
Suppression of Breast Cancer by Small Molecules That Block the Prolactin Receptor

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
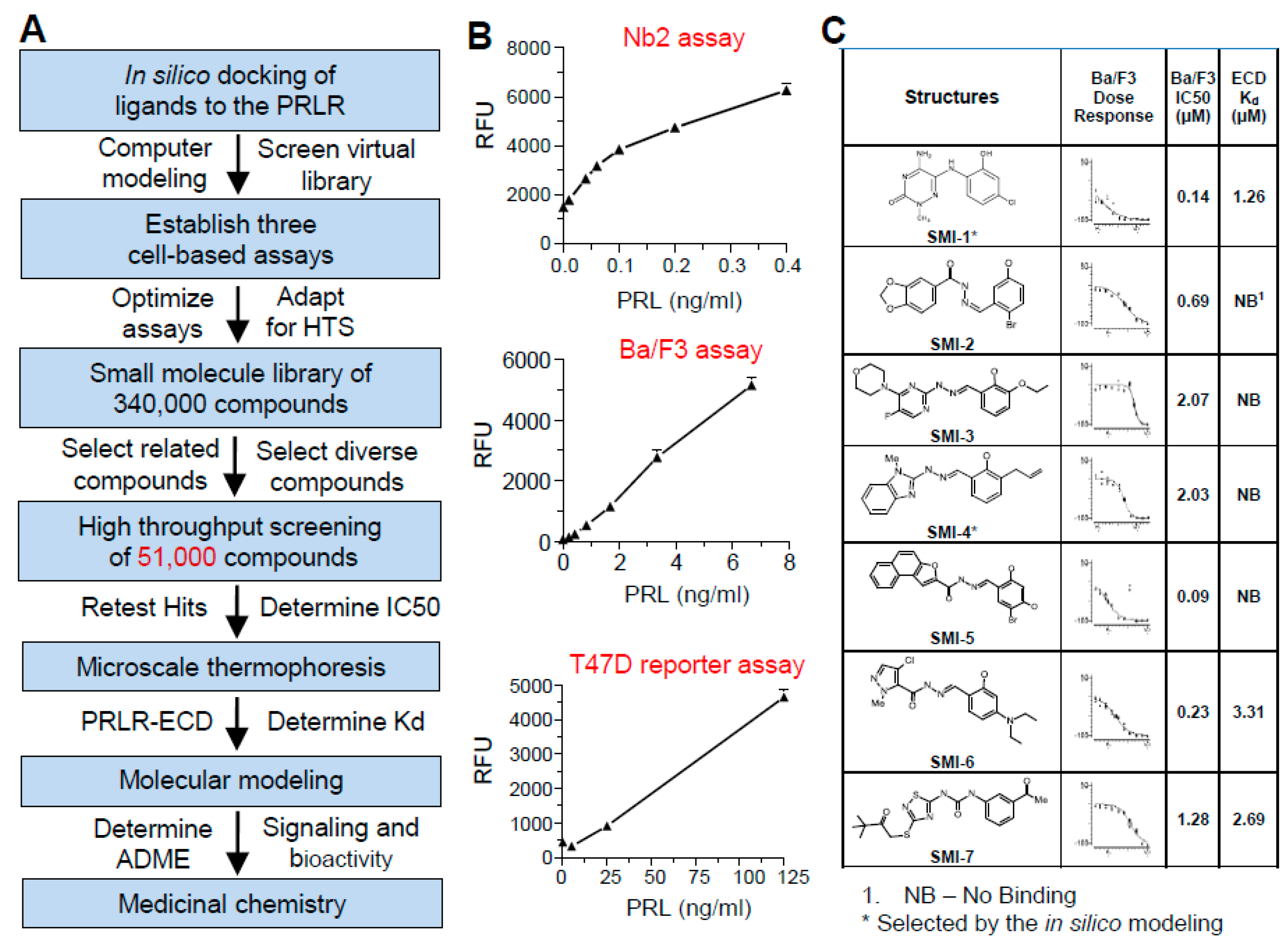
2.1. Strategy for HTS
2.2. Rationale for Using Three Sequential Bioassays
2.3. Analysis of Binding Affinity of the SMIs to PRLR-ECD by Microscale Thermophoresis (MST)
2.4. Selection of Compounds for Progression
2.5. Both SMIs Antagonize PRL-Induced Cell Invasion
2.6. Both SMIs Blocked Autocrine PRL-Induced Lymphocyte Proliferation
2.7. The SMIs Antagonize PRL-Induced JAK2 Phosphorylation
2.8. Progression with SMI-6
2.9. SMI-6 Has High Selectivity
2.10. Effects of SMI-6 on Various BCC and Normal Human Cells
2.11. Analysis of In Vitro and In Vivo Toxicity of the SMIs
2.12. Generation and Characterization of Doxycycline-Regulated hPRL-Producing MDA-MB-468 Cells
2.13. SMI-6 Causes Rapid and Dramatic Suppression of Tumor Growth In Vivo
3. Discussion
4. Methods
4.1. Animals
Mouse Xenograft Studies
4.2. Reagents
4.3. Cell Culture
4.4. Generation of Genetically-Modified Cell Lines
4.4.1. T47D-GAS/luc Cells Have a PRL-Responsive Luciferase Reporter
4.4.2. T47D PRLR−/− Cells Are PRLR Null
4.4.3. MDA-MB-468-CW Cells Are Doxycycline-Regulated PRL Expressing Cells
4.4.4. Culture Conditions
4.5. PRL Determination by Nb2 Cells
4.6. Small Molecule Compound Library and HTS Facilities
4.6.1. In Silico Modeling
4.6.2. Assay Adaptation for HTS
4.7. Microscale Thermophoresis for Analysis of Receptor Binding
4.8. Molecular Docking Simulation
4.9. Signaling Assays
4.10. Cell Proliferation
4.11. EdU Incorporation
4.12. Immunoprecipitation and Western Blots
4.13. Invasion Assay
4.14. Lactate Dehydrogenase (LDH) Cytotoxicity Assay
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cintolo-Gonzalez, J.A.; Braun, D.; Blackford, A.L.; Mazzola, E.; Acar, A.; Plichta, J.K.; Griffin, M.; Hughes, K.S. Breast cancer risk models: A comprehensive overview of existing models, validation, and clinical applications. Breast Cancer Res. Treat. 2017, 164, 263–284. [Google Scholar] [CrossRef]
- Zhou, Z.; Tang, D.H.; Xie, J.; Ayyagari, R.; Wu, E.; Niravath, P.A. Systematic Literature Review of the Impact of Endocrine Monotherapy and in Combination with Targeted Therapy on Quality of Life of Postmenopausal Women with HR+/HER2− Advanced Breast Cancer. Adv. Ther. 2017, 34, 2566–2584. [Google Scholar] [CrossRef]
- Witzel, I.; Oliveira-Ferrer, L.; Pantel, K.; Muller, V.; Wikman, H. Breast cancer brain metastases: Biology and new clinical perspectives. Breast Cancer Res. 2016, 18, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Ben-Jonathan, N.; LaPensee, C.R.; LaPensee, E.W. What can we learn from rodents about prolactin in humans? Endocr. Rev. 2008, 29, 1–41. [Google Scholar] [CrossRef]
- Marano, R.J.; Ben-Jonathan, N. Minireview: Extrapituitary prolactin: An update on the distribution, regulation, and functions. Mol. Endocrinol. 2014, 28, 622–633. [Google Scholar] [CrossRef]
- Gellersen, B.; Kempf, R.; Telgmann, R.; DiMattia, G.E. Nonpituitary human prolactin gene transcription is independent of Pit-1 and differentially controlled in lymphocytes and in endometrial stroma. Mol. Endocrinol. 1994, 8, 356–373. [Google Scholar] [PubMed]
- Brooks, C.L. Molecular mechanisms of prolactin and its receptor. Endocr. Rev. 2012, 33, 504–525. [Google Scholar] [CrossRef]
- Goffin, V.; Touraine, P. The prolactin receptor as a therapeutic target in human diseases: Browsing new potential indications. Expert Opin. Ther. Targets 2015, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chilton, B.S.; Hewetson, A. Prolactin and growth hormone signaling. Curr. Top. Dev. Biol. 2005, 68, 1–23. [Google Scholar]
- Faupel-Badger, J.M.; Duggan, M.A.; Sherman, M.E.; Garcia-Closas, M.; Yang, X.R.; Lissowska, J.; Brinton, L.A.; Peplonska, B.; Vonderhaar, B.K.; Figueroa, J.D. Prolactin receptor expression and breast cancer: Relationships with tumor characteristics among pre- and post-menopausal women in a population-based case-control study from Poland. Horm. Cancer 2014, 5, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Tworoger, S.S.; Eliassen, A.H.; Zhang, X.; Qian, J.; Sluss, P.M.; Rosner, B.A.; Hankinson, S.E. A 20-year prospective study of plasma prolactin as a risk marker of breast cancer development. Cancer Res. 2013, 73, 4810–4819. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.N.; Bu, W.; Hein, S.; García, S.; Camacho, L.; Xue, L.; Qin, L.; Nagi, C.; Hilsenbeck, S.G.; Kapali, J.; et al. Hyperprolactinemia-inducing antipsychotics increase breast cancer risk by activating JAK-STAT5 in precancerous lesions. Breast Cancer Res. 2018, 20, 42. [Google Scholar] [CrossRef] [PubMed]
- Ben-Jonathan, N.; Liby, K.; McFarland, M.; Zinger, M. Prolactin as an autocrine/paracrine growth factor in human cancer. Trends Endocrinol. Metab. 2002, 13, 245–250. [Google Scholar] [CrossRef]
- Clevenger, C.V.; Furth, P.A.; Hankinson, S.E.; Schuler, L.A. The role of prolactin in mammary carcinoma. Endocr. Rev. 2003, 24, 1–27. [Google Scholar] [CrossRef]
- Wu, Z.S.; Yang, K.; Wan, Y.; Qian, P.X.; Perry, J.K.; Chiesa, J.; Mertani, H.C.; Zhu, T.; Lobie, P.E. Tumor expression of human growth hormone and human prolactin predict a worse survival outcome in patients with mammary or endometrial carcinoma. J. Clin. Endocrinol. Metab. 2011, 96, E1619–E1629. [Google Scholar] [CrossRef]
- Zinger, M.; McFarland, M.; Ben-Jonathan, N. Prolactin expression and secretion by human breast glandular and adipose tissue explants. J. Clin. Endocrinol. Metab. 2003, 88, 689–696. [Google Scholar] [CrossRef][Green Version]
- Liby, K.; Neltner, B.; Mohamet, L.; Menchen, L.; Ben-Jonathan, N. Prolactin overexpression by MDA-MB-435 human breast cancer cells accelerates tumor growth. Breast Cancer Res. Treat. 2003, 79, 241–252. [Google Scholar] [CrossRef]
- LaPensee, E.W.; Ben-Jonathan, N. Novel roles of prolactin and estrogens in breast cancer: Resistance to chemotherapy. Endocr. Relat. Cancer 2010, 17, R91–R107. [Google Scholar] [CrossRef]
- Costa, R.; Santa-Maria, C.A.; Scholtens, D.M.; Jain, S.; Flaum, L.; Gradishar, W.J.; Clevenger, C.V.; Kaklamani, V.G. A pilot study of cabergoline for the treatment of metastatic breast cancer. Breast Cancer Res. Treat. 2017, 165, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Goffin, V.; Bernichtein, S.; Touraine, P.; Kelly, P.A. Development and potential clinical uses of human prolactin receptor antagonists. Endocr. Rev. 2005, 26, 400–422. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S.; Rendahl, K.G.; Karim, C.; Embry, M.G.; Ghoddusi, M.; Holash, J.; Fanidi, A.; Abrams, T.J.; Abraham, J.A. Neutralization of prolactin receptor function by monoclonal antibody LFA102, a novel potential therapeutic for the treatment of breast cancer. Mol. Cancer Ther. 2013, 12, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Lemech, C.; Woodward, N.; Chan, N.; Mortimer, J.; Naumovski, L.; Nuthalapati, S.; Tong, B.; Jiang, F.; Ansell, P.; Ratajczak, C.K.; et al. A first-in-human, phase 1, dose-escalation study of ABBV-176, an antibody-drug conjugate targeting the prolactin receptor, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1815–1825. [Google Scholar] [CrossRef]
- Jin, L.; Wang, W.; Fang, G. Targeting protein-protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Utama, F.E.; LeBaron, M.J.; Neilson, L.M.; Sultan, A.S.; Parlow, A.F.; Wagner, K.U.; Rui, H. Human prolactin receptors are insensitive to mouse prolactin: Implications for xenotransplant modeling of human breast cancer in mice. J. Endocrinol. 2006, 188, 589–601. [Google Scholar] [CrossRef]
- Rowe, R.C.; Cowden, E.A.; Faiman, C.; Friesen, H.G. Correlation of Nb2 bioassay and radioimmunoassay values for human serum prolactin. J. Clin. Endocrinol. Metab. 1983, 57, 942–946. [Google Scholar] [CrossRef]
- Ali, S.; Pellegrini, I.; Kelly, P.A. A prolactin-dependent immune cell line (Nb2) expresses a mutant form of prolactin receptor. J. Biol. Chem. 1991, 266, 20110–20117. [Google Scholar] [CrossRef]
- Goffin, V.; Bogorad, R.L.; Touraine, P. Identification of gain-of-function variants of the human prolactin receptor. Methods Enzymol. 2010, 484, 329–355. [Google Scholar] [PubMed]
- Meng, J.; Tsai-Morris, C.H.; Dufau, M.L. Human prolactin receptor variants in breast cancer: Low ratio of short forms to the long-form human prolactin receptor associated with mammary carcinoma. Cancer Res. 2004, 64, 5677–5682. [Google Scholar] [CrossRef][Green Version]
- Johnson, K.A.; Brown, P.H. Drug development for cancer chemoprevention: Focus on molecular targets. Semin. Oncol. 2010, 37, 345–358. [Google Scholar] [CrossRef]
- Dagil, R.; Knudsen, M.J.; Olsen, J.G.; O’Shea, C.; Franzmann, M.; Goffin, V.; Teilum, K.; Breinholt, J.; Kragelund, B.B. The WSXWS motif in cytokine receptors is a molecular switch involved in receptor activation: Insight from structures of the prolactin receptor. Structure 2012, 20, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Matera, L.; Cutufia, M.; Geuna, M.; Contarini, M.; Buttiglieri, S.; Galin, S.; Fazzari, A.; Cavaliere, C. Prolactin is an autocrine growth factor for the Jurkat human T-leukemic cell line. J. Neuroimmunol. 1997, 79, 12–21. [Google Scholar] [CrossRef]
- Goffin, V.; Kelly, P.A. The prolactin/growth hormone receptor family: Structure/function relationships. J. Mammary Gland. Biol. Neoplasia 1997, 2, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sun, D.; Jiang, J.; Deng, L.; Zhang, Y.; Yu, H.; Bahl, D.; Langenheim, J.F.; Chen, W.Y.; Fuchs, S.Y.; et al. The role of prolactin receptor in GH signaling in breast cancer cells. Mol. Endocrinol. 2013, 27, 266–279. [Google Scholar] [CrossRef] [PubMed]
- LaPensee, E.W.; Schwemberger, S.J.; LaPensee, C.R.; Bahassi, e.M.; Afton, S.E.; Ben-Jonathan, N. Prolactin confers resistance against cisplatin in breast cancer cells by activating glutathione-S-transferase. Carcinogenesis 2009, 30, 1298–1304. [Google Scholar] [CrossRef]
- O’Leary, K.A.; Rugowski, D.E.; Sullivan, R.; Schuler, L.A. Prolactin cooperates with loss of p53 to promote claudin-low mammary carcinomas. Oncogene 2014, 33, 3075–3082. [Google Scholar] [CrossRef] [PubMed]
- Ueda, E.K.; Huang, K.; Nguyen, V.; Ferreira, M.; Andre, S.; Walker, A.M. Distribution of prolactin receptors suggests an intraductal role for prolactin in the mouse and human mammary gland, a finding supported by analysis of signaling in polarized monolayer cultures. Cell Tissue Res. 2011, 346, 175–189. [Google Scholar] [CrossRef]
- Rider, L.; Oladimeji, P.; Diakonova, M. PAK1 regulates breast cancer cell invasion through secretion of matrix metalloproteinases in response to prolactin and three-dimensional collagen IV. Mol. Endocrinol. 2013, 27, 1048–1064. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.; Touraine, P.; Goffin, V. Prolactin and human tumourogenesis. J. Neuroendocrinol. 2010, 22, 771–777. [Google Scholar] [PubMed]
- Clevenger, C.V.; Gadd, S.L.; Zheng, J. New mechanisms for PRLr action in breast cancer. Trends Endocrinol. Metab. 2009, 20, 223–229. [Google Scholar] [CrossRef]
- O’Leary, K.A.; Rugowski, D.E.; Shea, M.P.; Sullivan, R.; Moser, A.R.; Schuler, L.A. Prolactin synergizes with canonical Wnt signals to drive development of ER+ mammary tumors via activation of the Notch pathway. Cancer Lett. 2021, 503, 231–239. [Google Scholar] [CrossRef]
- Shams, A.; Binothman, N.; Boudreault, J.; Wang, N.; Shams, F.; Hamam, D.; Tian, J.; Moamer, A.; Dai, M.; Lebrun, J.J.; et al. Prolactin receptor-driven combined luminal and epithelial differentiation in breast cancer restricts plasticity, stemness, tumorigenesis and metastasis. Oncogenesis 2021, 10, 10–18. [Google Scholar] [CrossRef]
- Gribe, M.J.; Zot, P.; Olex, A.L.; Hedrick, S.E.; Harrell, J.C.; Woock, A.E.; Idowu, M.O.; Clevenger, C.V. The human intermediate prolactin receptor is a mammary proto-oncogene. NPJ Breast Cancer 2021, 7, 37–48. [Google Scholar] [CrossRef]
- Steven, A.; Seliger, B. The Role of Immune Escape and Immune Cell Infiltration in Breast Cancer. Breast Care (Basel) 2018, 13, 16–21. [Google Scholar] [CrossRef]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef] [PubMed]
- Sozio, P.; Cerasa, L.S.; Abbadessa, A.; Di, S.A. Designing prodrugs for the treatment of Parkinson’s disease. Expert Opin. Drug Discov. 2012, 7, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, E.; Alexander, S.; Lieber, S.; Tarplin, S.; Jenkins, L.; Pang, L.; Heger, C.D.; Goldsmith, P.; Vonderhaar, B.K. Characterization of ductal and lobular breast carcinomas using novel prolactin receptor isoform specific antibodies. BMC Cancer 2010, 10, 678. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.T.; Mullan, R.J.; A Lane, M.; Hazem, A.; Prasad, C.; Gathaiya, N.W.; Fernández-Balsells, M.M.; Bagatto, A.; Coto-Yglesias, F.; Carey, J.; et al. Treatment of hyperprolactinemia: A systematic review and meta-analysis. Syst. Rev. 2012, 1, 33–42. [Google Scholar] [CrossRef]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009, 1, 15–20. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borcherding, D.C.; Hugo, E.R.; Fox, S.R.; Jacobson, E.M.; Hunt, B.G.; Merino, E.J.; Ben-Jonathan, N. Suppression of Breast Cancer by Small Molecules That Block the Prolactin Receptor. Cancers 2021, 13, 2662. https://doi.org/10.3390/cancers13112662
Borcherding DC, Hugo ER, Fox SR, Jacobson EM, Hunt BG, Merino EJ, Ben-Jonathan N. Suppression of Breast Cancer by Small Molecules That Block the Prolactin Receptor. Cancers. 2021; 13(11):2662. https://doi.org/10.3390/cancers13112662
Chicago/Turabian StyleBorcherding, Dana C., Eric R. Hugo, Sejal R. Fox, Eric M. Jacobson, Brian G. Hunt, Edward J. Merino, and Nira Ben-Jonathan. 2021. "Suppression of Breast Cancer by Small Molecules That Block the Prolactin Receptor" Cancers 13, no. 11: 2662. https://doi.org/10.3390/cancers13112662
APA StyleBorcherding, D. C., Hugo, E. R., Fox, S. R., Jacobson, E. M., Hunt, B. G., Merino, E. J., & Ben-Jonathan, N. (2021). Suppression of Breast Cancer by Small Molecules That Block the Prolactin Receptor. Cancers, 13(11), 2662. https://doi.org/10.3390/cancers13112662