
A Prospective Feasibility Trial to Challenge Patient–Derived Pancreatic Cancer Organoids in Predicting Treatment Response

 ,
,  , , , ,
, , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection and Ethics Statement
2.2. Tumor Specimen Collection
2.3. Isolation and Culture of Patient-Derived Organoids
2.4. Pharmacotyping
2.5. Immunostaining
2.6. Patients’ Evaluation of Therapeutic Response
2.7. Linear Support Vector Machines
2.8. Statistical Analysis
3. Results
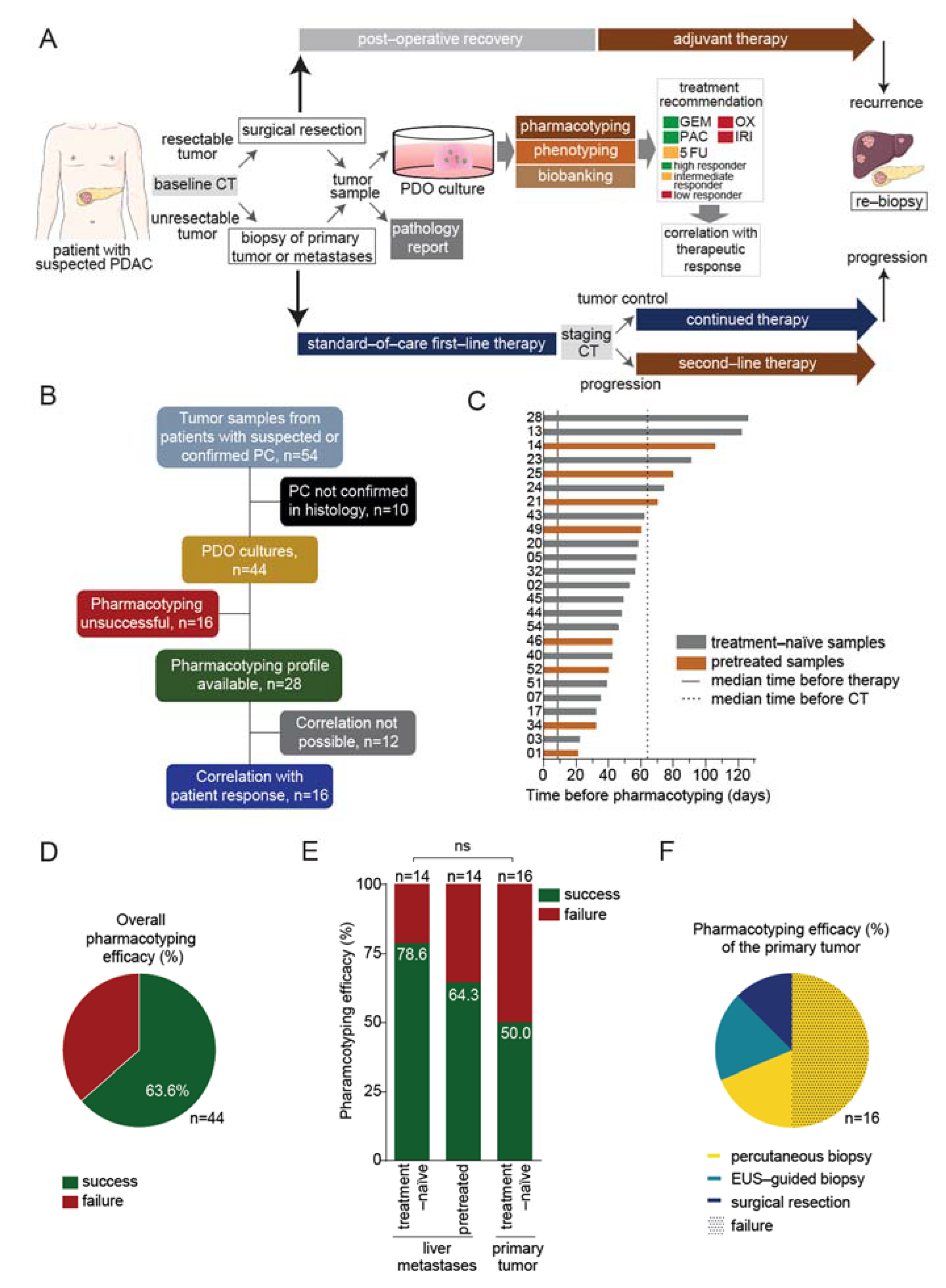
3.1. Prospective Feasibility Trial
3.2. Pharmacotyping of Patient-Derived Organoids Is Feasible in a Reasonable Time Frame
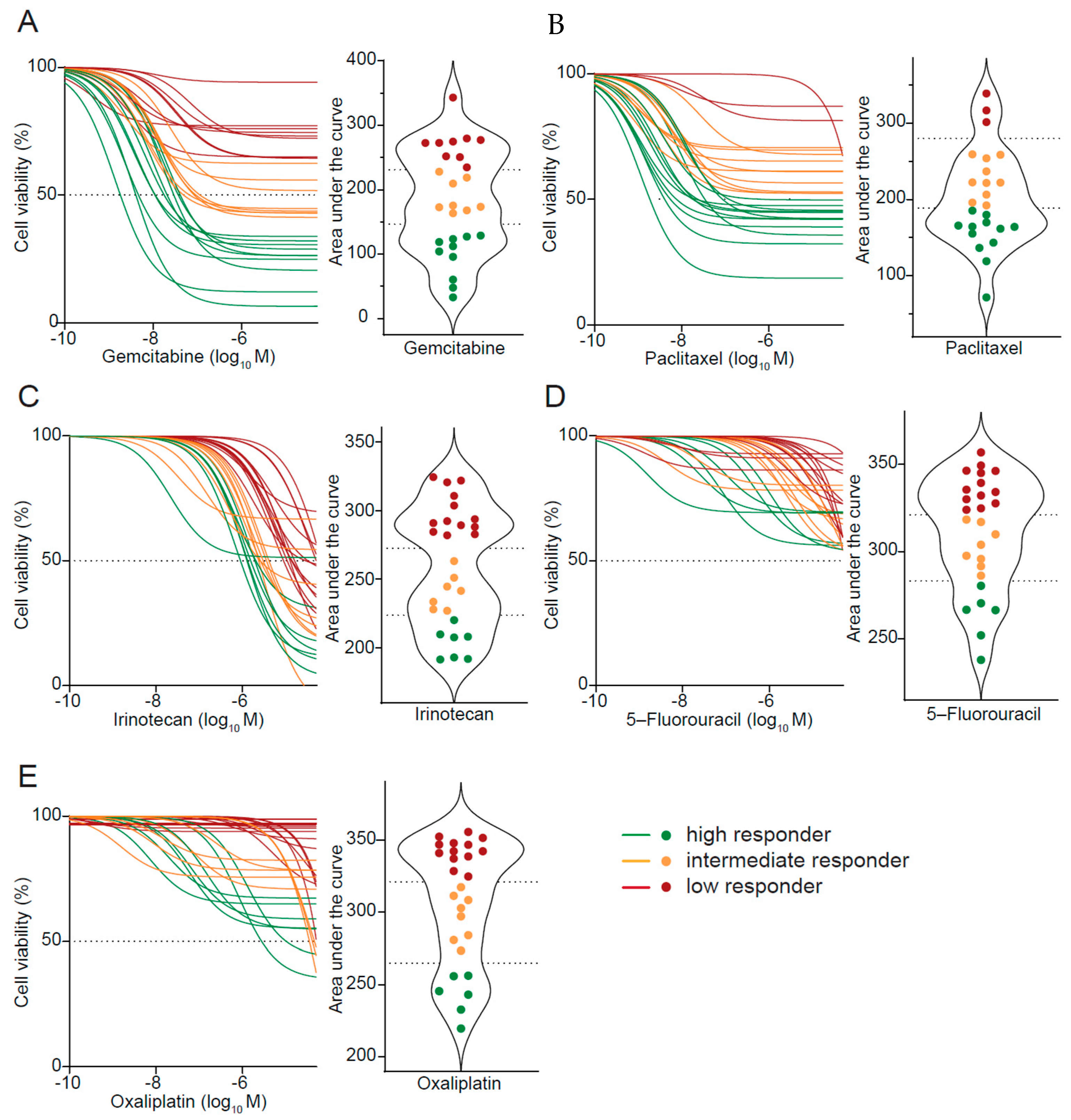
3.3. Drug Response Prediction in PDO Cultures
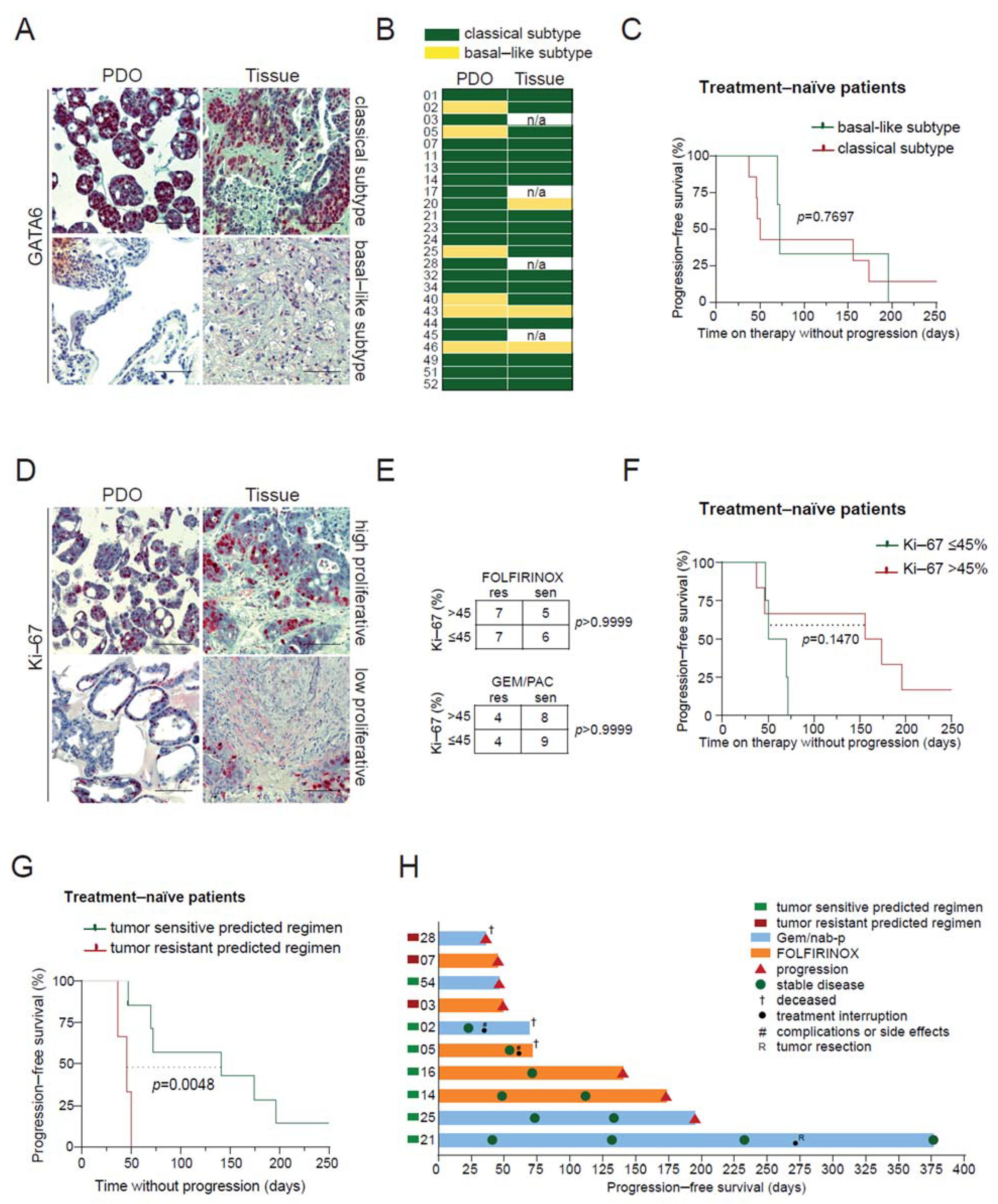
3.4. PDO-Based Pharmacotyping Predicts Drug Response to Aid Decision-Making in Patients
3.5. A Machine Learning Classifier as an Alternative Prediction Model to Guide Treatment
3.6. PDO Prediction and Potential Clinical Impact
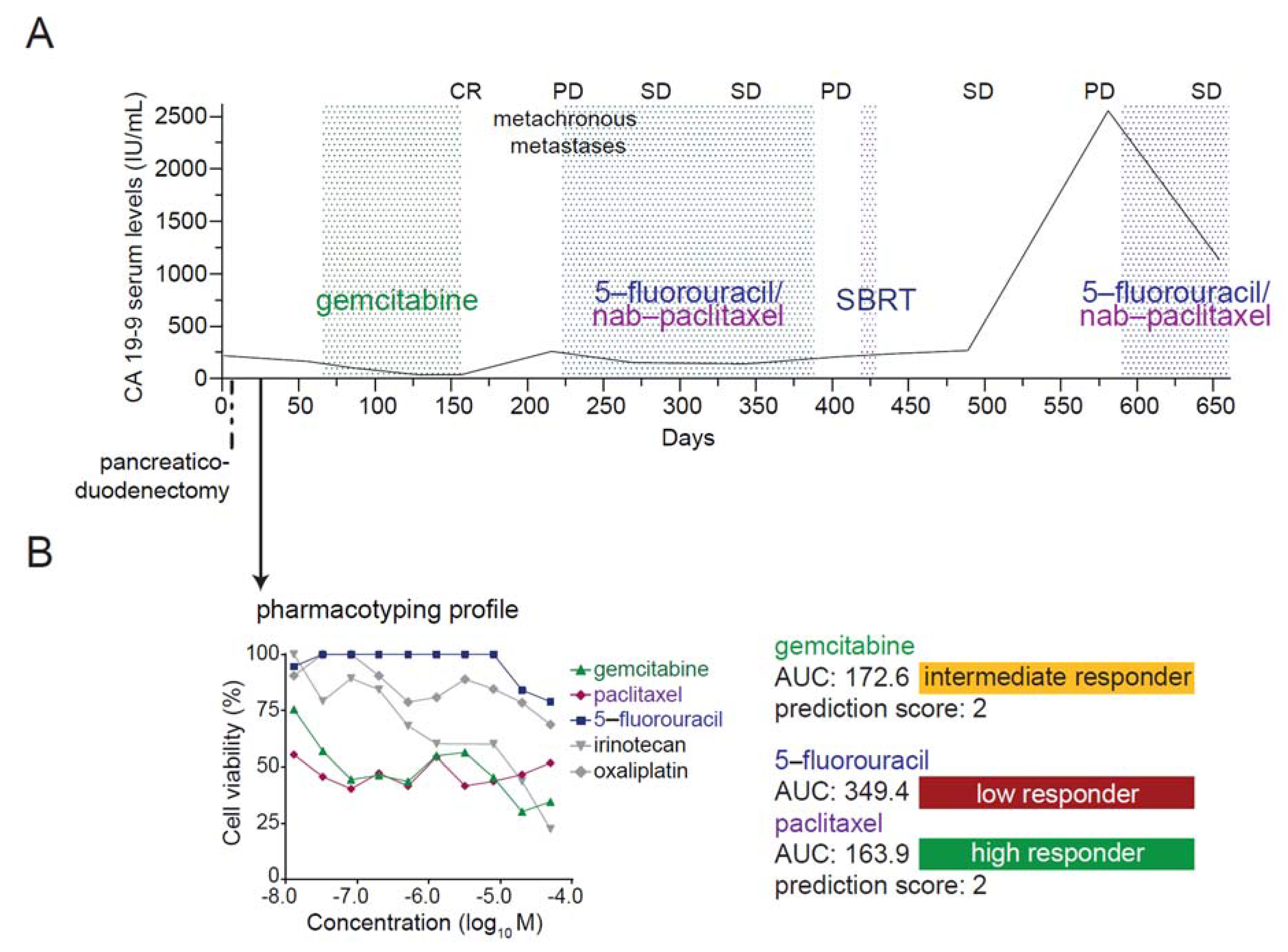
3.7. Case Report: Integration of Patient-Derived Organoids into Medical Care
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA A Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Mitry, E.; Lievre, A.; Lepere, C.; Vaillant, J.N.; Declety, G.; Parlier, H.; Emile, J.F.; Julie, C.; Rougier, P. Second- and third-line chemotherapy in patients with metastatic pancreatic adenocarcinoma: Feasibility and potential benefits in a retrospective series of 117 patients. Gastroentérol. Clin. Biol. 2009, 33, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.J.; Ko, A.H. Beyond first-line chemotherapy for advanced pancreatic cancer: An expanding array of therapeutic options? World J. Gastroenterol. 2014, 20, 2224–2236. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.; Li, C.P.; Wang-Gillam, A.; Bodoky, G.; Dean, A.; Jameson, G.; Macarulla, T.; Lee, K.H.; Cunningham, D.; Blanc, J.F.; et al. NAPOLI-1: Randomized Phase 3 Study of MM-398 (NAL-IRI), with or Without 5-Fluorouracil and Leucovorin, Versus 5-Fluorouracil and Leucovorin, in Metastatic Pancreatic Cancer Progressed on or Following Gemcitabine-Based Therapy. Ann. Oncol. 2014, 25, ii105. [Google Scholar] [CrossRef]
- Oettle, H.; Riess, H.; Stieler, J.M.; Heil, G.; Schwaner, I.; Seraphin, J.; Görner, M.; Mölle, M.; Greten, T.F.; Lakner, V.; et al. Second-Line Oxaliplatin, Folinic Acid, and Fluorouracil Versus Folinic Acid and Fluorouracil Alone for Gemcitabine-Refractory Pancreatic Cancer: Outcomes From the CONKO-003 Trial. J. Clin. Oncol. 2014, 32, 2423–2429. [Google Scholar] [CrossRef]
- De Jesus, V.H.F.; Camandaroba, M.P.G.; Calsavara, V.F.; Riechelmann, R.P. Systematic review and meta-analysis of gemcitabine-based chemotherapy after FOLFIRINOX in advanced pancreatic cancer. Ther. Adv. Med Oncol. 2020, 12, 1758835920905408. [Google Scholar] [CrossRef]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Asbun, H.; Bain, A.; Behrman, S.W.; Benson, A.B.; Binder, E.; Cardin, D.B.; Cha, C.; et al. Pancreatic Adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 1028–1061. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.C.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.C.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- Frappart, P.; Walter, K.; Gout, J.; Beutel, A.K.; Morawe, M.; Arnold, F.; Breunig, M.; Barth, T.F.; Marienfeld, R.; Schulte, L.; et al. Pancreatic cancer-derived organoids: A disease modeling tool to predict drug response. United Eur. Gastroenterol. J. 2020, 8, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Bockorny, B.; Paul, I.; Akshinthala, D.; Frappart, P.-O.; Gandarilla, O.; Bose, A.; Sanchez-Gonzalez, V.; Rouse, E.E.; Lehoux, S.D.; et al. PDX-derived organoids model in vivo drug response and secrete biomarkers. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Fichtner, I.; Slisow, W.; Gill, J.; Becker, M.; Elbe, B.; Hillebrand, T.; Bibby, M. Anticancer drug response and expression of molecular markers in early-passage xenotransplanted colon carcinomas. Eur. J. Cancer 2004, 40, 298–307. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Uson, M.; RajeshKumar, N.; Tan, A.C.; De Oliveira, E.; Karikari, C.; Villaroel, M.C.; Salomon, A.; Taylor, G.; Sharma, R.; et al. Tumor Engraftment in Nude Mice and Enrichment in Stroma- Related Gene Pathways Predict Poor Survival and Resistance to Gemcitabine in Patients with Pancreatic Cancer. Clin. Cancer Res. 2011, 17, 5793–5800. [Google Scholar] [CrossRef]
- Seppälä, T.T.; Zimmerman, J.W.; Sereni, E.; Plenker, D.; Suri, R.; Rozich, N.; Blair, A.; Thomas, D.L.; Teinor, J.; Javed, A.; et al. Patient-derived Organoid Pharmacotyping is a Clinically Tractable Strategy for Precision Medicine in Pancreatic Cancer. Ann. Surg. 2020, 272, 427–435. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Mathijssen, R.H.; Van Alphen, R.J.; Verweij, J.; Loos, W.J.; Nooter, K.; Stoter, G.; Sparreboom, A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin. Cancer Res. 2001, 7, 2182–2194. [Google Scholar] [PubMed]
- Pavillard, V.; Agostini, C.; Richard, S.; Charasson, V.; Montaudon, D.; Robert, J. Determinants of the cytotoxicity of irinotecan in two human colorectal tumor cell lines. Cancer Chemother. Pharmacol. 2002, 49, 329–335. [Google Scholar] [CrossRef]
- Dantes, Z.; Yen, H.-Y.; Pfarr, N.; Winter, C.; Steiger, K.; Muckenhuber, A.; Hennig, A.; Lange, S.; Engleitner, T.; Öllinger, R.; et al. Implementing cell-free DNA of pancreatic cancer patient-derived organoids for personalized oncology. JCI Insight 2020, 5, e137809. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.M.; Grünwald, B.T.; Jang, G.-H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4901–4910. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Initiative, A.P.C.G.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Kim, H.; Park, C.Y.; Lee, J.H.; Kim, J.C.; Cho, C.K.; Kim, H.J. Ki-67 and p53 expression as a predictive marker for early postoperative recurrence in pancreatic head cancer. Ann. Surg. Treat. Res. 2015, 88, 200–207. [Google Scholar] [CrossRef]
- Temraz, S.; Shamseddine, A.; Mukherji, D.; Charafeddine, M.; Tfayli, A.; Assi, H.; Hammoud, M.S.; Makki, I.; Nassif, S. Ki67 and P53 in Relation to Disease Progression in Metastatic Pancreatic Cancer: A Single Institution Analysis. Pathol. Oncol. Res. 2018, 25, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Pergolini, I.; Crippa, S.; Pagnanelli, M.; Belfiori, G.; Pucci, A.; Partelli, S.; Rubini, C.; Castelli, P.; Zamboni, G.; Falconi, M. Prognostic impact of Ki-67 proliferative index in resectable pancreatic ductal adenocarcinoma. BJS Open 2019, 3, 646–655. [Google Scholar] [CrossRef]
- Kim, Y.J.; Bang, S.; Park, J.Y.; Park, S.W.; Chung, J.B.; Song, S.Y. Phase II study of 5-fluorouracil and paclitaxel in patients with gemcitabine-refractory pancreatic cancer. Cancer Chemother. Pharmacol. 2008, 63, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Lacomb, J.F.; Plenker, D.; Tiriac, H.; Bucobo, J.C.; D’Souza, L.S.; Khokhar, A.S.; Patel, H.; Channer, B.; Joseph, D.; Wu, M.; et al. Single-Pass vs 2-Pass Endoscopic Ultrasound-Guided Fine-Needle Biopsy Sample Collection for Creation of Pancreatic Adenocarcinoma Organoids. Clin. Gastroenterol. Hepatol. 2021, 19, 845–847. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Bucobo, J.C.; Tzimas, D.; Grewel, S.; Lacomb, J.F.; Rowehl, L.M.; Nagula, S.; Wu, M.; Kim, J.; Sasson, A.; et al. Successful creation of pancreatic cancer organoids by means of EUS-guided fine-needle biopsy sampling for personalized cancer treatment. Gastrointest. Endosc. 2018, 87, 1474–1480. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Engle, D.D.; Tuveson, D.A.; Clevers, H. Model organoids provide new research opportunities for ductal pancreatic cancer. Mol. Cell. Oncol. 2015, 3, e1014757. [Google Scholar] [CrossRef]
- Baker, L.A.; Tiriac, H.; Tuveson, D.A. Generation and Culture of Human Pancreatic Ductal Adenocarcinoma Organoids from Resected Tumor Specimens. Methods Mol. Biol. 2018, 1882, 97–115. [Google Scholar] [CrossRef]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 35, 267–282.e7. [Google Scholar] [CrossRef]
- Thomas, R.M.; Truty, M.J.; Kim, M.; Kang, Y.; Zhang, R.; Chatterjee, D.; Katz, M.H.; Fleming, J.B. The Canary in the Coal Mine: The Growth of Patient-Derived Tumorgrafts in Mice Predicts Clinical Recurrence after Surgical Resection of Pancreatic Ductal Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 1884–1892. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.-J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 1–13. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; DuBois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: Phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Untreated | Treated | |
|---|---|---|
| Patients, no. | 30 | 14 |
| Age at diagnosis, mean (range) in years | 66.6 (41–81) | 59.0 (44–67) |
| Sex, no. (%) | ||
| Male | 18 (60) | 9 (64) |
| Female | 12 (40) | 5 (36) |
| Tumor stage, no. (%) | ||
| Metastasized | 18 (60) | 14 (100) |
| Locally advanced | 8 (27) | 0 (0) |
| Resectable | 4 (13) | 0 (0) |
| ECOG, no. (%) | ||
| 0 | 19 (63) | 9 (64) |
| 1 | 10 (33) | 5 (36) |
| 2 | 1 (3) | 0 (0) |
| Sampling method, no. (%) | ||
| US-guided biopsy | 25 (83) | 14 (100) |
| EUS-guided FNB | 3 (10) | 0 (0) |
| Surgical resection | 2 (7) | 0 (0) |
| Localization of biopsy, no. (%) | ||
| Primary tumor | 16 (53) | 0 (0) |
| Liver metastases | 14 (47) | 14 (100) |
| Pharmacotyping, no. (%) | ||
| Success | 19 (63) | 9 (64) |
| Failure | 11 (37) | 5 (36) |
| Prior systemic therapy lines, no. (%) | ||
| 1 | 0 (0) | 4 (29) |
| 2 | 0 (0) | 6 (43) |
| ≥3 | 0 (0) | 4 (29) |
| Prior therapy before PDO generation, no. (%) | ||
| Platinum-based (FOLFIRINOX, OFF, carboplatin/nab-p) | 0 (0) | 13 (93) |
| Gemcitabine/nab-p | 0 (0) | 11 (79) |
| Surgical resection | 0 (0) | 4 (29) |
| Radio(chemo)therapy | 0 (0) | 2 (14) |
| Nal-iri/5-FU | 0 (0) | 1 (7) |
| Treatment regimen after PDO generation, no. (%) | ||
| Platinum-based (FOLFIRINOX, FOLFOX, Doce/Ox) | 12 (40) | 1 (7) |
| Gemcitabine/(nab-p) | 11 (37) | 2 (14) |
| No (adjuvant/palliative) chemotherapy | 6 (20) | 4 (29) |
| Surgical resection | 4 (13) | 0 (0) |
| Nal-iri/5-FU | 0 (0) | 3 (21) |
| Clinical trial | 0 (0) | 3 (21) |
| Olaparib | 0 (0) | 1 (7) |
| Restaging, no. (%) | ||
| Response (CR, PR, SD) | 13 (43) | 4 (29) |
| No response (PD) | 9 (30) | 6 (43) |
| Not available | 8 (27) | 4 (29) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beutel, A.K.; Schütte, L.; Scheible, J.; Roger, E.; Müller, M.; Perkhofer, L.; Kestler, A.M.T.U.; Kraus, J.M.; Kestler, H.A.; Barth, T.F.E.; et al. A Prospective Feasibility Trial to Challenge Patient–Derived Pancreatic Cancer Organoids in Predicting Treatment Response. Cancers 2021, 13, 2539. https://doi.org/10.3390/cancers13112539
Beutel AK, Schütte L, Scheible J, Roger E, Müller M, Perkhofer L, Kestler AMTU, Kraus JM, Kestler HA, Barth TFE, et al. A Prospective Feasibility Trial to Challenge Patient–Derived Pancreatic Cancer Organoids in Predicting Treatment Response. Cancers. 2021; 13(11):2539. https://doi.org/10.3390/cancers13112539
Chicago/Turabian StyleBeutel, Alica K., Lena Schütte, Jeanette Scheible, Elodie Roger, Martin Müller, Lukas Perkhofer, Annika M. T. U. Kestler, Johann M. Kraus, Hans A. Kestler, Thomas F. E. Barth, and et al. 2021. "A Prospective Feasibility Trial to Challenge Patient–Derived Pancreatic Cancer Organoids in Predicting Treatment Response" Cancers 13, no. 11: 2539. https://doi.org/10.3390/cancers13112539
APA StyleBeutel, A. K., Schütte, L., Scheible, J., Roger, E., Müller, M., Perkhofer, L., Kestler, A. M. T. U., Kraus, J. M., Kestler, H. A., Barth, T. F. E., Lemke, J., Kornmann, M., Ettrich, T. J., Gout, J., Seufferlein, T., & Kleger, A. (2021). A Prospective Feasibility Trial to Challenge Patient–Derived Pancreatic Cancer Organoids in Predicting Treatment Response. Cancers, 13(11), 2539. https://doi.org/10.3390/cancers13112539