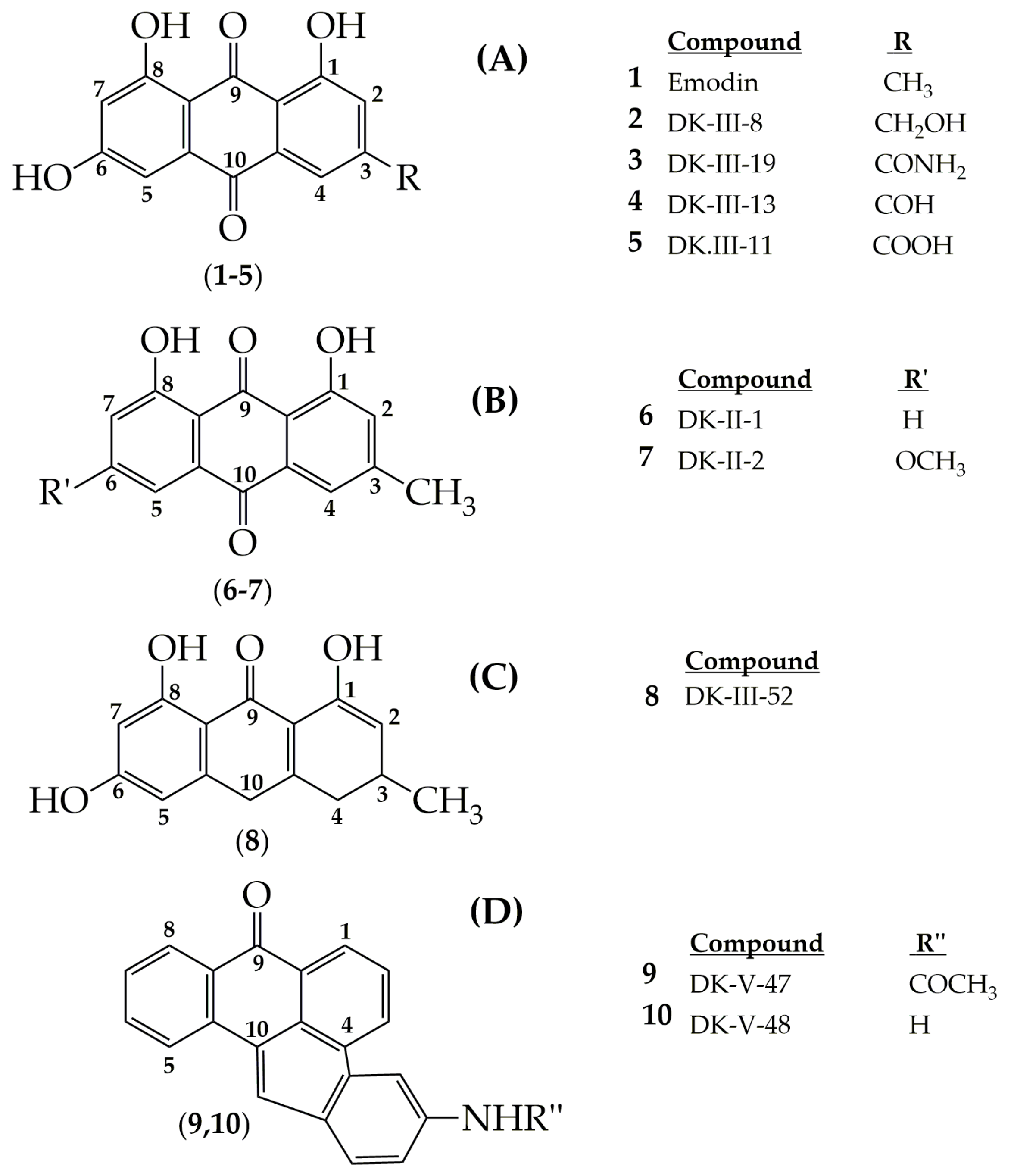
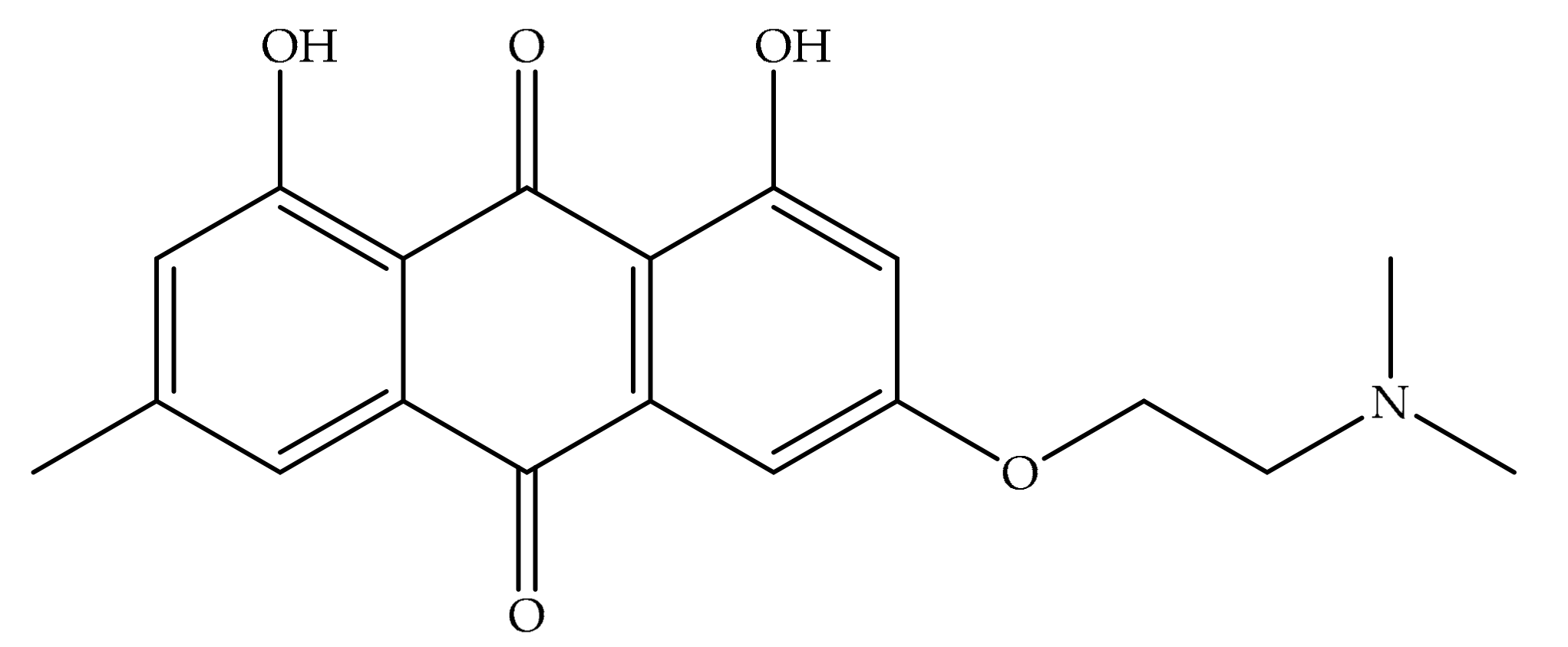
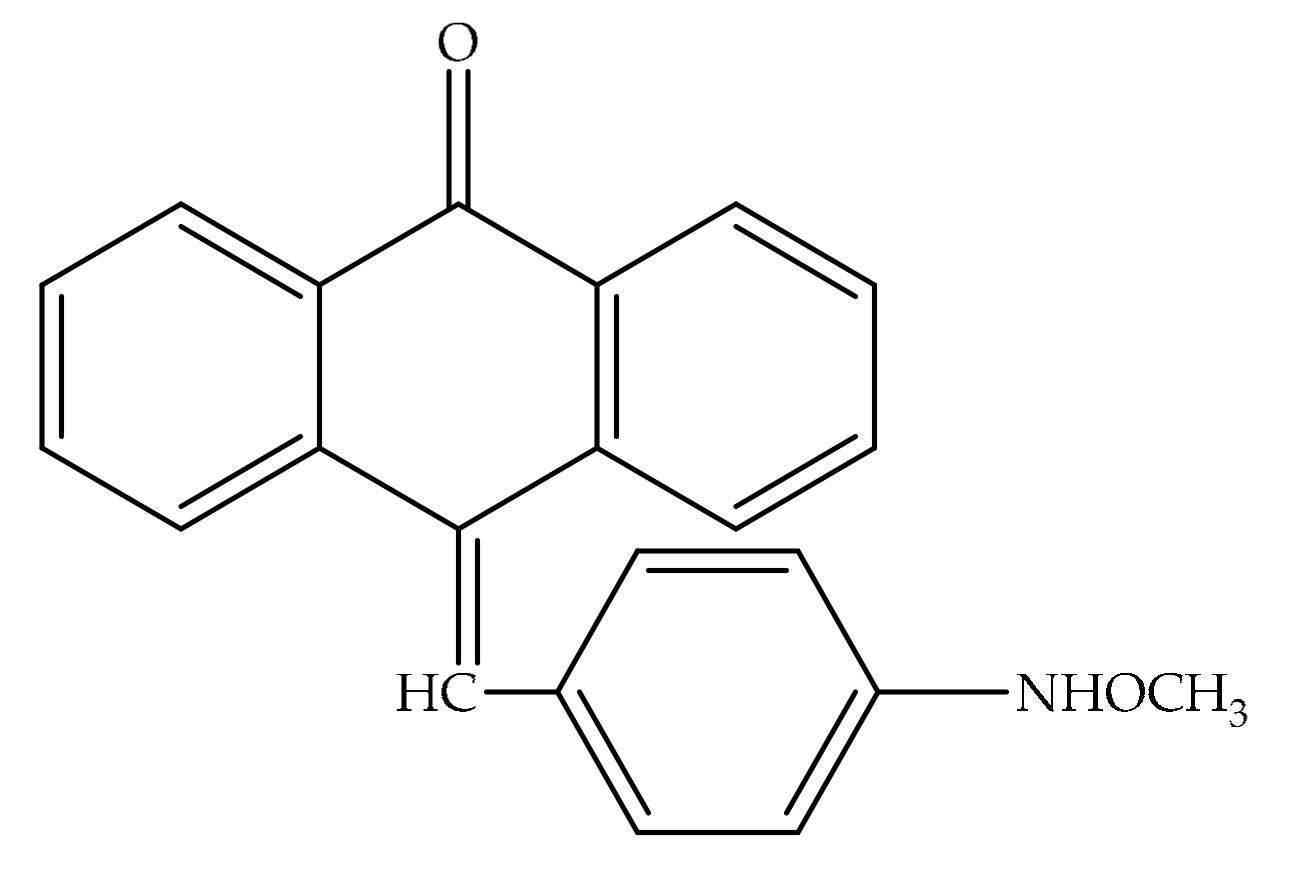
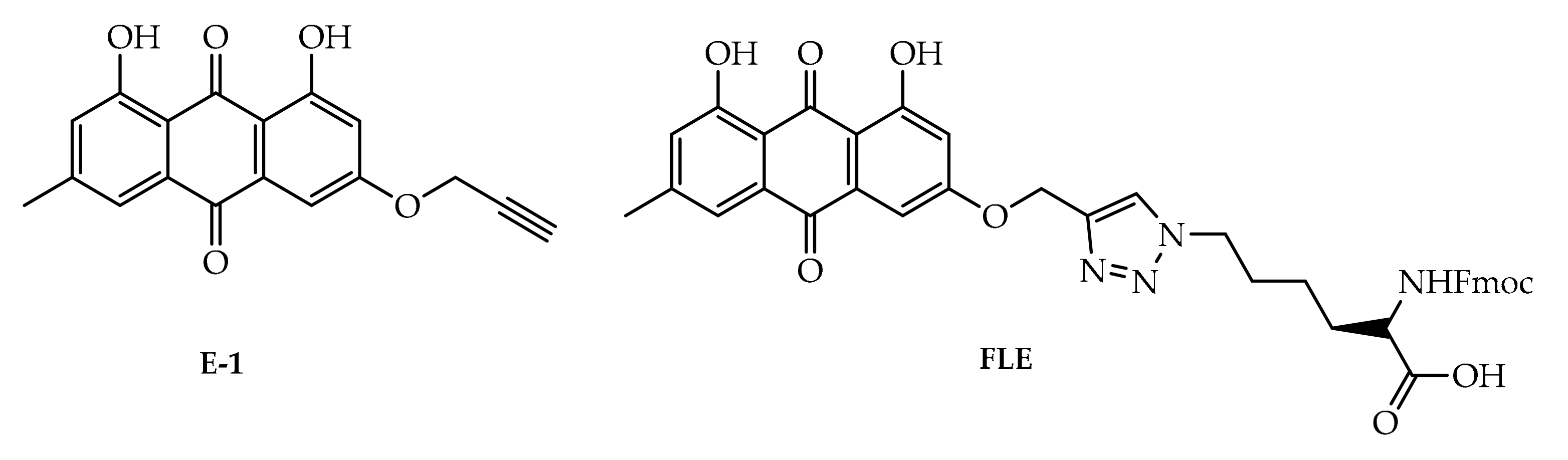
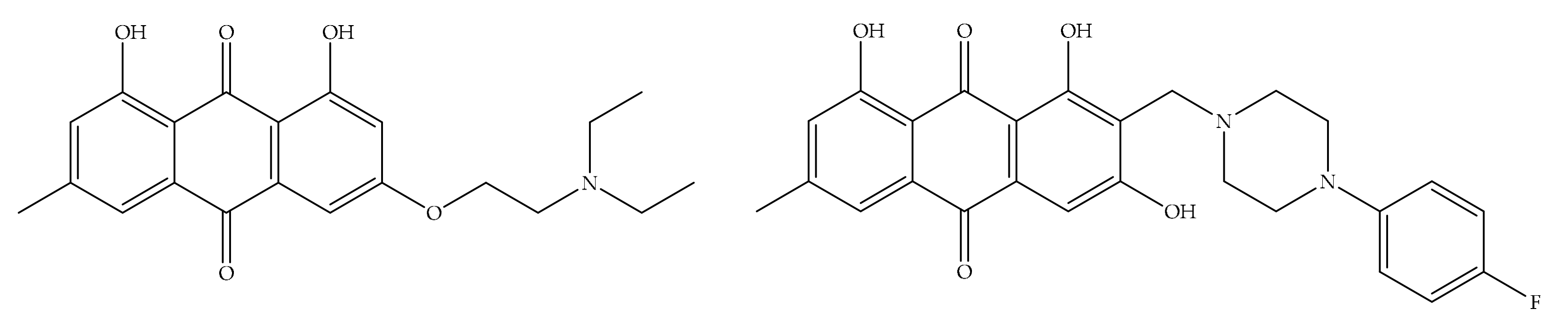
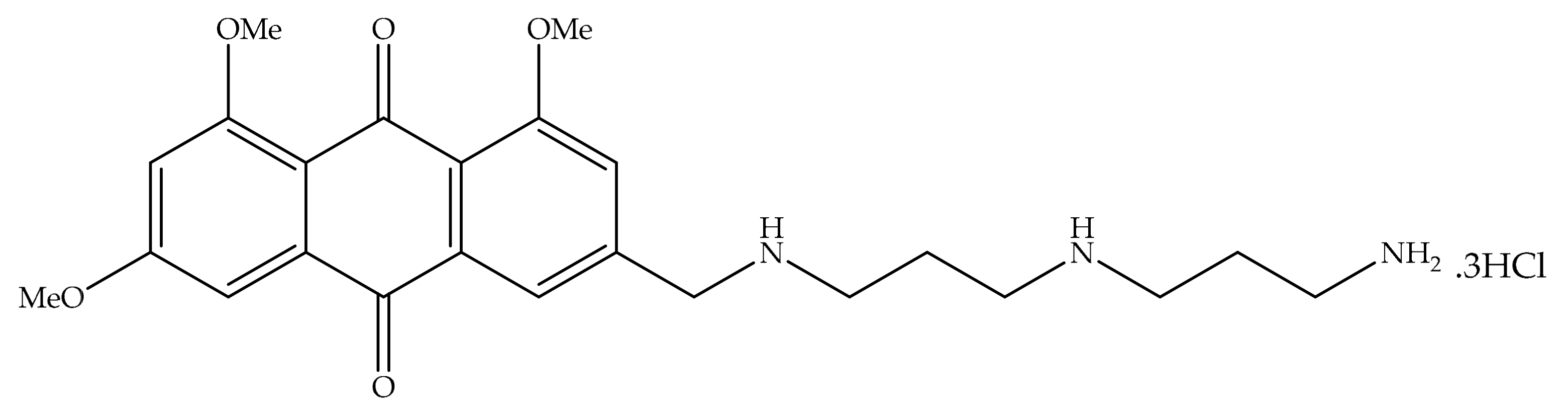
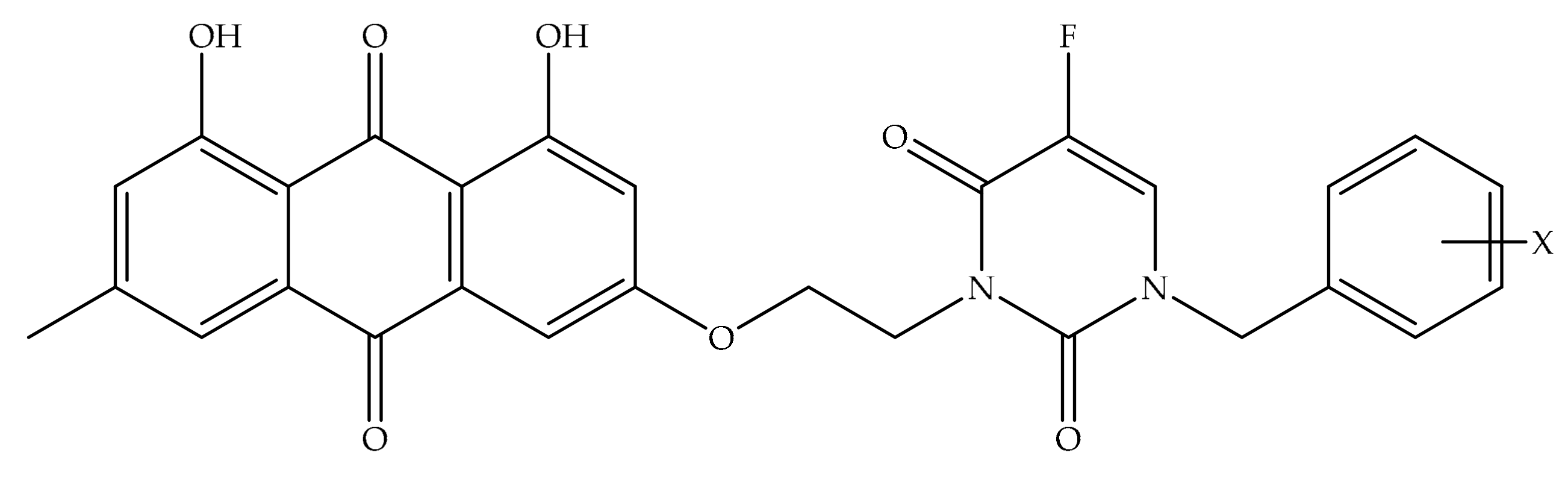
4. Potential of Synthesized Derivatives of Emodin
4.1. Anticancer and Antiproliferative Activities
4.2. Potential Matrix Metalloproteinases (MMPs) Activities
4.3. Bone Affinity Effects
4.4. Anti-Inflammatory Activities
5. Nano-Drug Delivery Strategies for Emodin
6. Toxicity of Emodin
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hsu, S.-C.; Chung, J.-G. Anticancer potential of emodin. Biomedicine 2012, 2, 108–116. [Google Scholar] [CrossRef]
- Sun, Y. Chemosensitization by emodin, a plant-derived anticancer agent: Mechanism of action. Cancer Biol. Ther. 2008, 7, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, G.; Babykutty, S.; Sathiadevan, P.P.; Srinivas, P. Molecular mechanism of emodin action: Transition from laxative ingredient to an antitumor agent. Med. Res. Rev. 2007, 27, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Arcamone, F.; Penco, S. Anthracycline and Anthracenedione-Based Anticancer Agents; Lown, J.W., Ed.; Elsevier: Amsterdam, The Netherlands, 1988. [Google Scholar]
- Chow, K.C.; Macdonald, T.L.; Ross, W.E. DNA binding by epipodophyllotoxins and N-acyl anthracyclines: Implications for mechanism of topoisomerase II inhibition. Mol. Pharmacol. 1988, 34, 467–473. [Google Scholar] [PubMed]
- Sharma, R.; Tiku, A.B. Emodin inhibits splenocyte proliferation and inflammation by modulating cytokine responses in a mouse model system. J. Immunotoxicol. 2016, 13, 20–26. [Google Scholar] [CrossRef]
- Han, J.W.; Shim, D.W.; Shin, W.Y.; Heo, K.H.; Kwak, S.B.; Sim, E.J.; Jeong, J.H.; Kang, T.B.; Lee, K.H. Anti-inflammatory effect of emodin via attenuation of NLRP3 inflammasome activation. Int. J. Mol. Sci. 2015, 16, 8102–8109. [Google Scholar] [CrossRef]
- Cao, F.; Peng, W.; Li, X.; Liu, M.; Li, B.; Qin, R.; Jiang, W.; Cen, Y.; Pan, X.; Yan, Z.; et al. Emodin is identified as the active component of ether extracts from Rhizoma Polygoni Cuspidati, for anti-MRSA activity. Can. J. Physiol. Pharmacol. 2015, 93, 485–493. [Google Scholar] [CrossRef]
- Li, W.Y.; Ng, Y.F.; Zhang, H.; Guo, Z.D.; Guo, D.J.; Kwan, Y.W.; Leung, G.P.H.; Lee, S.M.Y.; Yu, P.H.F.; Chan, S.W. Emodin elicits cytotoxicity in human lung adenocarcinoma A549 cells through inducing apoptosis. Inflammopharmacology 2014, 22, 127–134. [Google Scholar] [CrossRef]
- Yu, J.Q.; Bao, W.; Lei, J.C. Emodin regulates apoptotic pathway in human liver cancer cells. Phytother. Res. 2013, 27, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Cui, C.F.; Yang, L.; Wang, L.; Jiang, X.H. Emodin inhibits colon cancer cell invasion and migration by suppressing epithelial mesenchymal transition via the Wnt/beta-catenin pathway. Oncol. Res. 2019, 27, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhong, M.; Yin, H.; Chen, Y.; Cao, Q.; Wang, C. Emodin induces hepatocellular carcinoma cell apoptosis through MAPK and PI3K/AKT signaling pathways in vitro and in vivo. Oncol. Rep. 2016, 36, 961–967. [Google Scholar] [CrossRef]
- Su, Y.T.; Chang, H.L.; Shyue, S.K.; Hsu, S.L. Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem. Pharmacol. 2005, 70, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, G.; Anto, R.J.; Srinivas, P.; Vidhyalakshmi, S.; Senan, V.P.; Karunagaran, D. Emodin induces apoptosis of human cervical cancer cells through poly (ADP-ribose) polymerase cleavage and activation of caspase-9. Eur. J. Pharmacol. 2003, 473, 117–125. [Google Scholar] [CrossRef]
- Chen, Y.C.; Shen, S.C.; Lee, W.R.; Hsu, F.L.; Lin, H.Y.; Ko, C.H.; Tseng, S.W. Emodin induces apoptosis in human promyeloleukemic HL-60 cells accompanied by activation of caspase 3 cascade but independent of reactive oxygen species production. Biochem. Pharmacol. 2002, 64, 1713–1724. [Google Scholar] [CrossRef]
- Zhang, L.; Chang, C.J.; Bacus, S.S.; Hung, M.C. Suppressed transformation and induced differentiation of HER-2/neu-overexpressing breast cancer cells by emodin. Cancer Res. 1995, 55, 3890–3896. [Google Scholar]
- Xing, J.Y.; Song, G.P.; Deng, J.P.; Jiang, L.Z.; Xiong, P.; Yang, B.J. Antitumor effects and mechanism of novel emodin rhamnoside derivatives against human cancer cells in vitro. PLoS ONE 2015, 10, e0144781. [Google Scholar] [CrossRef]
- Li-Weber, M. Targeting apoptosis pathways in cancer by Chinese medicine. Cancer Lett. 2013, 332, 304–312. [Google Scholar] [CrossRef]
- Muto, A.; Hori, M.; Sasaki, Y.; Saitoh, A.; Yasuda, I.; Maekawa, T.; Uchida, T.; Asakura, K.; Nakazato, T.; Kaneda, T.; et al. Emodin has a cytotoxic activity against human multiple myeloma as a Janus-activated kinase 2 inhibitor. Mol. Cancer Ther. 2007, 6, 987–994. [Google Scholar] [CrossRef]
- Huang, Q.; Shen, H.M.; Shui, G.; Wenk, M.R.; Ong, C.N. Emodin inhibits tumor cell adhesion through disruption of the membrane lipid Raft-associated integrin signaling pathway. Cancer Res. 2006, 66, 5807–5815. [Google Scholar] [CrossRef]
- Kwak, H.-J.; Park, M.-J.; Park, C.-M.; Moon, S.-I.; Yoo, D.-H.; Lee, H.-C.; Lee, S.-H.; Kim, M.-S.; Lee, H.-W.; Shin, W.-S.; et al. Emodin inhibits vascular endothelial growth factor-A-induced angiogenesis by blocking receptor-2 (KDR/Flk-1) phosphorylation. Int. J. Cancer 2006, 118, 2711–2720. [Google Scholar] [CrossRef]
- Cha, T.L.; Qiu, L.; Chen, C.T.; Wen, Y.; Hung, C. Emodin down-regulates androgen receptor and inhibits prostate cancer cell growth. Cancer Res. 2005, 65, 2287–2295. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Park, M.J.; Kim, S.J.; Lee, C.H.; Yoo, H.; Shin, S.H.; Song, E.S.; Lee, S.H. Emodin suppresses hyaluronic acid-induced MMP-9 secretion and invasion of glioma cells. Int. J. Oncol. 2005, 27, 839–846. [Google Scholar]
- Lee, H.Z. Effects and mechanisms of emodin on cell death in human lung squamous cell carcinoma. Br. J. Pharmacol. 2001, 134, 11–20. [Google Scholar] [CrossRef]
- Guo, J.M.; Xiao, B.X.; Liu, Q.; Zhang, S.; Liu, D.H.; Gong, Z.H. Anticancer effect of aloe-emodin on cervical cancer cells involves G2/M arrest and induction of differentiation. Acta Pharmacol. Sin. 2007, 28, 1991–1995. [Google Scholar] [CrossRef]
- Shieh, D.E.; Chen, Y.Y.; Yen, M.H.; Chiang, L.C.; Lin, C.C. Emodin-induced apoptosis through p53-dependent pathway in human hepatoma cells. Life Sci. 2004, 74, 2279–2290. [Google Scholar] [CrossRef]
- Yi, J.; Yang, J.; He, R.; Gao, F.; Sang, H.; Tang, X.; Ye, R.D. Emodin enhances arsenic trioxide-induced apoptosis via generation of reactive oxygen species and inhibition of survival signaling. Cancer Res. 2004, 64, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Yang, J.; Cang, H.; Zou, Y.Q.; Yi, J. Gene expression alteration during redox-dependent enhancement of arsenic cytotoxicity by emodin inHeLa cells. Cell Res. 2005, 15, 511–1522. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chang, Y.C.; Lai, T.Y.; Yu, C.S.; Chen, H.Y.; Yang, J.S.; Chueh, F.S.; Lu, C.C.; Chiang, J.H.; Huang, W.W.; Ma, C.Y.; et al. Emodin induces apoptotic death in murine myelomoocytic leukemia WEHI-3 cells in vitro and enhances phagocytosis in leukemia mice in vivo. Evid. Based Complement. Altern. Med. 2011, 2011, 523596. [Google Scholar] [CrossRef]
- Wu, Y.; Tu, X.; Lin, G.; Xia, H.; Huang, H.; Wan, J.; Cheng, Z.; Liu, M.; Chen, G.; Zhang, H.; et al. Emodin mediated protection from acute myocardial infarction via inhibition of inflammation and apoptosis in local ischemic myocardium. Life Sci. 2007, 81, 1332–1338. [Google Scholar] [CrossRef]
- Yang, L.; Tan, J.; Wang, B.C.; Zhu, L.C. Synthesis, characterization, and anti-cancer activity of emodin-Mn (II) metal complex. Chin. J. Nat. Med. 2014, 12, 937–942. [Google Scholar] [CrossRef]
- Lin, S.-Z.; Wei, W.-T.; Chen, H.; Chen, K.-J.; Tong, H.-F.; Wang, Z.-H.; Ni, Z.-L.; Liu, H.-C.; Liu, D.-L. Antitumor activity of emodin against pancreatic cancer depends on its dual role: Promotion of apoptosis and suppression of angiogenesis. PLoS ONE 2012, 7, e42146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, H.; Liu, D.L.; Li, H.; Luo, J.; Zhang, J.H. Emodin sensitizes the gemcitabine-resistant cell line Bxpc-3/Gem to gemcitabine via downregulation of NF-kappaB and its regulated targets. Int. J. Oncol. 2013, 42, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, P.; Mao, H.; Wanga, A.; Zhang, X. Emodin sensitizes paclitaxel-resistant human ovarian cancer cells to paclitaxel-induced apoptosis in vitro. Oncol. Rep. 2009, 21, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lau, Y.K.; Xia, W.; Hortobagyi, G.N.; Hung, M.C. Tyrosine kinase inhibitor emodin suppresses growth of HER-2/neu-overexpressing breast cancer cells in athymic mice and sensitizes these cells to the inhibitory effect of paclitaxel. Clin. Cancer Res. 1999, 5, 343–353. [Google Scholar]
- Zhang, L.; Hung, M.C. Sensitization of HER-2/neu-overexpressing non-small cell lung cancer cells to chemotherapeutic drugs by tyrosine kinase inhibitor emodin. Oncogene 1996, 12, 571–576. [Google Scholar]
- Duval, J.; Pecher, V.; Poujol, M.; Lesellier, E. Research advances for the extraction, analysis and uses of anthraquinones: A review. Ind. Crop. Prod. 2016, 94, 812–833. [Google Scholar] [CrossRef]
- Seigler, D.S. Plant Secondary Metabolism; Springer Science and Business Media: New York, NY, USA, 1998. [Google Scholar]
- Diaz-Munoz, G.; Miranda, I.L.; Sartori, S.K.; de Rezende, D.C.; Diaz, M.A.N. Chapter 11-anthraquinones: An overview. Stud. Nat. Prod. Chem. 2018, 58, 313–338. [Google Scholar]
- Reddy, N.R.R.; Mehta, R.H.; Soni, P.H.; Makasana, J.; Gajbhiye, N.A.; Ponnuchamy, M.; Kumar, J. Next generation sequencing and transcriptome analysis predicts biosynthetic pathway of sennosides from Senna (Cassia angustifolia Vahl.), a non-model plant with potent laxative properties. PLoS ONE 2015, 10, e0129422. [Google Scholar] [CrossRef]
- Han, Y.S.; Van der Heijden, R.; Verpoorte, R. Biosynthesis of anthraquinones in cell cultures of the Rubiaceae. Plant Cell Tiss. Org. Cult. 2001, 67, 201–220. [Google Scholar] [CrossRef]
- Teuscher, E.; Lindequist, U. Biogene Gifte; Gustav Fischer Verlag: Stuttgart, Germany, 1994. [Google Scholar]
- Derksen, G.C.H.; Naayer, M.; van Beek, T.A.; Capelle, A.; Haaksman, I.K.; van Doren, H.A.; de Groot, A. Chemical and enzymatic hydrolysis of anthraquinone glycosides from madder roots. Phytochem. Anal. 2003, 14, 137–144. [Google Scholar] [CrossRef]
- Staunton, J.; Weissman, K.J. Polyketide biosynthesis: A millennium review. Nat. Prod. Rep. 2001, 18, 380–416. [Google Scholar] [CrossRef]
- Rawlings, B.J. Biosynthesis of polyketides (other than actinomycete macrolides). Nat. Prod. Rep. 1999, 16, 425–484. [Google Scholar] [CrossRef] [PubMed]
- Hopwood, D.A.; Sherman, D.H. Molecular genetics of polyketides and its comparison to fatty acid biosynthesis. Annu. Rev. Genet. 1990, 24, 37–66. [Google Scholar] [CrossRef] [PubMed]
- Lemli, J.A.J.M. Chemical assay of anthraquinone drugs. Pharmacology 1976, 14, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Andrade, F.I.; Purgato, G.A.; Maia, T.F.; Siqueira, R.P.; Lima, S.; Diaz, G.; Nogueira Diaz, M.A. Chemical constituents and an alternative medicinal veterinary herbal soap made from Senna macranthera. Evid. Based Complement. Alternat. Med. 2015, 2015, 217598. [Google Scholar]
- Malik, E.M.; Muller, C.E. Anthraquinones as pharmacological tools and drugs. Med. Res. Rev. 2016, 36, 705–748. [Google Scholar] [CrossRef]
- Chien, S.C.; Wu, Y.C.; Chen, Z.W.; Yang, W.C. Naturally occurring anthraquinones: Chemistry and therapeutic potential in autoimmune diabetes. Evid. Based Complement. Alternat. Med. 2015, 2015, 357357. [Google Scholar] [CrossRef]
- Izhaki, I. Emodin—A secondary metabolite with multiple ecological functions in higher plants. New Phytol. 2002, 155, 205–217. [Google Scholar] [CrossRef]
- Strotmann, H.; Brendel, K.; Boos, K.S.; Schlimme, E. Energy transfer inhibition in photosynthesis by anthraquinone dyes. FEBS Lett. 1982, 145, 11–15. [Google Scholar] [CrossRef]
- Hande, K.R. Topoisomerase II inhibitors. Update Cancer Ther. 2008, 3, 13–26. [Google Scholar] [CrossRef]
- Schrader, K.K.; Dayan, F.E.; Allen, S.N.; de Regt, M.Q.; Tucker, C.S.; Paul, J. 9,10-Anthraquinone reduces the photosynthetic efficiency of oscillatoria perornata and modifies cellular inclusions. Int. J. Plant Sci. 2000, 161, 265–270. [Google Scholar] [CrossRef]
- Huang, Q.; Lu, G.; Shen, H.M.; Chung, M.C.M.; Choon, N.O. Anti-cancer properties of anthraquinones from rhubarb. Med. Res. Rev. 2007, 27, 609–630. [Google Scholar] [CrossRef]
- Sharma, R.; Tiku, A.B.; Giri, A. Pharmacological properties of emodin anthraquinone derivatives. J. Nat. Prod. Resour. 2017, 3, 97–101. [Google Scholar]
- Lee, H.Z. Protein kinase C involvement in aloe-emodin- and emodin-induced apoptosis in lung carcinoma cell. Br. J. Pharmacol. 2001, 134, 1093–1103. [Google Scholar] [CrossRef]
- Goulart, M.O.; Falkowski, P.; Ossowski, T.; Liwo, A. Electrochemical study of oxygen interaction with lapachol and its radical anions. Bioelectrochemistry 2003, 59, 85–87. [Google Scholar] [CrossRef]
- Watanabe, N.; Forman, H.J. Autoxidation of extracellular hydroquinones is a causative event for the cytotoxicity of menadione and DMNQ in A549-S cells. Arch. Biochem. Biophys. 2003, 411, 145–157. [Google Scholar] [CrossRef]
- Sugawara, M.; Sugawara, Y.; Wen, K.; Giulivi, C. Generation of oxygen free radicals in thyroid cells and inhibition of thyroid peroxidase. Exp. Biol. Med. 2002, 227, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.Y.; Han, J.X.; Huang, H.Y. Effects of emodin on gene expression profile in small cell lung cancer NCI-H446 cells. Chin. Med. J. 2007, 120, 1710–1715. [Google Scholar] [CrossRef]
- Chen, R.S.; Jhan, J.Y.; Su, Y.J.; Lee, W.T.; Cheng, C.M.; Ciou, S.C.; Lin, S.T.; Chuang, S.M.; Ko, J.C.; Lin, Y.W. Emodin enhances gefitinib-induced cytotoxicity via Rad51 downregulation and ERK1/2 inactivation. Exp. Cell Res. 2009, 315, 2658–2672. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.M.; Chang, J.T.; Wen, C.L.; Hsu, S.L. Emodin induces a reactive oxygen species-dependent and ATM-p53-Bax mediated cytotoxicity in lung cancer cells. Eur. J. Pharmacol. 2009, 623, 1–9. [Google Scholar] [CrossRef]
- Ko, J.C.; Su, Y.J.; Lin, S.T.; Jhan, J.Y.; Ciou, S.C.; Cheng, C.M.; Lin, Y.W. Suppression of ERCC1 and Rad51 expression through ERK1/2 inactivation is essential in emodin-mediated cytotoxicity in human non-small cell lung cancer cells. Biochem. Pharmacol. 2010, 79, 655–664. [Google Scholar] [CrossRef]
- Ko, J.C.; Tsai, M.S.; Kuo, Y.H.; Chiu, Y.F.; Weng, S.H.; Su, Y.C.; Lin, Y.W. Modulation of Rad51, ERCC1, and thymidine phosphorylase by emodin result in synergistic cytotoxic effect in combination with capecitabine. Biochem. Pharmacol. 2011, 81, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.C.; Su, Y.J.; Lin, S.T.; Jhan, J.Y.; Ciou, S.C.; Cheng, C.M.; Chiu, Y.F.; Kuo, Y.H.; Tsai, M.S.; Lin, Y.W. Emodin enhances cisplatin-induced cytotoxicity via down-regulation of ERCC1 and inactivation of ERK1/2. Lung Cancer. 2010, 69, 155–164. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Bi, J.J.; Guo, Q.; Yu, Y.; Ye, X.F. Effects of emodin extracted from Chinese herbs on proliferation of non-small cell lung cancer and underlying mechanisms. Asian Pac. J. Cancer Prev. 2012, 13, 1505–1510. [Google Scholar] [CrossRef]
- Ok, S.; Kim, S.M.; Kim, C.; Nam, D.; Shim, B.S.; Kim, S.H.; Ahn, K.S.; Choi, S.H.; Ahn, K.S. Emodin inhibits invasion and migration of prostate and lung cancer cells by downregulating the expression of chemokine receptor CXCR4. Immunopharmacol. Immunotoxicol. 2012, 34, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Hintzpeter, J.; Seliger, J.M.; Hofman, J.; Martin, H.J.; Wsol, V.; Maser, E. Inhibition of human anthracycline reductases by emodin—A possible remedy for anthracycline resistance. Toxicol. Appl. Pharmacol. 2016, 293, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Zhao, S.; Wu, J.; Zheng, F.; Yang, L.; Hu, J.H.; Han, S.S. Inhibition of integrin-linked kinase expression by emodin through crosstalk of AMPKα and ERK1/2 signaling and reciprocal interplay of Sp1 and c-Jun. Cell. Signal. 2015, 27, 1469–1477. [Google Scholar] [CrossRef]
- Tang, Q.; Wu, J.; Zheng, F.; Hann, S.S.; Chen, Y. Emodin increases expression of insulin-like growth factor binding protein 1 through activation of MEK/ERK/AMPKα and interaction of PPARγ and Sp1 in lung cancer. Cell. Physiol. Biochem. 2017, 41, 339–357. [Google Scholar] [CrossRef]
- Jelassi, B.; Anchelin, M.; Chamouton, J.; Cayuela, M.L.; Clarysse, L.; Li, J.; Goré, J.; Jiang, L.H.; Roger, S. Anthraquinone emodin inhibits human cancer cell invasiveness by antagonizing P2X7 receptors. Carcinogenesis 2013, 34, 1487–1496. [Google Scholar] [CrossRef]
- Su, J.; Yan, Y.; Qu, J.; Xue, X.; Liu, Z.; Cai, H. Emodin induces apoptosis of lung cancer cells through ER stress and the TRIB3/NF-κB pathway. Oncol. Rep. 2017, 37, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Dar, R.A.; Majeed, R.; Ali Sheikh, A.; Rehman, S.; Hamid, A.; Hassan, Q.P. Emodin, isolated and characterized from an endophytic fungus Polyporales sp., induces apoptotic cell death in human lung cancer cells through the loss of mitochondrial membrane potential. J. Phytopharmacol. 2017, 6, 288–292. [Google Scholar]
- Haque, E.; Kamil, M.; Irfan, S.; Sheikh, S.; Hasan, A.; Nazir, A.; Mir, S.S. Blocking mutation independent p53 aggregation by emodin modulates autophagic cell death pathway in lung cancer. Int. J. Biochem. Cell Biol. 2018, 96, 90–95. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Z.; Zhang, J. Emodin enhances antitumor effect of paclitaxel on human non-small-cell lung cancer cells in vitro and in vivo. Drug Des. Devel. Ther. 2019, 13, 1145–1153. [Google Scholar] [CrossRef]
- Li, Z.; Lin, Y.; Zhang, S.; Zhou, L.; Yan, G.; Wang, Y.; Zhang, M.; Wang, M.; Lin, H.; Tong, Q.; et al. Emodin regulates neutrophil phenotypes to prevent hypercoagulation and lung carcinogenesis. J. Transl. Med. 2019, 17, 90. [Google Scholar] [CrossRef]
- Peng, S.; Wang, J.; Lu, C.; Xu, Z.; Chai, J.; Ke, Q.; Deng, X.Z. Emodin enhances cisplatin sensitivity in non-small cell lung cancer through Pgp downregulation. Oncol. Lett. 2021, 21, 230. [Google Scholar] [CrossRef]
- Li, B.; Zhao, X.; Zhang, L.; Cheng, W. Emodin interferes with AKT1-mediated DNA damage and decreases resistance of breast cancer cells to doxorubicin. Front. Oncol. 2021, 10, 588533. [Google Scholar] [CrossRef]
- Wahi, D.; Soni, D.; Grover, A.A. Double-Edged Sword: The anti-cancer effects of emodin by inhibiting the redox-protective protein MTH1 and augmenting ROS in NSCLC. J. Cancer 2021, 12, 652–681. [Google Scholar] [CrossRef]
- Su, Y.J.; Tsai, M.S.; Kuo, Y.H.; Chiu, Y.F.; Cheng, C.M.; Lin, S.T.; Lin, Y.W. Role of Rad51 down-regulation and extracellular signal-regulated kinases 1 and 2 inactivation in emodin and mitomycin C-induced synergistic cytotoxicity in human non-small-cell lung cancer cells. Mol. Pharmacol. 2010, 77, 633–643. [Google Scholar] [CrossRef]
- Li, M.; Jin, S.; Cao, Y.; Xu, J.; Zhu, S.; Li, Z. Emodin regulates cell cycle of non-small lung cancer (NSCLC) cells through hyaluronan synthase 2 (HA2)-HA-CD44/receptor for hyaluronic acid-mediated motility (RHAMM) interaction-dependent signaling pathway. Cancer Cell Int. 2021, 21, 19. [Google Scholar] [CrossRef]
- Zhang, L.; Lau, Y.K.; Xi, L.; Hong, R.L.; Kim, D.S.; Chen, C.F.; Hortobagyi, G.N.; Chang, C.J.; Hung, M.C. Tyrosine kinase inhibitors, emodin and its derivative repress HER-2/neu-induced cellular transformation and metastasis-associated properties. Oncogene 1998, 16, 2855–2863. [Google Scholar] [CrossRef]
- Kim, Y.G.; Park, Y.H.; Yang, E.Y.; Park, W.S.; Park, K.S. Inhibition of tamoxifen’s therapeutic effects by emodin in estrogen receptor-positive breast cancer cell lines. Ann. Surg. Treat. Res. 2019, 97, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Shen, H.M.; Ong, C.N. Inhibitory effect of emodin on tumor invasion through suppression of activator protein-1 and nuclear factor-kappaB. Biochem. Pharmacol. 2004, 68, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, G.; Shi, P. Emodin-induced apoptosis in human breast cancer BCap-37 cells through the mitochondrial signaling pathway. Arch. Pharm. Res. 2008, 31, 742–748. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, G.; Shi, P. Effects of emodin on the gene expression profiling of human breast carcinoma cells. Cancer Detect. Prev. 2009, 32, 286–291. [Google Scholar] [CrossRef]
- Kalkhoran, R.M.; Kazerouni, F.; Omrani, M.D.; Rahimipour, A.; Shanaki, M.; Dehghan-Nayeri, N.; Ahmadi, F.; Younesian, O.; Cheshmi, F. Cytotoxic effect of emodin on growth of SKBR3 breast cancer cells. Int. J. Cancer Manag. 2017, 10, e8094. [Google Scholar] [CrossRef]
- Yan, Y.Y.; Zheng, L.S.; Zhang, X.; Chen, L.K.; Singh, S.; Wang, F.; Zang, J.Y.; Liang, Y.J.; Dai, C.L.; Gu, L.Q.; et al. Blockade of Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone derivative induces proteasomal degradation of Her2/neu. Mol. Pharm. 2011, 8, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.Y.; Fu, L.W.; Zhang, W.; Ma, H.S.; Ma, C.G.; Liang, Y.J.; Liu, B.Y.; Yu, J.Z.; Wu, Q.Z.; Dong, Y.M. Emodin azide methyl anthraquinone derivative induced G0/ G1 arrest in HER2/neu-overexpressing MDA-MB-453 breast cancer cells. J. BUON 2014, 19, 650–655. [Google Scholar] [PubMed]
- Fu, J.M.; Zhou, J.; Shi, J.; Xie, J.S.; Huang, L.; Yip, A.; Loo, W.; Chow, L.; Ng, E. Emodin affects ERCC1 expression in breast cancer cells. J. Transl. Med. 2012, 10, S1–S7. [Google Scholar] [CrossRef]
- Huang, P.H.; Huang, C.Y.; Chen, M.C.; Lee, Y.T.; Yue, C.H.; Wang, H.Y.; Lin, H. Emodin and aloe-emodin suppress breast cancer cell proliferation through ER α inhibition. Evid. Based Complement. Alternat. Med. 2013, 2013, 376123. [Google Scholar] [CrossRef]
- Li, W.Y.; Chan, R.Y.; Yu, P.H.; Chan, S.W. Emodin induces cytotoxic effect in human breast carcinoma MCF-7 cell through modulating the expression of apoptosis-related genes. Pharm. Biol. 2013, 51, 1175–1181. [Google Scholar] [CrossRef]
- Sui, J.-Q.; Xie, K.-P.; Zou, W.; Xie, M.-J. Emodin inhibits breast cancer cell proliferation through the ERα-MAPK/Akt-Cyclin D1/Bcl-2 signaling pathway. Asian Pac. J. Cancer Prev. 2014, 15, 6247–6251. [Google Scholar] [CrossRef]
- Jia, X.; Iwanowycz, S.; Wang, J.; Saaoud, F.; Yu, F.; Wang, Y.; Ju, J.; Chatterjee, S.; Wang, Q.; Fan, D. Emodin attenuates systemic and liver inflammation in hyperlipidemic mice administrated with lipopolysaccharides. Exp. Biol. Med. 2014, 239, 1025–1035. [Google Scholar] [CrossRef]
- Jia, X.; Yu, F.; Wang, J.; Iwanowycz, S.; Saaoud, F.; Wang, Y.; Hu, J.; Wang, Q.; Fan, D. Emodin suppresses pulmonary metastasis of breast cancer accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res. Treat. 2014, 148, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wang, Y.; Wang, X.; Li, J.; Hu, F. Emodin inhibits ATP-induced IL-1beta secretion, ROS production and phagocytosis attenuation in rat peritoneal macrophages via antagonizing P2X receptor. Pharm. Biol. 2014, 52, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Wenxin, F. The effects of lipopolysaccharides, imiquimod, and emodin on adherence of macrophages to breast cancer cells and the production of tumor necrosis factor alpha (TNFα) by macrophages. SCJAS 2015, 109. Available online: https://scholarexchange.furman.edu/scjas/2015/all/109 (accessed on 3 March 2021).
- Iwanowycz, S.; Wang, J.; Hodge, J.; Wang, Y.; Yu, F.; Fan, D. Emodin inhibits breast cancer growth by blocking the tumor-promoting feedforward loop between cancer cells and macrophages. Mol. Cancer Ther. 2016, 15, 1931–1942. [Google Scholar] [CrossRef]
- Liu, Q.; Hodge, J.; Wang, J.; Wang, Y.; Wang, L.; Singh, U.P.; Li, Y.; Yao, Y.; Wang, D.; Ai, W.; et al. Emodin reduces breast cancer lung metastasis by suppressing macrophage-induced breast cancer cell epithelial-mesenchymal transition and cancer stem cell formation. Theranostics 2020, 10, 8365–8381. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhou, X.; Qin, Y.; Yang, J.; Wang, Y.; Sun, Z.; Yu, K.; Zhang, S.; Liu, S. Emodin inhibits epithelial mesenchymal transition and metastasis of triple negative breast cancer via antagonism of CC chemokine ligand 5 secreted from adipocytes. Int. J. Mol. Med. 2018, 42, 579–588. [Google Scholar]
- Zu, C.; Qin, G.; Yang, C.; Liu, N.; He, A.; Zhang, M.; Zheng, X. Low dose Emodin induces tumor senescence for boosting breast cancer chemotherapy via silencing NRARP. Biochem. Biophys. Res. Commun. 2018, 505, 973–978. [Google Scholar] [CrossRef]
- Ponnusamy, L.; Kothandan, G.; Manoharan, R. Berberine and emodin abrogates breast cancer growth and facilitates apoptosis through inactivation of SIK3-induced mTOR and Akt signaling pathway. Biochim. Biophys. Acta 2020, 1866, 165897. [Google Scholar] [CrossRef]
- Bhattacharjee, M.; Upadhyay, P.; Sarker, S.; Basu, A.; Das, S.; Ghosh, A.; Ghosh, S.; Adhikary, A. Combinatorial therapy of Thymoquinone and Emodin synergistically enhances apoptosis, attenuates cell migration and reduces stemness efficiently in breast cancer. Biochim. Biophys. Acta 2020, 1864, 129695. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, J.; Sheng, A.; Huang, S.; Tang, Y.; Ma, S.; Hong, G. Emodin inhibits the proliferation of MCF-7 human breast cancer cells through activation of Aaryl hydrocarbon receptor (AhR). Front. Pharmacol. 2021, 11, 622046. [Google Scholar] [CrossRef]
- Chihara, T.; Shimpo, K.; Beppu, H.; Yamamoto, N.; Kaneko, T.; Wakamatsu, K.; Sonoda, S. Effects of aloe-emodin and emodin on proliferation of the MKN45 human gastric cancer cell line. Asian Pac. J. Cancer Prev. 2015, 16, 3887–3891. [Google Scholar] [CrossRef]
- Huang, L.; Wang, X.B.; Yu, Q.M.; Luo, Q.Y.; Zhang, Z.Z. Synergistic cancer growth-inhibitory effect of emodin and low-dose cisplatin on gastric cancer cells in vitro. Trop. J. Pharm. Res. 2015, 14, 1427–1434. [Google Scholar] [CrossRef]
- Sun, Z.H.; Bu, P. Downregulation of phosphatase of regenerating liver-3 is involved in the inhibition of proliferation and apoptosis induced by emodin in the SGC-7901 human gastric carcinoma cell line. Exp. Ther. Med. 2012, 3, 1077–1081. [Google Scholar] [CrossRef][Green Version]
- Jiang, X.H.; Ma, M.X.; Li, M.J.; Shao, S.H.; Yuan, H.; Hu, F.Q.; Liu, J.; Huang, X. Preparation and evaluation of novel emodin-loaded stearic acid-g-chitosan oligosaccharide nanomicelles. Nanoscale Res. Lett. 2020, 15, 93. [Google Scholar] [CrossRef]
- Liu, A.; Chen, H.; Wei, W.; Ye, S.; Liao, W.; Gong, J.; Jiang, Z.; Wang, L.; Lin, S. Antiproliferative and antimetastatic effects of emodin on human pancreatic cancer. Oncol. Rep. 2011, 26, 81–89. [Google Scholar] [PubMed]
- Li, N.; Wang, C.; Zhang, P.; You, S. Emodin inhibits pancreatic cancer EMT and invasion by up-regulating microRNA-1271. Med. Rep. 2018, 18, 3366–3374. [Google Scholar] [CrossRef]
- Hu, L.; Cui, R.; Liu, H.; Wang, F. Emodin and rhein decrease levels of hypoxia-inducible factor-1 α in human pancreatic cancer cells and attenuate cancer cachexia in athymic mice carrying these cells. Oncotarget 2017, 8, 88008–88020. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, R.S.; Wang, X.W.; Sun, Q.F.; Ye, Z.J.; Liu, J.W.; Zhou, D.H.; Tang, Y. Anticancer effects of emodin on HepG2 cell: Evidence from bioinformatic analysis. Biomed. Res. Int. 2019, 3065818. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.G.; Wu, J.F.; Tang, R.F.; Sun, C.; Ji, J.W.; Yin, Z.L.; Ma, G.; Yang, W. Emodin, a natural anthraquinone, suppresses liver cancer in vitro and in vivo by regulating VEGFR(2) and miR-34a. Investig. New Drugs 2020, 38, 229–245. [Google Scholar] [CrossRef]
- Yang, N.; Li, C.; Li, H.; Liu, M.; Cai, X.; Cao, F.; Wang, X. Emodin induced SREBP1-dependent and SREBP1-independent apoptosis in hepatocellular carcinoma cells. Front. Pharmacol. 2019, 10, 709. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.X.; Li, M.H.; Tao, L.; Ruan, L.Y.; Hong, W.; Chen, C.; Zhao, W.L.; Xu, H.; Chen, J.F.; Wang, J.S. Anti-cancer effects of emodin on HepG2 cells as revealed by 1H NMR based metabolic profiling. J. Proteome Res. 2018, 17, 1943–1952. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, Y.M.; Oh, T.I.; Shin, D.H.; Kim, G.H.; Kan, S.Y.; Kang, H.; Kim, J.H.; Kim, B.M.; Yim, W.J.; et al. Emodin sensitizes hepatocellular carcinoma cells to the anti-cancer effect of sorafenib through suppression of cholesterol metabolism. Int. J. Mol. Sci. 2008, 19, 3127. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Ni, B.R.; Fu, J.; Yin, X.B.; You, L.T.; Leng, X.; Liang, X.; Ni, J. Emodin induces apoptosis in human hepatocellular carcinoma HepaRG cells via the mitochondrial caspase-dependent pathway. Oncol. Rep. 2018, 40, 1985–1993. [Google Scholar] [CrossRef]
- Dong, H.; Wu, G.; Xu, H.; Zhang, C.; Wang, J.; Gao, M.; Pang, Y.; Zhang, H.; Zhang, B.; Tian, Y.; et al. N-acetylaminogalactosyl-decorated biodegradable PLGA-TPGS copolymer nanoparticles containing emodin for the active targeting therapy of liver cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; He, D.; Li, K.; Liu, H.L.; Wang, B.T.; Zheng, L.F.; Li, J.Z. Emodin targets mitochondrial cyclophilin D to induce apoptosis in HepG2 cells. Biomed. Pharmacother. 2017, 90, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhong, M.; Liang, S.; Chen, Y.; Liu, D.; Yin, Z.; Cao, Q.; Wang, C.; Ling, C. Emodin inhibits migration and invasion of MHCC-97H human hepatocellular carcinoma cells. Exp. Ther. Med. 2016, 12, 3369–3374. [Google Scholar] [CrossRef][Green Version]
- Cui, Y.; Lu, P.; Song, G.; Liu, Q.; Zhu, D.; Liu, X. Involvement of PI3K/Akt, ERK and p38 signaling pathways in emodin-mediated extrinsic and intrinsic human hepatoblastoma cell apoptosis. Food Chem. Toxicol. 2016, 92, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Y.P.; Zhang, T.; Zhang, Y.M. Inhibitory effect of emodin on human hepatoma cell line SMMC-7721 and its mechanism. Afr. Health Sci. 2015, 15, 97–100. [Google Scholar] [CrossRef]
- Subramaniam, A.; Shanmugam, M.K.; Ong, T.H.; Li, F.; Perumal, E.; Chen, L.X.; Vali, S.; Abbasi, T.; Kapoor, S.; Ahn, K.S.; et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. Br. J. Pharmacol. 2013, 170, 807–821. [Google Scholar] [CrossRef]
- Hsu, C.M.; Hsu, Y.A.; Tsai, Y.; Shieh, F.K.; Huang, S.H.; Wan, L.; Tsai, F.J. Emodin inhibits the growth of hepatoma cells: Finding the common anti-cancer pathway using Huh7, Hep3B, and HepG2 cells. Biochem. Biophys. Res. Commun. 2010, 392, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.B.; Ueki, N.; Cheng, J.D.; Imanishi, H.; Hada, T. Induction of apoptosis in hepatocellular carcinoma cell lines by emodin. Jpn. J. Cancer Res. 2002, 93, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, Y.; Li, X.; Li, H.; Chen, Y.; Tian, Y.; Wang, J. Emodin potentiates the anticancer effect of cisplatin on gallbladder cancer cells through the generation of reactive oxygen species and the inhibition of survivin expression. Oncol. Rep. 2011, 26, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, Y.P.; Huang, X.Z.; He, M.; Chen, Y.Y.; Shi, G.Y.; Li, H.; Yi, J.; Wang, J. Emodin enhances sensitivity of gallbladder cancer cells to platinum drugs via glutathion depletion and MRP1 downregulation. Biochem. Pharmacol. 2010, 79, 1134–1140. [Google Scholar] [CrossRef]
- Li, X.X.; Dong, Y.; Wang, W.; Wang, H.L.; Chen, Y.Y.; Shi, G.Y.; Yi, J.; Wang, J. Emodin as an effective agent in targeting cancer stem-like side population cells of gallbladder carcinoma. Stem Cells Dev. 2013, 22, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.S.; Weng, S.W.; Lin, M.W.; Lu, C.C.; Chiang, J.H.; Yang, J.S.; Lai, K.C.; Lin, J.P.; Tang, N.Y.; Lin, J.G.; et al. Antitumor effects of emodin on LS1034 human colon cancer cells in vitro and in vivo: Roles of apoptotic cell death and LS1034 tumor xenografts model. Food Chem. Toxicol. 2012, 50, 1271–1278. [Google Scholar] [CrossRef]
- Yan-Xin, Z.; Wen-Gang, S.; Feng-Lan, G. IInfluence of emodin on VEGF-C and MMP-9 of mice with colon cancer. Her. Med. 2013, 32, 583–585. [Google Scholar]
- Xie, M.-J.; Ma, Y.-H.; Miao, L.; Wang, Y.; Wang, H.-Z.; Xing, Y.-Y.; Xi, T.; Lu, Y.-Y. Emodin-provoked oxidative stress induces apoptosis in human colon cancer HCT116 cells through a p53-mitochondrial apoptotic pathway. Asian Pac. J. Cancer Prev. 2014, 15, 5201–5205. [Google Scholar] [CrossRef]
- Ma, L.; Li, W. Emodin inhibits LOVO colorectal cancer cell proliferation via the regulation of the Bcl-2/Bax ratio and cytochrome c. Exp. Ther. Med. 2014, 8, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Lee, M.S.; Cha, E.Y.; Sul, J.Y.; Lee, J.S.; Kim, J.S.; Park, J.B.; Kim, J.Y. Inhibitory effect of emodin on fatty acid synthase, colon cancer proliferation and apoptosis. Mol. Med. Rep. 2017, 15, 2163–2173. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xu, J.; Zhao, J.; Zhang, R. Inhibitory effect of emodin on human colon cancer SW620 cells and possible mechanisms. Biomed. Res. 2017, 28, 9686–9690. [Google Scholar]
- Pooja, T.; Karunagaran, D. Emodin suppresses Wnt signaling in human colorectal cancer cells SW480 and SW620. Eur. J. Pharmacol. 2014, 742, 55–64. [Google Scholar] [CrossRef]
- Saunders, I.T.; Mir, H.; Kapur, N.; Singh, S. Emodin inhibits colon cancer by altering BCL-2 family proteins and cell survival pathways. Cancer Cell Int. 2019, 19, 98. [Google Scholar] [CrossRef]
- Dai, G.; Ding, K.; Cao, Q.; Xu, T.; He, F.; Liu, S.; Ju, W. Emodin suppresses growth and invasion of colorectal cancer cells by inhibiting VEGFR2. Eur. J. Pharmacol. 2019, 859, 172525. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, Q.; He, X.; Wei, H.; Wang, T.; Shao, J.; Jiang, X. Emodin induces apoptosis of colon cancer cells via induction of autophagy in a ROS-dependent manner. Oncol. Res. 2018, 26, 889–899. [Google Scholar] [CrossRef]
- Höhn, P.; Braumann, C.; Freiburger, M.; Koplin, G.; Dubiel, W.; Luu, A. Anti-tumorigenic effects of emodin and its’ homologue BTB14431 on vascularized colonic cancer in a rat model. Asian Pac. J. Cancer Prev. 2020, 21, 205–210. [Google Scholar] [CrossRef]
- Li, T.; Si, W.; Zhu, J.; Yin, L.; Zhong, C. Emodin reverses 5-Fu resistance in human colorectal cancer via downregulation of PI3K/Akt signaling pathway. Am. J. Trans. Res. 2020, 12, 1851–1861. [Google Scholar]
- Zhang, Y.; Pu, W.; Bousquenaud, M.; Cattin, S.; Zaric, J.; Sun, L.K.; Rüegg, C. Emodin inhibits inflammation, carcinogenesis, and cancer progression in the AOM/DSS model of colitis-associated intestinal tumorigenesis. Front. Oncol. 2021, 10, 564674. [Google Scholar] [CrossRef]
- Yang, J.; Tang, X.M.; Li, H.; Shi, G.Y.; Zhu, P.; Jin, H.F.; Yi, J. Emodin sensitizes HeLa cell to arsenic trioxide induced apoptosis via the reactive oxygen species-mediated signaling pathways. Shi Yan Sheng Wu Xue Bao 2003, 36, 465–475. [Google Scholar] [PubMed]
- Moreira, T.F.; Sorbo, J.M.; Souza, F.O.; Fernandes, B.C.; Ocampos, F.; de Oliveira, D.; Arcaro, C.A.; Assis, R.P.; Barison, A.; Miguel, O.G.; et al. Emodin, physcion, and crude extract of Rhamnus sphaerosperma var. pubescens induce mixed cell death, increase in oxidative stress, DNA damage, and inhibition of AKT in cervical and oral squamous carcinoma cell lines. Oxid. Med. Cell. Longev. 2018, 2018, 2390234. [Google Scholar] [CrossRef]
- Liu, R.; Liu, J.; Wang, S.; Wang, Y.; Zhang, T.; Liu, Y.; Geng, X.; Wang, F. Combined treatment with emodin and a telomerase inhibitor induces significant telomere damage/dysfunction and cell death. Cell Death Dis. 2019, 10, 527. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.B.; Bjorling-Poulsen, M.; Guerra, B. Emodin negatively affects the phosphoinositide 3-kinase/AKT signalling pathway: A study on its mechanism of action. Int. J. Biochem. Cell Biol. 2007, 39, 227–237. [Google Scholar] [CrossRef]
- Yaoxian, W.; Hui, Y.; Yunyan, Z.; Yanqin, L.; Xin, G.; Xiaoke, W. Emodin induces apoptosis of human cervical cancer hela cells via intrinsic mitochondrial and extrinsic death receptor pathway. Cancer Cell Int. 2013, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Thacker, P.C.; Karunagaran, D. Curcumin and emodin down-regulate TGF-β signaling pathway in human cervical cancer cells. PLoS ONE 2015, 10, e0120045. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, H.; Zhang, J.; Ge, X.; Gao, J.; Zhang, Y.; Lou, G. Anti-tumor effect of emodin on gynecological cancer cells. Cell. Oncol. 2015, 38, 353–363. [Google Scholar] [CrossRef]
- Trybus, W.; Król, T.; Trybus, E.; Kopacz-Bednarska, A.; Król, G.; Karpowicz, E. Changes in the lysosomal system of cervical cancer cells induced by emodin action. Anticancer Res. 2017, 37, 6087–6096. [Google Scholar]
- Trybus, W.; Król, T.; Trybus, E.; Stachurska, A.; Król, G.; Kopacz-Bednarska, A. Emodin induces death in human cervical cancer cells through mitotic catastrophe. Anticancer Res. 2019, 39, 679–686. [Google Scholar] [CrossRef]
- Kurokawa, T.; He, G.; Siddik, Z.H. Protein kinase inhibitors emodin and dichloro-ribofuranosylbenzimidazole modulate the emodin and dichloro-ribofuranosylbenzimidazole modulate the cellular accumulation and cytotoxicity of cisplatin in a schedule-dependent manner. Cancer Chemother. Pharmacol. 2010, 65, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Chen, Y.; Cai, X.; Zhao, L.; He, A.; Guo, K.; Zheng, X. The combined effect of survivin-targeted shRNA and emodin on the proliferation and invasion of ovarian cancer cells. Anti-Cancer Drugs 2013, 24, 937–944. [Google Scholar] [CrossRef]
- Ma, J.; Yang, J.; Wang, C.; Zhang, N.; Dong, Y.; Wang, C.; Wang, Y.; Lin, X. Emodin augments cisplatin cytotoxicity in platinum-resistant ovarian cancer cells via ROS-dependent MRP1 downregulation. Biomed. Res. Int. 2014, 107671. [Google Scholar] [CrossRef]
- Nam, D.Y.; Lee, D.U. Efficacy of emodin/paclitaxel versus paclitaxel for the treatment of ovarian cancer in vivo. Int. J. Pharmacol. 2016, 12, 743–748. [Google Scholar] [CrossRef]
- Lu, J.; Xu, Y.; Wei, X.; Zhao, Z.; Xue, J.; Liu, P. Emodin inhibits the epithelial to mesenchymal transition of epithelial ovarian cancer cells via ILK/GSK-3β/Slug signaling pathway. BioMed Res. Int. 2016, 2016, 6253280. [Google Scholar] [CrossRef]
- Lu, J.; Xu, Y.; Zhao, Z.; Ke, X.; Wei, X.; Kang, J.; Zong, X.; Mao, H.; Liu, P. Emodin suppresses proliferation, migration and invasion in ovarian cancer cells by down regulating ILK in vitro and in vivo. Onco Targets Ther. 2017, 10, 3579–3589. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Dong, T.; Li, R.; Lu, J.; Wei, X.; Liu, P. Emodin inhibits epithelial to mesenchymal transition in epithelial ovarian cancer cells by regulation of GSK-3β/β-catenin/ZEB1 signaling pathway. Oncol. Rep. 2016, 35, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Giraud, F.; Akué-Gédu, R.; Nauton, L.; Candelon, N.; Debiton, E.; Théry, V.; Anizon, F.; Moreau, P. Synthesis and biological activities of 4-substituted pyrrolo[2,3-a]carbazole Pim kinase inhibitors. Eur. J. Med. Chem. 2012, 56, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Ju, X.; Meng, Q.; Yu, Z.J.; Ma, L.B. Emodin inhibits the proliferation of PC3 prostate cancer cells in vitro via the Notch signaling pathway. Mol. Med. Rep. 2015, 12, 4427–4433. [Google Scholar] [CrossRef]
- Masaldan, S.; Iyer, V.V. Exploration of effects of emodin in selected cancer cell lines: Enhanced growth inhibition by ascorbic acid and regulation of LRP1 and AR under hypoxia-like conditions. J. Appl. Toxicol. 2014, 34, 95–104. [Google Scholar] [CrossRef]
- Yu, C.X.; Zhang, X.Q.; Kang, L.D.; Zhang, P.J.; Chen, W.W.; Liu, W.W.; Liu, Q.W.; Zhang, J.Y. Emodin induces apoptosis in human prostate cancer cell LNCaP. Asian J. Androl. 2008, 10, 625–634. [Google Scholar] [CrossRef]
- Huang, X.Z.; Wang, J.; Huang, C.; Chen, Y.Y.; Shi, G.Y.; Hu, Q.S.; Yi, J. Emodin enhances cytotoxicity of chemotherapeutic drugs in prostate cancer cells: The mechanisms involve ROS-mediated suppression of multidrug resistance and hypoxia inducible factor-1. Cancer Biol. Ther. 2008, 7, 468–475. [Google Scholar] [CrossRef]
- Brown, M.; Bellon, M.; Nicot, C. Emodin and DHA potently increase arsenic trioxide interferon-α-induced cell death of HTLV-I-transformed cells by generation of reactive oxygen species and inhibition of Akt and AP-1. Blood 2007, 109, 1653–1659. [Google Scholar] [CrossRef]
- Wang, C.-G.; Yang, J.-Q.; Liu, B.-Z.; Dan-Ting, J.; Chong, W.; Liang, Z.; Dan, Z.; Yan, W. Anti-tumor activity of emodin against human chronic myelocytic leukemia K562 cell lines in vitro and in vivo. Eur. J. Pharmacol. 2010, 627, 33–41. [Google Scholar]
- Wei, W.T.; Lin, S.Z.; Liu, D.L.; Wang, Z.H. The distinct mechanisms of the antitumor activity of emodin in different types of cancer (Review). Oncol. Rep. 2013, 30, 2555–2562. [Google Scholar] [CrossRef]
- Lin, S.Y.; Lai, W.W.; Ho, C.C.; Yu, F.S.; Chen, G.W.; Yang, J.S.; Liu, K.C.; Lin, M.L.; Wu, P.P.; Fan, M.J.; et al. Emodin induces apoptosis of human tongue squamous cancer SCC-4 cells through reactive oxygen species and mitochondria-dependent pathways. Anticancer Res. 2009, 29, 327–335. [Google Scholar]
- Chen, Y.Y.; Chiang, S.Y.; Lin, J.G.; Ma, Y.S.; Liao, C.L.; Weng, S.W.; Lai, T.Y.; Chung, J.G. Emodin, aloe-emodin and rhein inhibit migration and invasion in human tongue cancer SCC-4 cells through the inhibition of gene expression of matrix metalloproteinase-9. J. Oncol. 2010, 36, 1113–1120. [Google Scholar]
- Cichewicz, R.H.; Lim, K.C.; McKerrow, J.H.; Nair, M.G. Kwanzoquinones A-G and other constituents of Hemerocallis fulva ‘Kwanzo’ roots and their activity against the human pathogenic trematode Schistosoma mansoni. Tetrahedron 2002, 58, 8597–8606. [Google Scholar] [CrossRef]
- Cichewicz, R.H.; Zhang, Y.; Seeram, N.P.; Nair, M.G. Inhibition of human tumor cell proliferation by novel anthraquinones from daylilies. Life Sci. 2004, 74, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F.; Gericke, K.M.; Güntner, C. First total synthesis of the bioactive anthraquinone kwanzoquinone C and related natural products by a diels—Alder approach. Eur. J. Org. Chem. 2006, 21, 4910–4915. [Google Scholar] [CrossRef]
- Kim, K.; Min, M.; Hong, S. Efficient synthesis of anthraquinones from diaryl carboxylic acids via palladium (II)-catalyzed and visible light-mediated transformations. Adv. Synth. Catal. 2017, 359, 848–852. [Google Scholar] [CrossRef]
- Narender, T.; Sukanya, P.; Sharma, K.; Bathula, S.R. Apoptosis and DNA intercalating activities of novel emodin derivatives. RSC Adv. 2013, 3, 6123. [Google Scholar] [CrossRef]
- Tan, J.H.; Zhang, Q.X.; Huang, Z.S.; Chen, Y.; Wang, X.D.; Gu, L.Q.; Wu, J.Y. Synthesis, DNA binding and cytotoxicity of new pyrazole emodin derivatives. Eur. J. Med. Chem. 2006, 41, 1041–1047. [Google Scholar] [CrossRef]
- Koerner, S.K.; Hanai, J.I.; Bai, S.; Jernigan, F.E.; Oki, M.; Komaba, C.; Shuto, E.; Sukhatme, V.P.; Sun, L. Design and synthesis of emodin derivatives as novel inhibitors of ATPcitrate lyase. Eur. J. Med. Chem. 2017, 126, 920–928. [Google Scholar] [CrossRef]
- Lev-Goldman, V.; Mester, B.; Ben-Aroya, N.; Koch, Y.; Weiner, L.; Fridkin, M. Synthesis and active oxygen generation by new emodin derivatives and their gonadotropin-releasing hormone conjugates. Bioconjugate 2006, 17, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Bai, Z.; Zhang, F.; Wang, C.; Yuan, Y.; Shao, J. Synthesis and biological activity evaluation of emodin quaternary ammonium salt derivatives as potential anticancer agents. Eur. J. Med. Chem. 2012, 56, 320–331. [Google Scholar] [CrossRef]
- Gecibesler, I.H.; Disli, F.; Bayindir, S.; Toprak, M.; Tufekci, A.R.; Sahin Yaglioglu, A.; Altun, M.; Kocak, A.; Demirtas, I.; Adem, S. The isolation of secondary metabolites from Rheum ribes L. and the synthesis of new semi-synthetic anthraquinones: Isolation, synthesis and biological activity. Food Chem. 2021, 342, 128378. [Google Scholar] [CrossRef]
- Narender, T.; Sukanya, P.; Sharma, K.; Bathula, S.R. Preparation of novel antiproliferative emodin derivatives and studies on their cell cycle arrest, caspase dependent apoptosis and DNA binding interaction. Phytomedicine 2013, 20, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, F.; Chen, T.; Zhang, L.; Wang, Z.; Su, Q.; Feng, L. Design, synthesis, molecular docking, and biological evaluation of new emodin anthraquinone derivatives as potential antitumor substances. Chem. Biodivers. 2020, 17, e2000328. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gan, Y.; Li, S.; Luo, T.; Zhang, Y.; Zhao, J. Potent P-glycoprotein inhibition of emodin derivative: Synthesis and biological evaluation. Med. Chem. Res. 2014, 23, 2106–2112. [Google Scholar] [CrossRef]
- Wang, X.; Xu, W. Facile synthesis of emodin derivatives as potential MMPIs. Bull. Korean Chem. Soc. 2005, 26, 1923–1924. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Y.; Leng, L.; Cheng, M.S.; Yu, P.F.; Zhang, J.Z. A new series of emodin derivatives with bone affinity. Chin. Chem. Lett. 2007, 18, 141–144. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, Q.; Yang, Y.; Ai, X.; Chen, S.; Song, Y. Synthesis and anti-inflammatory effects of novel emodin derivatives bearing azole moieties. Arch. Pharm. Chem. Life Sci. 2020, 353, 1900264. [Google Scholar] [CrossRef]
- Jeetah, R.; Bhaw-Luximon, A.; Jhurry, D. Nanopharmaceutics: Phytochemical-based controlled or sustained drug-delivery systems for cancer treatment. J. Biomed. Nanotechnol. 2014, 10, 1810–1840. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, C.; Zhou, G.; Wu, Y.; Chen, J. Improving the controlled release of water-insoluble emodin from amino-functionalized mesoporous silica. Appl. Surf. Sci. 2012, 258, 6366–6372. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Shen, J.; Wang, J.; Yang, X.; Dong, S.; Lu, S. Nanoparticle-based drug delivery system: A patient-friendly chemotherapy for oncology. Dose-Response 2020, 18, 1559325820936161. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, T.; Chen, R.; Hu, Y.; Chen, M.; Wang, Y. Emodin loaded solid lipid nanoparticles: Preparation, characterization and antitumor activity studies. Int. J. Pharm. 2012, 430, 238–246. [Google Scholar] [CrossRef]
- Shi, Y.; Li, J.; Ren, Y.; Wang, H.; Cong, Z.; Wu, G.; Du, L.; Li, H.; Zhang, X. Pharmacokinetics and tissue distribution of emodin loaded nanoemulsion in rats. J. Drug Deliv. Sci. Technol. 2015, 30, 242–249. [Google Scholar] [CrossRef]
- Zhang, T.P.; Dong, D.; Lu, D.Y.; Wang, S.; Wu, B.J. Cremophor EL-based nanoemulsion enhances transcellular permeation of emodin through glucuronidation reduction in UGT1A1-overexpressing MDCKII cells. Int. J. Pharm. 2016, 501, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Gobin, A.S.; Rhea, R.; Newman, R.A.; Mathur, A.B. Silk-fibroin-coated liposomes for long-term and targeted drug delivery. Int. J. Nanomed. 2006, 1, 81–87. [Google Scholar] [CrossRef]
- Cheema, S.K.; Gobin, A.S.; Rhea, R.; Lopez-Berestein, G.; Newman, R.A.; Mathur, A.B. Silk fibroin mediated delivery of liposomal emodin to breast cancer cells. Int. J. Pharm. 2007, 341, 221–229. [Google Scholar] [CrossRef]
- Yücel, Ç.; Aktaş, Y.; Değim, Z.; Yılmaz, Ş.; Arsoy, T.; Altıntaş, L.; Çokçalışkan, C.; Sözmen, M. Novel approach to the treatment of diabetes: Embryonic stem cell and insulin-loaded liposomes and nanocochleates. J. Nanosci. Nanotechnol. 2019, 19, 3706–3719. [Google Scholar] [CrossRef]
- Wang, T.; Yin, X.; Lu, Y.; Shan, W.; Xiong, S. Formulation, antileukemia mechanism, pharmacokinetics, and biodistribution of a novel liposomal emodin. Int. J. Nanomed. 2012, 7, 2325–2337. [Google Scholar]
- Fu, M.; Tang, W.; Liu, J.J.; Gong, X.Q.; Kong, L.; Yao, X.M.; Jing, M.; Cai, F.Y.; Li, X.T.; Lu, R.J. Combination of targeted daunorubicin liposomes and targeted emodin liposomes for treatment of invasive breast cancer. J. Drug Target. 2020, 28, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Xie, S.; Han, S.; Zhang, J.; Chang, X.; Chao, J.; Huang, Q.; Yuan, Q.; Lin, H.; Xu, L.; et al. Preparation of a nano emodin transfersome and study on its anti-obesity mechanism in adipose tissue of diet-induced obese rats. J. Transl. Med. 2014, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Pourhajibagher, M.; Etemad-Moghadam, S.; Alaeddini, M.; Bahador, A. Modulation of the triggered apoptosis by nano emodin transfersome-mediated sonodynamic therapy on head and neck squamous cell carcinoma cell lines. Photodiagn. Photodyn. Ther. 2021, 9, 102253. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gao, M.; Xu, H.; Guan, X.; Lv, L.; Deng, S.; Zhang, C.; Tian, Y. A promising emodin-loaded poly (Lactic-Co-Glycolic Acid)-d-α-tocopheryl polyethylene glycol 1000 succinate nanoparticles for liver cancer therapy. Pharm. Res. 2016, 33, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Luo, J.; Tan, S.; Otieno, B.O.; Zhang, Z. The applications of vitamin E TPGS in drug delivery. Eur. J. Pharm. Sci. 2013, 49, 175–186. [Google Scholar] [CrossRef]
- Wang, B.; Chen, L.; Sun, Y.; Zhu, Y.; Sun, Z.; An, T.; Li, Y.; Lin, Y.; Fan, D.; Wang, Q. Development of phenylboronic acid-functionalized nanoparticles for emodin delivery. Mater. Chem. B 2015, 3, 3840–3847. [Google Scholar] [CrossRef]
- Chen, X.; Yang, Z.; Sun, R.; Mo, Z.; Jin, G.; Wei, F.; Hu, J.; Guan, W.; Zhong, N. Preparation of lung-targeting, emodin-loaded polylactic acid microspheres and their properties. Int. J. Mol. Sci. 2014, 15, 6241–6251. [Google Scholar] [CrossRef]
- Krajnović, T.; Maksimović-Ivanić, D.; Mijatović, S.; Drača, D.; Wolf, K.; Edeler, D.; Wessjohann, L.A.; Kaluđerović, G.N. Drug delivery system for emodin based on mesoporous silica SBA-15. Nanomaterials 2018, 8, 322. [Google Scholar] [CrossRef]
- Song, Y.; Sheng, Z.; Xu, Y.; Dong, L.; Xu, W.; Li, F.; Wang, J.; Wu, Z.; Yang, Y.; Su, Y.; et al. Magnetic liposomal emodin composite with enhanced killing efficiency against breast cancer. Biomater. Sci. 2019, 7, 867–875. [Google Scholar] [CrossRef]
- Radiac Brkanac, S.; Geric, M.; Gajski, G.; Vujcic, V.; Garaj-Vrhovac, V.; Kremer, D.; Domijan, A.M. Toxicity and antioxidant capacity of Frangula alnus Mill. bark and its active component emodin. Regul. Toxicol. Pharmacol. 2015, 73, 923–929. [Google Scholar] [CrossRef]
- Jiang, L.L.; Jiang, Y.; Zhao, D.S.; Fan, Y.X.; Yu, Q.; Li, P.; Li, H.J. CYP3A activation and glutathione depletion aggravate emodin-induced liver injury. Chem. Res. Toxicol. 2018, 31, 1052–1060. [Google Scholar] [CrossRef]
- Dong, X.; Fu, J.; Yin, X.; Cao, S.; Li, X.; Lin, L.; Ni, J. Emodin: A review of its pharmacology, toxicity and pharmacokinetics. Phytother. Res. 2016, 30, 1207–1218. [Google Scholar] [CrossRef]
- Chang, M.H.; Huang, F.J.; Chan, W.H. Emodin induces embryonic toxicity in mouse blastocysts through apoptosis. Toxicology 2012, 299, 25–32. [Google Scholar] [CrossRef]
- Luo, T.; Li, N.; He, Y.Q.; Weng, S.Q.; Wang, T.; Zou, Q.X.; Zeng, X.H. Emodin inhibits human sperm functions by reducing sperm [Ca2+]i and tyrosine phosphorylation. Reprod. Toxicol. 2015, 51, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gao, J.; Wang, T.S.; Guo, C.; Yan, Y.J.; Mao, C.Y.; Gu, L.W.; Yang, Y.; Li, Z.F.; Liu, A. NMR-based metabolomic techniques identify the toxicity of emodin in HepG2 cells. Sci. Rep. 2018, 8, 9379. [Google Scholar] [CrossRef]
- Yi, T.; Zhang, H.; Cai, Z. Analysis of Rhizoma Polygoni Cuspidati by HPLC and HPLC-ESI/MS. Phytochem. Anal. 2007, 18, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Leung, K.S.Y.; Lu, G.-H.; Zhang, H.; Chan, K. Identification and determination of the major constituents in traditional Chinese medicinal plant Polygonum multiflorum Thunb by HPLC coupled with PAD and ESI/MS. Phytochem. Anal. 2007, 18, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Jiafu, F.; Huazhong, R.; Qiufen, G.; Lin, Z.; Hong, J.; Tao, Y. Comparative analysis of the major constituents in three related polygonaceous medicinal plants using pressurized liquid extraction and HPLC-ESI/MS. Anal. Methods 2016, 8, 1557–1564. [Google Scholar] [CrossRef]
- Li, M.G.; Nie, Y.; Zhang, J.; Guo, C.-Y.; Zhang, L.Y. Extracting resveratrol, piceid and emodin from Polygonum cuspidatum by enzymolsis. Fine Chem. Corpus 2008, 5, 99817311. [Google Scholar]
- Tian, T.; Sun, Q.; Shen, J.; Zhang, T.; Gao, P.; Sun, Q. Microbial transformation of polydatin and emodin-8-β-D-glucoside of Polygonum cuspidatum Sieb. Et Zucc into resveratrol and emodin respectively by Rhizopus microsporus. World J. Microbiol. Biotechnol. 2008, 24, 861–866. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Cell Type | Concentration | Mechanism | Reference |
|---|---|---|---|---|
| A549 | Lung adenocarcinoma | 50 µM | Increased cyt c, activation of caspase-2, -3, -9, and mitochondrial Bax Inactivation of ERK and AKT, formation of ROS, disruption of ∆Ψm, reduction of mitochondrial Bcl-2 | [14] |
| CH27 | Lung squamous cell carcinoma | 50 µM | Morphological change, sub-G1 formation, disruption of focal adhesion kinase Increased expression of Bak and Bax proteins, activation of caspase-3, -8, -9 | [25] |
| CH27 | Lung squamous cell carcinoma | 50 µM | Internucleosomal DNA fragmentation Increase in cyt c, activation of caspase-3, expression of PKCa Decreased expression of PKCδ and ε | [58] |
| H460 | Non-small human lung carcinoma | |||
| NCI-H446 | Small cell lung cancer | 20 μmol/L | Increased caspase-3 activation Up-regulated NACA, p8, and PQBP1 genes Downregulated B2M, HLA-E, and CD1D genes | [62] |
| A549 H1650 | Lung adenocarcinoma | 2–10 μM (Emodin) 2 μM (Gefitinib) | Triggered Rad51 protein instability Decrease in phospho-ERK1/2 and Rad51 protein levels | [63] |
| A549 | Lung adenocarcinoma | 50 µM | Activation of the ATM-p53-Bax signaling pathway, ROS formation | [64] |
| H1650 A549 | Lung adenocarcinoma | 25–100 µM | Inactivation of ERK1/2 Down-regulation of Rad51 and ERCC1 | [65] |
| H520 | Human bronchioloalveolar cell carcinoma | |||
| H1703 | Lung squamous cell carcinoma | |||
| H1650 A549 | Lung adenocarcinoma | 25–200 µM (capecitabine) 50 µM (emodin) | Inactivation of ERK1/2 Down-regulation of Rad51 and ERCC1 Increased mRNA and protein expression of TP | [66] |
| H520 | Human bronchioloalveolar cell carcinoma | |||
| H1703 | Lung squamous cell carcinoma | |||
| H1703 A549 | Lung squamous cell carcinoma Lung adenocarcinoma | 1 µg/mL (cisplatin) 8.1; 16.2; 24.3 µg/mL (emodin) | Instability of the ERCC1 protein Inactivation of ERK1/2 | [67] |
| SK-MES | Lung squamous cell carcinoma | 40 μmol/L (SK-MES) 70 μmol/L (A549) | Decreased expression of ERCC1 and Rad51 protein and mRNA | [68] |
| A549 | Lung adenocarcinoma | |||
| A549 | Lung adenocarcinoma | 100 μM | Decreased expression of CXCR4 and HER2 | [69] |
| A549 PC9 | Lung adenocarcinoma | 50 μM | Increase in the phosphorylation of AMPKα and ERK1/2 Decrease in the expression of ILK | [71] |
| H1299 H1650 H1975 | Non-small cell lung carcinoma | |||
| A549 | Lung adenocarcinoma | 5 to 50 μM (emodin) 50 to 2000 nM (daunorubicin) | Inhibition of anthracycline reductase enzymes Sensitized cells to daunorubicin | [70] |
| A549 | Lung adenocarcinoma | 50 μM | Arrest in G2/M phase Invasion inhibition Increased PPARγ protein and luciferase reporter activity | [72] |
| H1975 | Non-small cell lung carcinoma | |||
| A549 | Lung adenocarcinoma | 1 nM to 10 µM | Prevention of ATP-induced increases in P2X7 Invasion inhibition | [73] |
| A549 H1299 | Lung adenocarcinoma | 80 µmol/L | Increased caspase 3 activation, TRIB3 expression | [74] |
| A549 | Lung adenocarcinoma | 30 to 100 µM/mL | Arrest in G1 and G2/M phase Loss of ∆Ψm Increased release of cyt c | [75] |
| A549 | Lung adenocarcinoma | 10,15, and 20 µM | Inhibition of p53 protein aggregates | [76] |
| A549 | Lung adenocarcinoma | Emodin 10 μM Paclitaxel 4 μM | Increased Bax and active caspase 3 expressions Decreased Bcl-2, p-Akt, and p-ERK levels | [77] |
| A549 | Lung adenocarcinoma | 5 µM emodin, 5–10 µM cisplatin (A549) 2.5 µM emodin, 5–10 µM cisplatin (H460) | Increased DNA damage Decreased Pgp expression | [79] |
| H460 | Non-small human lung carcinoma | |||
| NCI-H-520 | Human bronchioloalveolar cell carcinoma | 50 µM | Inhibition of MTH1, arrest in G2/M Increased expression of Bax, survivin, P-21, cleaved caspase 3, cleaved PARP (A549) Decreased Bcl-2, p-65 NFκβ (A549) Reduced integrin β1 and vimentin protein expression (A549) | [81] |
| NCI-H-460 | Non-small human lung carcinoma | |||
| A-549 | Lung adenocarcinoma | |||
| H1703 | Lung squamous cell carcinoma | 60 μM | Decreased levels of mitomycin C-derived Rad51 mRNA and protein | [82] |
| A549 | Lung adenocarcinoma | |||
| A549 | Lung adenocarcinoma | 30 µM | Suppressed the secretion of HA Augmented cells in G1/G0 phase and reduced cells in S and G2/M phase (A549) Decreased cyclin A and B, increased cyclin C,D,E (A549) | [83] |
| H520 | Human bronchioloalveolar cell carcinoma | |||
| H1975 | Non-small cell lung carcinoma | |||
| H1299 | Lung adenocarcinoma | |||
| H460 | Non-small human lung carcinoma |
| Cell Line | Concentration | Mechanism | Reference |
|---|---|---|---|
| MDA-MB453 BT-483 AU-565 | 40 µM | Suppressing the autophosphorylation and transphosphorylation activities of HER-2/neu tyrosine kinase | [17] |
| MDA-MB453 MCF-7 B104-1-1 | 40 µM emodin 20 µM potent derivative: 10-(4-acetamidobenzylidene)-9-anthrone) | Suppressing the tyrosine phosphorylation of p185neu, inhibiting the proliferation and transformation of HER-2/neu-overexpressing human breast cancer cells Cell cycle arrest at G0/G1 phase in MDA-MB 453 | [84] |
| MDA-MB-231 MCF-7 | 110 µM emodin 5 µM doxorubicin | Increased γH2Ax expression and the DNA damage Decreased expression of the AKT1, p53, PARP1, RAD51, and XRCC1 signaling pathway | [80] |
| ZR-75-1 MCF-7 | 60 µM emodin 4 µM tamoxifen | Up-regulation of cyclin D1 and p-ERK | [85] |
| MDA-MB-231 | 10–40 µM | Inhibition of TPA-stimulated MMP-9 activity Reduced transcriptional activity of AP-1 and NF-kB | [86] |
| BCap-37 | 50 µM | Increased the percentage of cells in the sub-G0/1 phase Decreased Bcl-2, Increased Bax level, and cytosolic cyt c level | [87] |
| BCap-37 | 50 µM | Up-regulation of P21 Down-regulation of IGF-2 Induced gene expression of p53 and caspase 3 | [88] |
| SKBR3 | 25 and 50 μM | Increased caspase 3, 8, and 9 mRNA levels Increased Bax level Reduced Bcl 2 level | [89] |
| MDA-MB-453 MCF-7/ADR MCF-7 | 2.3 to 9.2 µg/mL emodin azide methyl anthraquinone derivative | Lowered the Her2/neu protein Inhibited the downstream MAPK and PI3K-Akt signaling pathway by inhibiting p-Akt, and p-ERK1/2 in the MDA-MB-453 | [90] |
| MDA-MB-453 | Arrested the cell cylce in the G0/G1 phase Inhibited the expressions of: Cyclin D1, c/Myc, CDK4, and p-Rb | [91] | |
| MCF-7/Adr | 20 μg/mL | DecreasedERCC1 expression | [92] |
| MCF-7 MDA-MB-453 | 25 μM | Effect on nuclear ERα distribution | [93] |
| MCF-7 | 35 µM | Single-stranded DNA breaking, DNA fragmentation, up-regulation of FASL gene expression Down-regulation of the expression of CCND1, C-MYC, and MCL1 | [94] |
| MDA-MB-435s | 1 to 10 µM | Inhibition of ATP-induced increase in [Ca2+] | [73] |
| MCF-7 | 20 and 40 µM | Arrested the cell cycle in the G0/G1 phase Blocking the effect of estrogen on ERα expression and transcriptional activity Down-regulation of cyclin D1 and Bcl-2 protein expression, decreased PI3K/Akt protein expression | [95] |
| EO771-GFP | 10 µM and 30 µM | Reduced adhesion between macrophages and cancer cells | [99] |
| 4T1 | 100 µM | Decreased macrophage migration | [97] |
| EO771 4T1 | 0 to 50 μmol/L | Inhibition of the adherence of macrophages to the monolayer of tumor cells | [100] |
| 4T1 | 25 µM | Decreased TGF- β1 production Inhibited the formation of EMT and CSC | [101] |
| MDA-MB-231 MDA-MB-453 | 25 μM | Inhibition ofcell migration and invasion Decreased CCL5 levels Inhibition of the phosphorylation of AKT, activation of GSK3β, downregulation of the expression of β-catenin, Vimentin and snail Increased expression ofE-cadherin | [102] |
| MCF-7 | 20 μM emodin 40 μM 5-FU | Up-regulation of p21, p16, p27 protein Down-regulation of E2F1 and NRARP protein | [103] |
| MCF-10A MCF-7 MDA-MB-231 | Berberine + emodin (5, 10, 20 μM) | Suppressed SIK3 activity Cell cycle arrest (G0/G1) | [104] |
| MCF-7 T47D | Emodin 10 µg/mL and thymoquinone 2 µg/mL | Cell arrest in the sub G0/G1 phase, an increase in p53, Bax, and cleaved caspase 3 expression levels, a decrease in Bcl-2 protein, induction of ROS formation, and Cyt C releasein MCF7 cells Inhibition of cell migration and FAK, pFAK, and integrin β1 proteins were down-regulated in both cell lines | [105] |
| MCF7 | 25–100 µmol/L | Increase in CYP1A1 expression Regulating the expression of AhR and CYP1A1 proteins | [106] |
| Cell Line | Concentration and/or Method of Application | Efficacy and Mechanism | Reference |
|---|---|---|---|
| MKN45 | 0.05 mM emodin | Inhibition of proliferation Arrest in the G0/G1 and G2/M phase | [107] |
| SNU-5 | Combination with cisplatin (25 μM emodin + 3.0 μM CDDP) | Increase the apoptotic effect on cells and cell cycle arrest | [108] |
| SGC-7901 | Emodin (10 μM) | Down-regulation of PRL-3 mRNA | [109] |
| SGC-7901 | Emodin-loaded nanomicelles | Cytotoxic effect | [77] |
| BGC823 MGC803 | Emodin loaded stearic acid-chitosan oligosaccharide | Antitumoral effect | [110] |
| Cell Line | Concentration | Mechanism | Reference |
|---|---|---|---|
| SW1990 | Dose-dependent manner (10, 20, 40 µmol/L) | Prevention of migration and incursion Down-regulation NF-κB DNA binding capacity, survivin, and MMP-9 Up-regulation cleaved caspase-3 expression | [111] |
| SW1990 | Dose- and time-dependent manner (0, 20, 40 µmol/L) | Increase the expression level of miR-1271 Block proliferation capacity | [112] |
| MiaPaCa2 | Dose-dependent manner (Varied from 50 μM to 100 μM) | Inhibition of the expression of HIF-1α | [113] |
| In Vitro (Cell Line)/ In Vivo Studies | Concentration and/or Method of Application | Mechanism | Reference |
|---|---|---|---|
| SMMC-7721 | Dose-dependent manner (<50 µmol/L emodin) | Block the proliferation Stimulation of apoptosis Induction of phosphorylation of ERK and p38 Supression of AKT activation and expression of JNK | [122] |
| SMMC-7721 | 20, 40, and 80 µmol/L | Antiproliferation activity | [124] |
| HepG2 | Different emodin doses (79.01, 51.39, 33.13 µM) | Trigger intracellular ROS formation, reduction of the expression of proteins and genes involved in glycolysis, and disruption of cell cycle progression | [117] |
| HepG2 | Dose-dependent manner (50, 100 µM emodin) | Stimulation of mitochondrial dysfunction Stimulation of apoptosis by up-regulation of cyclophilin D Trigger ERK and ROS-associated cyclophilin D expression | [121] |
| HepG2 | 120 µM emodin | Induction of apoptosis and cause cell accumulation in the G1 phase Enhance release of cytochrome c Up-regulation caspase-8 and 9 expressions | [11] |
| PLC/PRF/5, HepG2/C3A, SK-HEP-1 | 60 µM emodin | Stimulation of apoptosis Arrest in the G2/M phase Increase in caspase-3, p53, Fas, and p21 signals | [27] |
| HCC (Bel-7402) with SREBP1 targeting | 100 μmol/Lemodin | Stimulation of apoptosis Decrease in mitochondrial membrane potential Activation of the expression of cytochrome C, caspases 9 and 3, endonuclease G, Bax, and Bcl-2 related proteins | [116] |
| HCC (PLC/PRF5, SK-HEP-1, HepG2, Huh7, and Hep3B) | Emodin and sorafenib | Inhibition of transcriptional activity of SREBP-2 Stimulation of cell cycle arrest in G1 phase | [118] |
| HepaRG | 80 µM emodin | Inhibition of cell cycle progression in G2/M and S phases Stimulation apoptosis Up-regulation of cyclin E, p21, p53, Bax, cleaved PARP, cleaved caspase-3, 8, and 9 Down-regulation of protein expression of Bcl-2 | [119] |
| HepG2/PLC mouse model | GalNAc-PLGA-sTPGS nanoparticles with emodin | Increase the antitumoral effect | [120] |
| BALB/c nude mice model | Dose-dependent manner (1 mg/kg or 10 mg/kg emodin) | Up-regulation miR-34a Inhibition of ERK1/2, AKT, and VEGFR2 Supression of SMAD2 and SMAD4 expression | [115] |
| HepG2 /Orthotopic mouse model | Dose-dependent manner (max. inhibition occurring at around 50 μM) | Inhibition of c-Src, JAK1 and JAK2 protein kinases | [125] |
| Cell Line | Concentration | Mechanism | Reference |
|---|---|---|---|
| LS1034 | 30 µM | Cell cycle arrest in the G2/M phase Decrease in ∆Ψm Increase in ROS production, protein levels of cyt c, caspase-9, and the ratio of Bax/Bcl-2 | [131] |
| HCT116 | 40 μmol/L | Increased ROS generation, overexpression of p53, up-regulated Bax expression | [133] |
| LOVO | 0–40 µM | Decreased Bcl-2 and increased Bax levels Increased cyt c release, reduction in ∆Ψm | [135] |
| HCT-116 | 25 µM | Decreased protein levels of FASN | [135] |
| HCT-116 | 25 µM emodin 100 µM cerulenin | Increase in apoptotic cell death and ERK1/2 phosphorylation Reduction in the PI3K/Akt phosphorylation | [135] |
| SW620 | 20–160 µmol/L | Cell cycle arrest in the G0/G1 phase Decreased Bcl2, increased Bax and p53 expression | [136] |
| HepG2 | 10, 20, and 40 μM | Inhibition of the expression of glycolysis-related proteins Increase in the percentage of the Sub-G1 phase | [117] |
| SW480 SW620 | 50 µM | Inhibition of the transcriptional activity of β-catenin/TCF Reduction inc-Myc and CCD1 levels Inhibited SNAI1 and vimentin levels Inhibition of the Wnt signal | [137] |
| DLD-1 COLO 201 | 18 µM 15 µM | Negatively regulated NF-κβ, PI3K/AKT, MAPK/JNK, and STAT pathways Activation of caspases, modulation of Bcl-2 proteins, and reduction in ∆Ψm | [138] |
| RKO | 5–20 µmol/L | Inhibited MMP-7, MMP-9, and VEGF proteins Increase in E-cadherin mRNA levels Decrease in Snail, N-cadherin, and β-catenin expressions Downregulated TCF4, cyclin D1, and c-Myc genes | [12] |
| HCT116 | 60 mg/mL | Inhibition of VEGFR2 expression Decreased PI3K and-AKT expression Suppressive effect on the growth, adhesion, and migration | [139] |
| HCT116 LOVO | 20 and 40 µM | Increased LC3-2 accumulation and changes in p62 and Beclin-1 levels Mitochondrial dysfunction | [140] |
| SW480 | 12 μg/mL 5-Fu 9 µM emodin | Reduced invasion and migration Inhibiting Bcl-2 and activating cleaved caspase-3 and Bax | [141] |
| Cell Line | Concentration | Mechanism | Reference |
|---|---|---|---|
| Bu 25TK | 56.7 μM | Activation of caspase-3 and -9 Induction of nuclear condensation, DNA fragmentation, and cleavage of poly (ADP-ribose) polymerase | [15] |
| HeLa | 10 µmol/L | Increased ROS levels Activation of transcription factors NF-κB Reduction in ∆Ψm | [144] |
| SiHa C33A | 46.3–185.0 μM | Increased intracellular ROS and DNA damage Decreased Bcl-2 expression, and increased Bax2 in SiHa cells, and decreased AKT activation in both SiHa and C33A cells | [145] |
| HeLa1.2.11 | 1–10 μM 5–20 μM | Genomic DNA damage S phase cell cycle arrest, and telomere damage | [146] |
| HeLa | 40 µM 60 μM | Down-regulation of AKT kinase Inhibition of the catalytic activity of mTOR kinase Reduction in the phosphorylation level of AKT protein | [147] |
| HeLa | 20 to 80 µM | Increased mRNA expression of caspase-9, -8 and -3, cyt c, and Apaf-1 Up-regulated FADD Fas, and FasL Down-regulatedpro-caspase-9, -8 and -3 | [148] |
| SiHa HeLa | 40 μM | Reduction in the expression of P-Smad3, Smad4, and TGF-β Receptor II Inhibition of TGF-β induced migration and invasion | [149] |
| Hela JAR HO-8910 | 5, 10, and 15 μM | Increased MMP-9 mRNA expression Increased caspase-9 and related activation of cleaved caspase-3, DNA damage, reduction of ΔΨM, reduction of Bcl-2 Cell cycle arrest in the G0/G1 phase | [150] |
| HeLa | 1–100 μM | Increased lysosomal membrane damage | [151] |
| HeLa | 1–100 μM | G2/M phase cell cycle arrest Enhanced mitotic catastrophe | [152] |
| Cell Line | Concentration | Mechanism | Reference |
|---|---|---|---|
| A2780 | 1 μM paclitaxel 10 μM emodin | Down-regulation P-gp, XIAP and survivin | [35] |
| A2780 | 1 μM emodin 100 μM cisplatin | Reduction in both intracellular platinum levels and DNA adducts | [153] |
| Sur-shRNA plasmid transfected SKOV3 and HO8910 | 60 µmol/L | Knockdown of survivin Proliferation inhibition, apoptosis induction, and reduction of invasion | [154] |
| COC1 cisplatin-resistant COC1/DDP | 33 μM cisplatin 50 μM emodin | Increased ROS formation Down-regulatedMRP1 gene Induction of apoptosis | [155] |
| SK-OV-3 A2780 | 5 to 80 μM | Decreased the β-catenin, p-GSK-3β, ILK, and Slug expression Up-regulation of E-cadherin and claudin Downregulation of N-cadherin and vimentin | [157] |
| A2780 SK-OV-3 | 20 μM | Down-regulation of the GSK-3β/-catenin/ZEB1 pathway Up-regulation of E-cadherin and keratin Down-regulation of N-cadherin, vimentin, MMP-9, and MMP-2 | [159] |
| In Vitro (Cell Line)/ In Vivo Studies | Concentration and/or Method of Application | Efficacy and Mechanism | Reference |
|---|---|---|---|
| PC3 | 125, 250, 500 μg/mL emodin | Stimulation of the Notch signaling pathway Induction of apoptosis Inhibition of the G2/M phase | [161] |
| DU145 | 100 μM emodin | Inhibition of NF-κB Reduction of CXCR4 activation | [69] |
| DU145 | Emodin plus cisplatin | Increase ROS levels Decrease MDR1 expression and HIF-1 transactivation | [164] |
| LNCaP | Dose-dependent manner (Maximum at 40 µmol/L emodin) | Enhance p53 and p21 expression Stimulation of ROS-mediated growth inhibition | [162] |
| C3(1)/SV40 transgenic mice model | Dose-dependent manner (Low-10 μmol/L and high-40 μmol/L concentration) | Suppression of androgen-dependent transactivation of AR Reduction of the relation between AR and heat shock protein 90 Increase the association of AR with the E3 ligase MDM2 | [23] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akkol, E.K.; Tatlı, I.I.; Karatoprak, G.Ş.; Ağar, O.T.; Yücel, Ç.; Sobarzo-Sánchez, E.; Capasso, R. Is Emodin with Anticancer Effects Completely Innocent? Two Sides of the Coin. Cancers 2021, 13, 2733. https://doi.org/10.3390/cancers13112733
Akkol EK, Tatlı II, Karatoprak GŞ, Ağar OT, Yücel Ç, Sobarzo-Sánchez E, Capasso R. Is Emodin with Anticancer Effects Completely Innocent? Two Sides of the Coin. Cancers. 2021; 13(11):2733. https://doi.org/10.3390/cancers13112733
Chicago/Turabian StyleAkkol, Esra Küpeli, Iffet Irem Tatlı, Gökçe Şeker Karatoprak, Osman Tuncay Ağar, Çiğdem Yücel, Eduardo Sobarzo-Sánchez, and Raffaele Capasso. 2021. "Is Emodin with Anticancer Effects Completely Innocent? Two Sides of the Coin" Cancers 13, no. 11: 2733. https://doi.org/10.3390/cancers13112733
APA StyleAkkol, E. K., Tatlı, I. I., Karatoprak, G. Ş., Ağar, O. T., Yücel, Ç., Sobarzo-Sánchez, E., & Capasso, R. (2021). Is Emodin with Anticancer Effects Completely Innocent? Two Sides of the Coin. Cancers, 13(11), 2733. https://doi.org/10.3390/cancers13112733