
Emerging Next-Generation Target for Cancer Immunotherapy Research: The Orphan Nuclear Receptor NR2F6


Abstract
Simple Summary
Abstract
1. Introduction—T Lymphocytes in Anti-Tumor Immunity
2. Current State of Tumor Immunology
2.1. Cancer Vaccination: A Strategy to Enhance T Lymphocyte-Mediated Anti-Tumor Immunity
2.2. Modulating T Lymphocyte Activation: Checkpoint Inhibitors, Co-Stimulatory Receptors and CAR-T
3. Beyond Current Immune Checkpoint Therapies
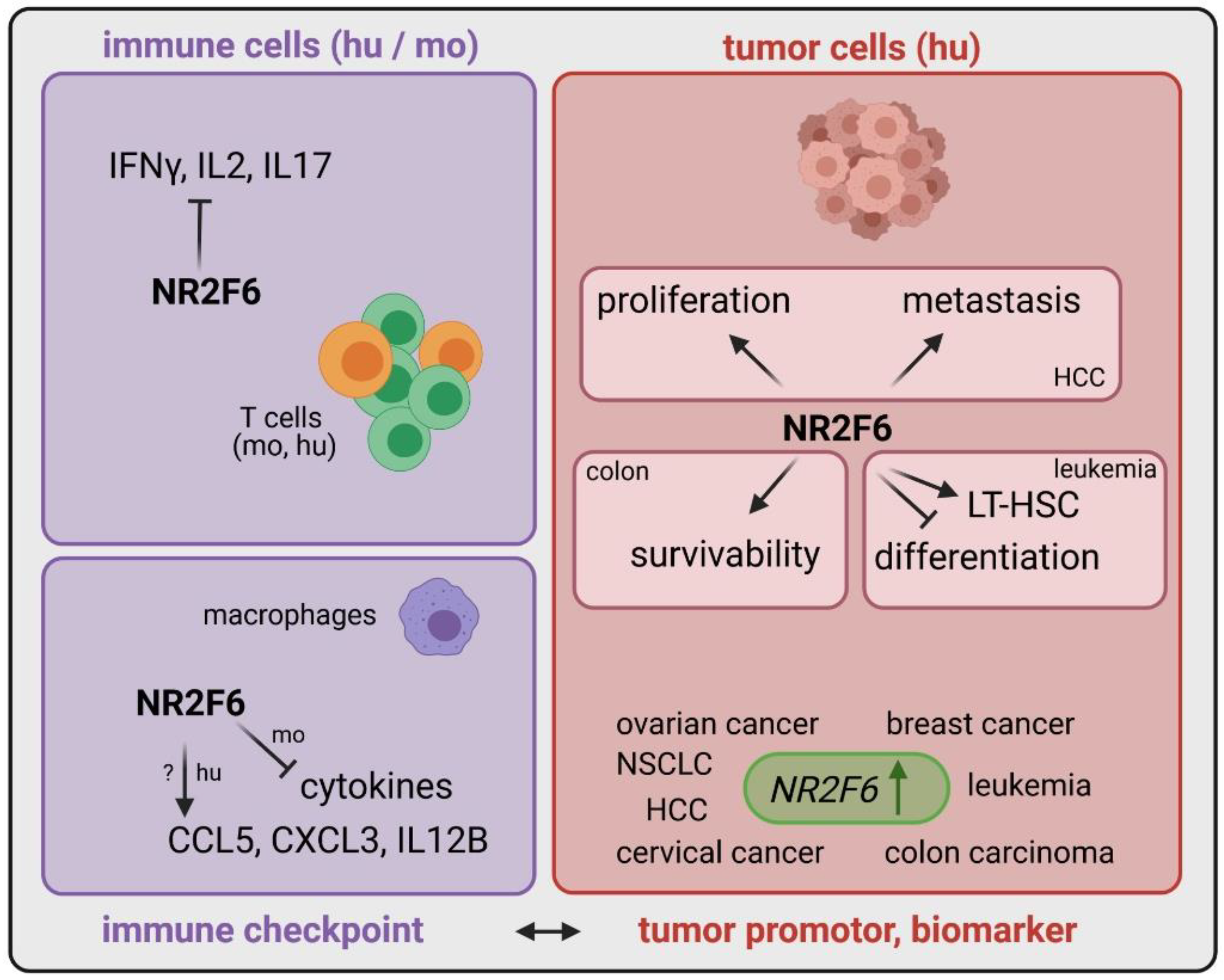
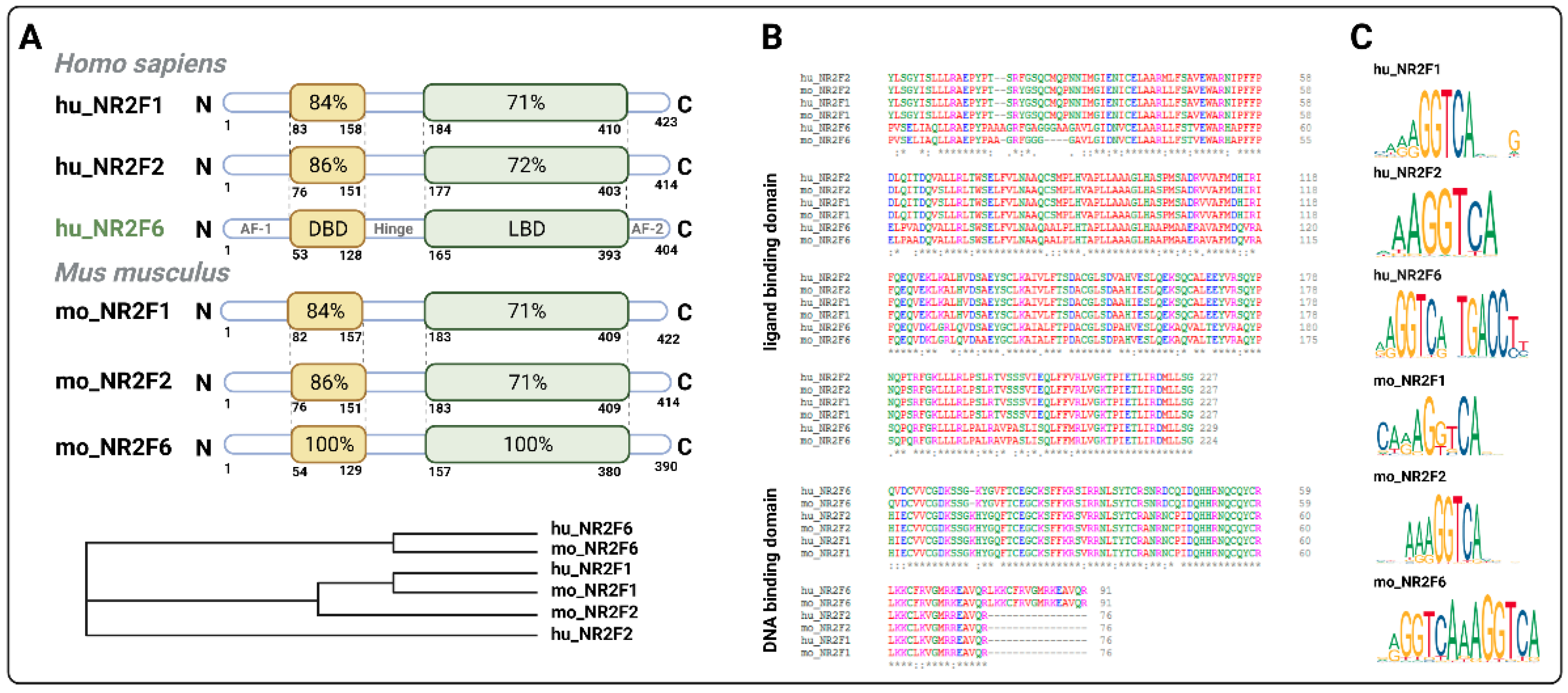
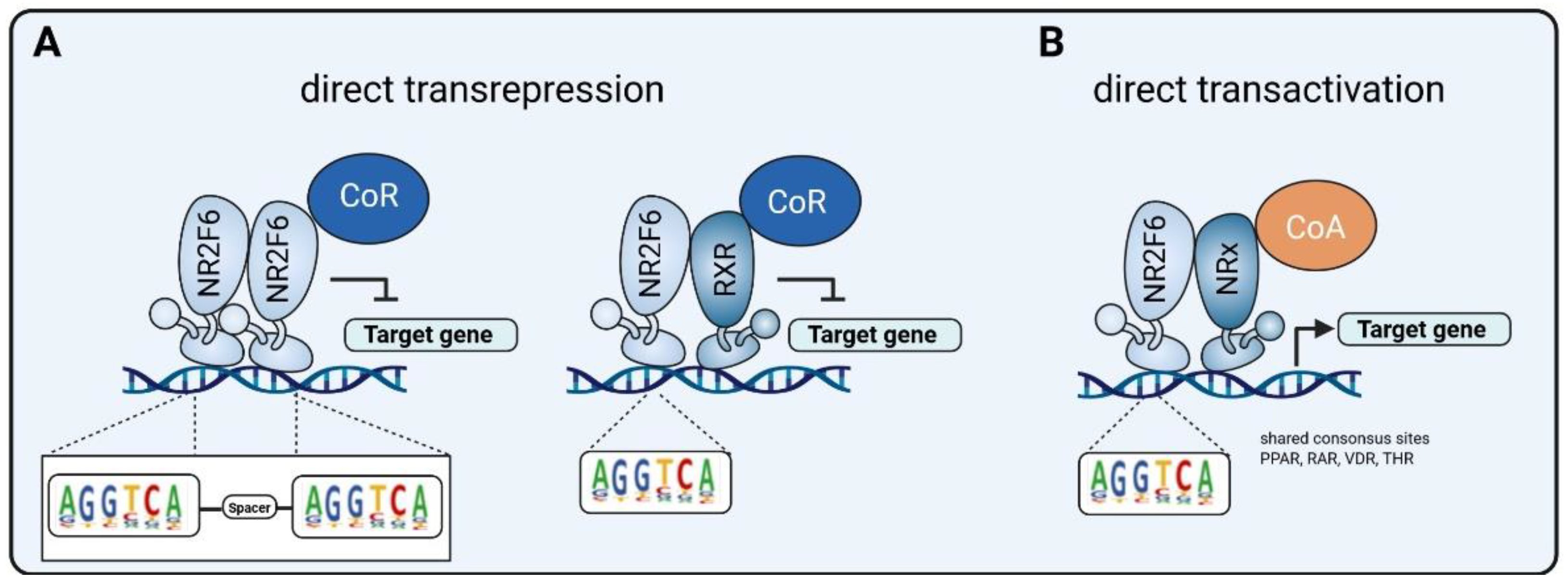
3.1. Intracellular Target NR2F6 in Both Immune Cells and Tumor Cells
3.2. Inducible Immune Checkpoint at the Tumor Site May Boost a Localized Effector T Cell Response with Fewer Systemic Irae
3.3. Double Score Principle of NR2F6 Antagonists
4. Outlook
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Eyles, J.; Puaux, A.L.; Wang, X.; Toh, B.; Prakash, C.; Hong, M.; Tan, T.G.; Zheng, L.; Ong, L.C.; Jin, Y.; et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Investig. 2010, 120, 2030–2039. [Google Scholar] [CrossRef]
- Romero, I.; Garrido, C.; Algarra, I.; Collado, A.; Garrido, F.; Garcia-Lora, A.M. T lymphocytes restrain spontaneous metastases in permanent dormancy. Cancer Res. 2014, 74, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Lacroix-Triki, M.; Joensuu, H.; Dieci, M.V.; Badve, S.; Demaria, S.; et al. Tumor-infiltrating lymphocytes and prognosis: A pooled individual patient analysis of early-stage triple-negative breast cancers. J. Clin. Oncol. 2019, 37, 559–569. [Google Scholar] [CrossRef]
- Oh, D.Y.; Kwek, S.S.; Raju, S.S.; Li, T.; McCarthy, E.; Chow, E.; Aran, D.; Ilano, A.; Pai, C.C.S.; Rancan, C.; et al. Intratumoral CD4+ T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 2020, 181, 1612–1625.e13. [Google Scholar] [CrossRef]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef]
- Fauskanger, M.; Haabeth, O.A.W.; Skjeldal, F.M.; Bogen, B.; Tveita, A.A. Tumor killing by CD4+ T cells is mediated via induction of inducible nitric oxide synthase-dependent macrophage cytotoxicity. Front. Immunol. 2018, 9, 1684. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Jordanova, E.S.; Gorter, A.; Ayachi, O.; Prins, F.; Durrant, L.G.; Kenter, G.G.; Van Der Burg, S.H.; Fleuren, G.J. Human leukocyte antigen class I, MHC class I chain-related molecule A, and CD8+/regulatory T cell ratio: Which variable determines survival of cervical cancer patients? Clin. Cancer Res. 2008, 14, 2028–2035. [Google Scholar] [CrossRef]
- Onda, M.; Kobayashi, K.; Pastan, I. Depletion of regulatory T cells in tumors with an anti-CD25 immunotoxin induces CD8 T cell-mediated systemic antitumor immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 4575–4582. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.P.; Gubin, M.M.; Schreiber, R.D.; States, U. Therapeutically Induced Immune Responses to Cancer. Adv. Immunol. 2016, 130, 25–74. [Google Scholar] [CrossRef] [PubMed]
- Jou, J.; Harrington, K.J.; Zocca, M.B.; Ehrnrooth, E.; Cohen, E.E.W. The changing landscape of therapeutic cancer vaccines-novel platforms and neoantigen identification. Clin. Cancer Res. 2021, 27, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Hu, Z.; Leet, D.E.; Allesøe, R.L.; Oliveira, G.; Li, S.; Luoma, A.M.; Liu, J.; Forman, J.; Huang, T.; Iorgulescu, J.B.; et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 2021, 27. [Google Scholar] [CrossRef] [PubMed]
- D’Alise, A.M.; Leoni, G.; Cotugno, G.; Troise, F.; Langone, F.; Fichera, I.; De Lucia, M.; Avalle, L.; Vitale, R.; Leuzzi, A.; et al. Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Poran, A.; Scherer, J.; Bushway, M.E.; Besada, R.; Balogh, K.N.; Wanamaker, A.; Williams, R.G.; Prabhakara, J.; Ott, P.A.; Hu-Lieskovan, S.; et al. Combined TCR Repertoire Profiles and Blood Cell Phenotypes Predict Melanoma Patient Response to Personalized Neoantigen Therapy plus Anti-PD-1. Cell Rep. Med. 2020, 1, 100141. [Google Scholar] [CrossRef]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Choi, Y.; Shi, Y.; Haymaker, C.L.; Naing, A.; Ciliberto, G.; Hajjar, J. T cell agonists in cancer immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Skokos, D.; Waite, J.C.; Haber, L.; Crawford, A.; Hermann, A.; Ullman, E.; Slim, R.; Godin, S.; Ajithdoss, D.; Ye, X.; et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 2020, 12, eaaw7888. [Google Scholar] [CrossRef]
- Mitchison, N. Studies on the immunological response to foreign tumor transplants in the mouse. J. Exp. Med. 1955, 102, 157–177. [Google Scholar] [CrossRef]
- Fefer, A. Immunotherapy and Chemotherapy of Moloney Sarcoma Virus-induced Tumors in Mice. Cancer Res. 1969, 29, 2177–2183. [Google Scholar]
- Claus, C.; Ferrara, C.; Xu, W.; Sam, J.; Lang, S.; Uhlenbrock, F.; Albrecht, R.; Herter, S.; Schlenker, R.; Hösser, T.; et al. Tumor-Targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-The-shelf therapy. Sci. Transl. Med. 2019, 11, eaaav5989. [Google Scholar] [CrossRef]
- You, G.; Lee, Y.; Kang, Y.W.; Park, H.W.; Park, K.; Kim, H.; Kim, Y.M.; Kim, S.; Kim, J.H.; Moon, D.; et al. B7-H3×4-1BB bispecific antibody augments antitumor immunity by enhancing terminally differentiated CD8+ tumor-infiltrating lymphocytes. Sci. Adv. 2021, 7, eaax3160. [Google Scholar] [CrossRef] [PubMed]
- Avanzi, M.P.; Brentjens, R.J. Emerging role of CAR-T cells in Non-Hodgkin’s Lymphoma. JNCCN J. Natl. Compr. Cancer Netw. 2017, 15, 1429–1437. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Long, H.; Zheng, Q.; Bo, X.; Xiao, X.; Li, B. Circular RNA circRHOT1 promotes hepatocellular carcinoma progression by initiation of NR2F6 expression. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef]
- Li, X.; Jiao, S.; Sun, H.; Xue, J.; Zhao, W.; Fan, L.; Wu, G.; Fang, J. The orphan nuclear receptor EAR2 is overexpressed in colorectal cancer and it regulates survivability of colon cancer cells. Cancer Lett. 2011, 309, 137–144. [Google Scholar] [CrossRef]
- Hermann-Kleiter, N.; Gruber, T.; Lutz-Nicoladoni, C.; Thuille, N.; Fresser, F.; Labi, V.; Schiefermeier, N.; Warnecke, M.; Huber, L.; Villunger, A.; et al. The Nuclear Orphan Receptor NR2F6 Suppresses Lymphocyte Activation and T Helper 17-Dependent Autoimmunity. Immunity 2008, 29, 205–216. [Google Scholar] [CrossRef]
- Ichim, C.V.; Atkins, H.L.; Iscove, N.N.; Wells, R.A. Identification of a role for the nuclear receptor EAR-2 in the maintenance of clonogenic status within the leukemia cell hierarchy. Leuk. Off. J. Leuk. Soc. Am. Leuk. Res. Fund. UK 2011, 25, 1687–1696. [Google Scholar] [CrossRef]
- Niu, C.; Sun, X.; Zhang, W.; Li, H.; Xu, L.; Li, J.; Xu, B.; Zhang, Y. NR2F6 expression correlates with pelvic lymph node metastasis and poor prognosis in early-stage cervical cancer. Int. J. Mol. Sci. 2016, 17, 1694. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, T.; Liu, X.L. DDA1 is induced by NR2F6 in ovarian cancer and predicts poor survival outcome. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1206–1213. [Google Scholar]
- Li, H.; Zhang, W.; Niu, C.; Lin, C.; Wu, X.; Jian, Y.; Li, Y.; Ye, L.; Dai, Y.; Ouyang, Y.; et al. Nuclear orphan receptor NR2F6 confers cisplatin resistance in epithelial ovarian cancer cells by activating the Notch3 signaling pathway. Int. J. Cancer 2019, 145, 1921–1934. [Google Scholar] [CrossRef] [PubMed]
- Muscat, G.E.O.; Eriksson, N.A.; Byth, K.; Loi, S.; Graham, D.; Jindal, S.; Davis, M.J.; Clyne, C.; Funder, J.W.; Simpson, E.R.; et al. Research resource: Nuclear receptors as transcriptome: Discriminant and prognostic value in breast cancer. Mol. Endocrinol. 2013, 27, 350–365. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Wang, X.; Fredericksen, Z.S.; McGuffog, L.; Tarrell, R.; Sinilnikova, O.M.; Healey, S.; Morrison, J.; Kartsonaki, C.; Lesnick, T.; et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat. Genet. 2010, 42, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Xiao, L.; Zhou, Z.; Zhu, Y.; Tian, G.; Ren, S. MiR-142-3p suppresses the proliferation, migration and invasion through inhibition of NR2F6 in lung adenocarcinoma. Hum. Cell 2019, 32, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Hermann-Kleiter, N.; Meisel, M.; Fresser, F.; Thuille, N.; Müller, M.; Roth, L.; Katopodis, A.; Baier, G. Nuclear orphan receptor NR2F6 directly antagonizes NFAT and RORγt binding to the Il17a promoter. J. Autoimmun. 2012, 39, 428–440. [Google Scholar] [CrossRef]
- Hermann-Kleiter, N.; Klepsch, V.; Wallner, S.; Siegmund, K.; Klepsch, S.; Tuzlak, S.; Villunger, A.; Kaminski, S.; Pfeifhofer-Obermair, C.; Gruber, T.; et al. The Nuclear Orphan Receptor NR2F6 Is a Central Checkpoint for Cancer Immune Surveillance. Cell Rep. 2015, 12, 2072–2085. [Google Scholar] [CrossRef] [PubMed]
- Klepsch, V.; Hermann-Kleiter, N.; Do-Dinh, P.; Jakic, B.; Offermann, A.; Efremova, M.; Sopper, S.; Rieder, D.; Krogsdam, A.; Gamerith, G.; et al. Nuclear receptor NR2F6 inhibition potentiates responses to PD-L1/PD-1 cancer immune checkpoint blockade. Nat. Commun. 2018, 9, 1538. [Google Scholar] [CrossRef]
- Santoso, C.S.; Li, Z.; Lal, S.; Yuan, S.; Gan, K.A.; Agosto, L.M.; Liu, X.; Carrasco, P.S.; Sewell, J.A.; Henderson, A.; et al. Comprehensive mapping of the human cytokine gene regulatory network. Nucleic Acids Res. 2020, 48, 12055–12073. [Google Scholar] [CrossRef] [PubMed]
- Ichim, C.V.; Dervovic, D.D.; Chan, L.S.A.; Robertson, C.J.; Chesney, A.; Reis, M.D.; Wells, R.A. The orphan nuclear receptor EAR-2 (NR2F6) inhibits hematopoietic cell differentiation and induces myeloid dysplasia in vivo. Biomark. Res. 2018, 6. [Google Scholar] [CrossRef]
- Warnecke, M.; Oster, H.; Revelli, J.; Alvarez-bolado, G.; Eichele, G. Abnormal development of the locus impairs the functionality of the forebrain clock and affects nociception. Genes Dev. 2005, 2, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Jia, L.; Zhang, Z.; Xiang, L.; Yuan, Y.; Zheng, P.; Liu, B.; Ren, X.; Bian, H.; Xie, L.; et al. The Nuclear Orphan Receptor NR2F6 Promotes Hepatic Steatosis through Upregulation of Fatty Acid Transporter CD36. Adv. Sci. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, X.; Sigmund, C.D. Identification of a nuclear orphan receptor (Ear2) as a negative regulator of renin gene transcription. Circ. Res. 2003, 92, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Eckerle, S.; Brune, V.; Döring, C.; Tiacci, E.; Bohle, V.; Sundström, C.; Kodet, R.; Paulli, M.; Falini, B.; Klapper, W.; et al. Gene expression profiling of isolated tumour cells from anaplastic large cell lymphomas: Insights into its cellular origin, pathogenesis and relation to Hodgkin lymphoma. Leuk. Off. J. Leuk. Soc. Am. Leuk. Res. Fund. UK 2009, 23, 2129–2138. [Google Scholar] [CrossRef]
- Gu-Trantien, C.; Loi, S.; Garaud, S.; Equeter, C.; Libin, M.; De Wind, A.; Ravoet, M.; Buanec, H.L.; Sibille, C.; Manfouo-Foutsop, G.; et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. J. Clin. Investig. 2013, 123, 2873–2892. [Google Scholar] [CrossRef]
- Bassi, C.; Li, Y.T.; Khu, K.; Mateo, F.; Baniasadi, P.S.; Elia, A.; Mason, J.; Stambolic, V.; Pujana, M.A.; Mak, T.W.; et al. The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell Death Differ. 2016, 23, 1198–1208. [Google Scholar] [CrossRef]
- Cregan, S.; McDonagh, L.; Gao, Y.; Barr, M.P.; O’Byrne, K.J.; Finn, S.P.; Cuffe, S.; Gray, S.G. KAT5 (Tip60) is a potential therapeutic target in malignant pleural mesothelioma. Int. J. Oncol. 2016, 48, 1290–1296. [Google Scholar] [CrossRef]
- Shiota, M.; Yokomizo, A.; Masubuchi, D.; Tada, Y.; Inokuchi, J.; Eto, M.; Uchiumi, T.; Fujimoto, N.; Naito, S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 2010, 70, 540–554. [Google Scholar] [CrossRef]
- Yoest, J. Clinical features, predictive correlates, and pathophysiology of immune-related adverse events in immune checkpoint inhibitor treatments in cancer: A short review. ImmunoTargets Ther. 2017, 6, 73–82. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.; Hudson, M.; Fifi-Mah, A.; Ye, C. Immune-related adverse events associated with cancer immunotherapy: A review for the practicing rheumatologist. J. Rheumatol. 2020, 47, 166–175. [Google Scholar] [CrossRef]
- Adam, K.; Iuga, A.; Tocheva, A.S.; Mor, A. A novel mouse model for checkpoint inhibitor-induced adverse events. PLoS ONE 2021, 16, 1–14. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, C.M.; Qin, J.; Xu, M.; Kao, C.Y.; Shi, J.; You, E.; Gong, W.; Rosa, L.P.; Chase, P.; et al. Small-molecule inhibitor targeting orphan nuclear receptor COUP-TFII for prostate cancer treatment. Sci. Adv. 2020, 6, eaaz8031. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.D.; Sanchez, R.; Rahman, T.; Rodriguez-Tirado, C.; Moritsch, S.; Martinez, A.R.; Miles, B.; Farias, E.; Mezei, M.; Cheung, J.F.; et al. A specific agonist of the orphan nuclear receptor NR2F1 suppresses metastasis through the induction of cancer cell dormancy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory Action of Glucocorticoids—New Mechanisms for Old Drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef]
- Tang, K.; Tsai, S.Y.; Tsai, M.J. COUP-TFs and eye development. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 201–209. [Google Scholar] [CrossRef]
- Klepsch, V.; Pommermayr, M.; Humer, D.; Brigo, N.; Hermann-Kleiter, N.; Baier, G. Targeting the orphan nuclear receptor NR2F6 in T cells primes tumors for immune checkpoint therapy. Cell Commun. Signal. 2020, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Hutcheson, J.; Scatizzi, J.C.; Siddiqui, A.M.; Haines, G.K.; Wu, T.; Li, Q.Z.; Davis, L.S.; Mohan, C.; Perlman, H. Combined Deficiency of Proapoptotic Regulators Bim and Fas Results in the Early Onset of Systemic Autoimmunity. Immunity 2008, 28, 206–217. [Google Scholar] [CrossRef]
- Olson, W.J.; Jakic, B.; Labi, V.; Schoeler, K.; Kind, M.; Klepsch, V.; Baier, G.; Hermann-Kleiter, N. Orphan Nuclear Receptor NR2F6 Suppresses T Follicular Helper Cell Accumulation through Regulation of IL-21. Cell Rep. 2019, 28, 2878–2891.e5. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Expression of NR2F6 | Role of NR2F6 | Ref |
|---|---|---|---|
| Leukemia | upregulated in patients | elevated population of LT-HSC | [41] |
| Hepatocellular carcinoma | upregulated in patients | NR2F6 induces proliferation and metastasis via circRHOT1 and TIP60 | [38] |
| Colon carcinoma | upregulated in patients | Nr2f6 increases survivability via XIAP | [39] |
| Cervical cancer | upregulated in patients | correlation between metastasis, poor prognosis and NR2F6 expression | [42] |
| Ovarian cancer | upregulated in patients | DDA1 is induced by NR2F6 and predicts poor outcome | [43,44] |
| Breast cancer | upregulated in patients | n.d. | [45,46] |
| Lung cancer | upregulated in patients | MiR-142-3p inhibits proliferation, migration and invasion via NR2F6 inhibition | [47] |
| Effector T cells (mo and hu) | upregulated upon stimulation | transcriptional repressor directly antagonizing key cytokine gene loci | [40,48,49,50] |
| Macrophages (mo) | n.d. | transcriptional repressor of cytokines | [51] |
| Macrophages (hu) | n.d. | transcriptional activator of chemokines | [51] |
| Tumor cells | upregulated | important for proliferation, metastasis, survivability | [38,39,42,47,52] |
| Neurons (locus coeruleus) | n.d. | control of circadian clock | [53] |
| Hepatocytes (mo and hu) | upregulated | hepatic steatosis promoted by NR2F6 | [54] |
| Kidney | n.d. | NR2F6 as a negative regulator of renin gene transcription | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klepsch, V.; Siegmund, K.; Baier, G. Emerging Next-Generation Target for Cancer Immunotherapy Research: The Orphan Nuclear Receptor NR2F6. Cancers 2021, 13, 2600. https://doi.org/10.3390/cancers13112600
Klepsch V, Siegmund K, Baier G. Emerging Next-Generation Target for Cancer Immunotherapy Research: The Orphan Nuclear Receptor NR2F6. Cancers. 2021; 13(11):2600. https://doi.org/10.3390/cancers13112600
Chicago/Turabian StyleKlepsch, Victoria, Kerstin Siegmund, and Gottfried Baier. 2021. "Emerging Next-Generation Target for Cancer Immunotherapy Research: The Orphan Nuclear Receptor NR2F6" Cancers 13, no. 11: 2600. https://doi.org/10.3390/cancers13112600
APA StyleKlepsch, V., Siegmund, K., & Baier, G. (2021). Emerging Next-Generation Target for Cancer Immunotherapy Research: The Orphan Nuclear Receptor NR2F6. Cancers, 13(11), 2600. https://doi.org/10.3390/cancers13112600