
Challenges of Neoantigen Targeting in Lynch Syndrome and Constitutional Mismatch Repair Deficiency Syndrome

 ,
,  , , , and
, , , and 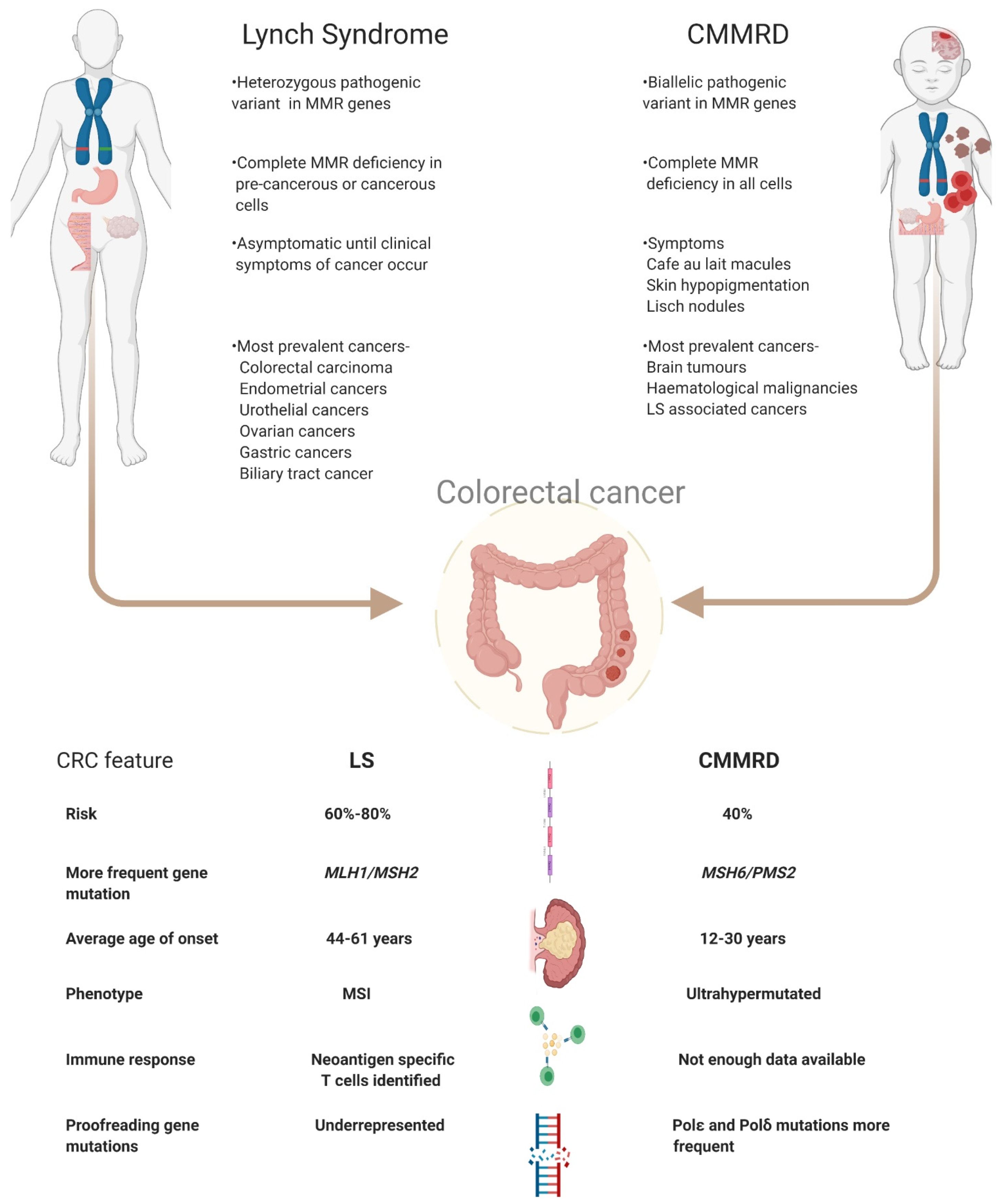{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
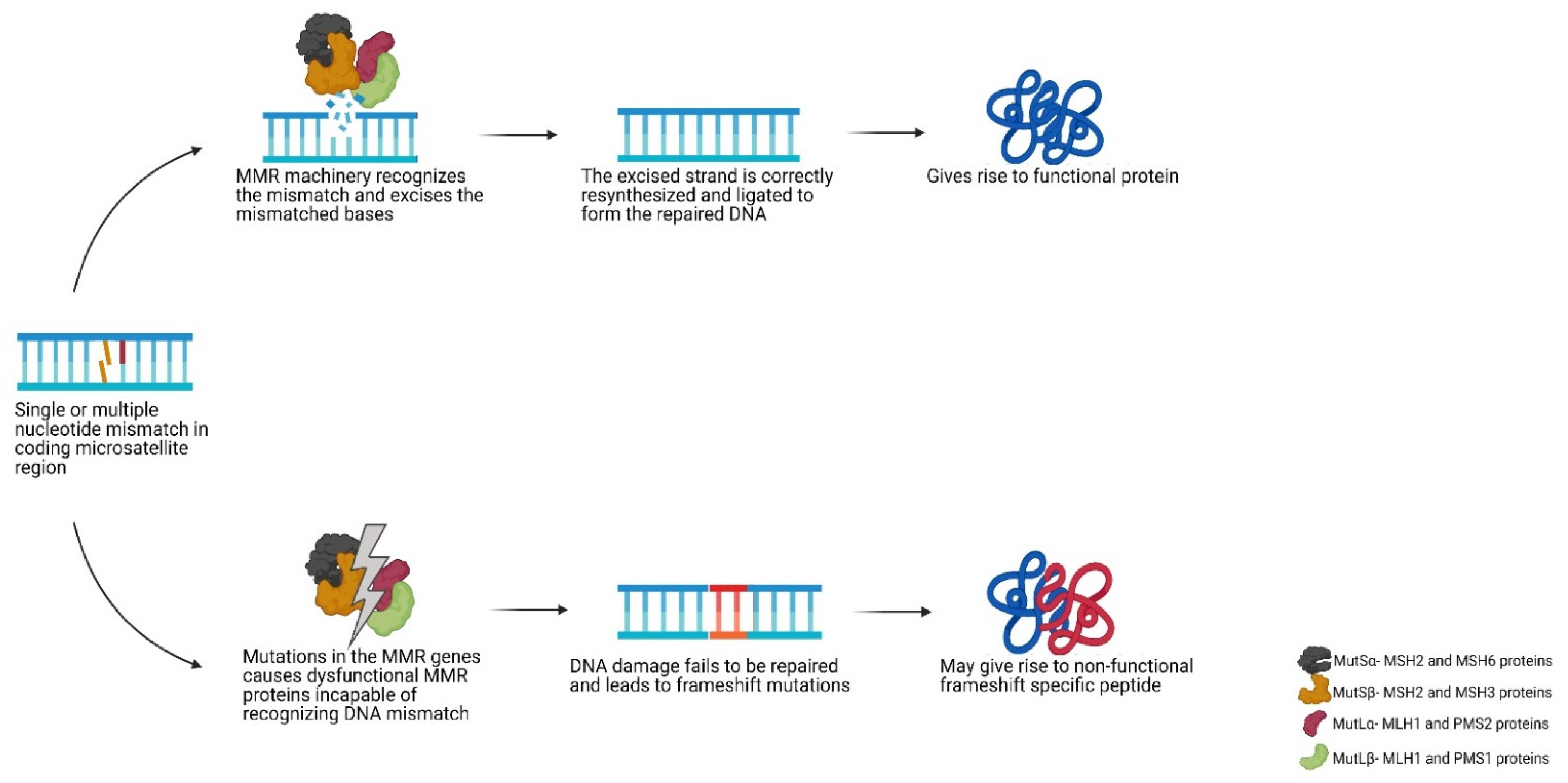
2. Mismatch Repair Deficiency and Microsatellite Instability
3. Clinical Management of LS and CMMRD
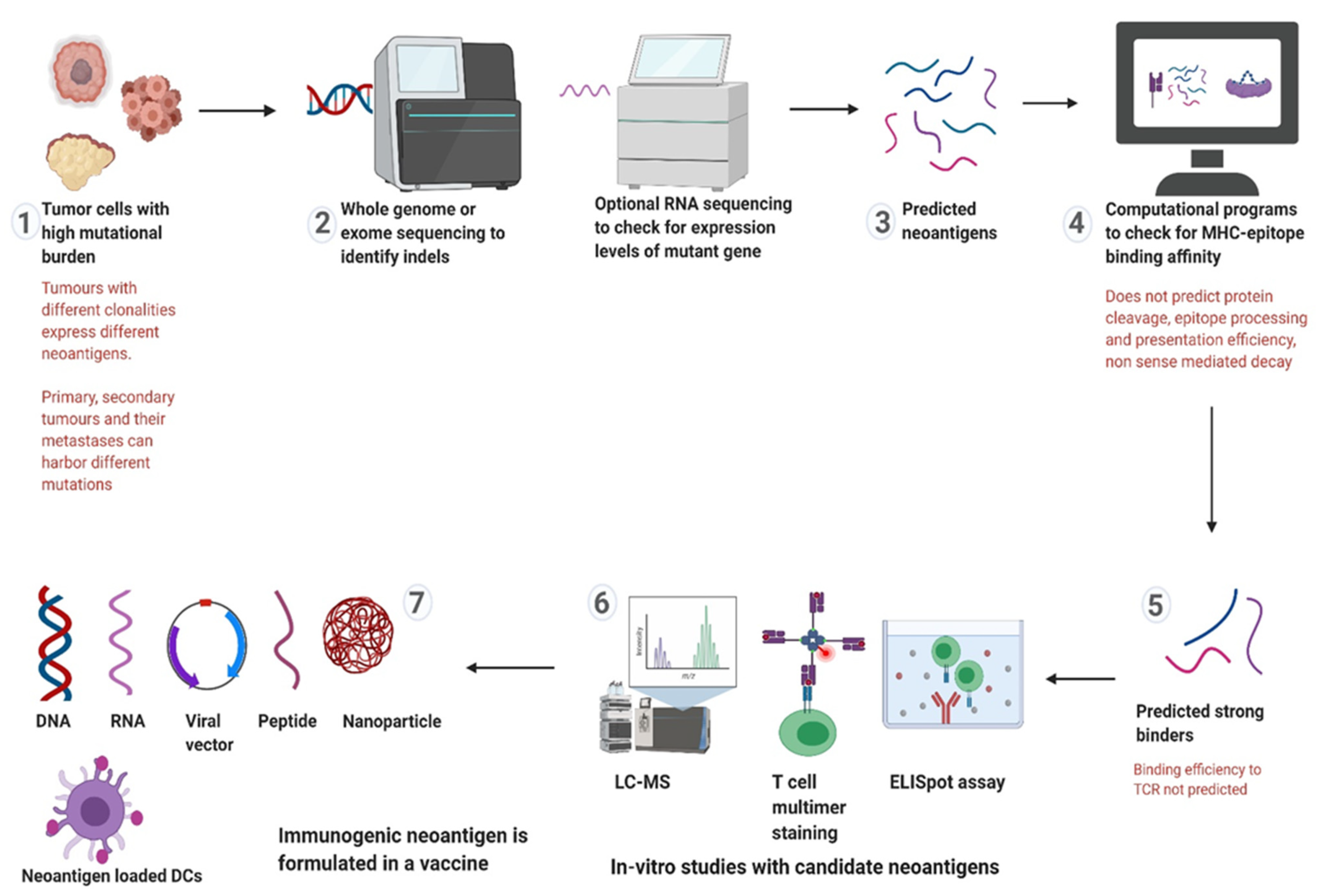
4. Targeting Neoantigens in LS and CMMRD
5. Challenges of Neoantigen Targeting in LS and CMMRD
5.1. Tumour Heterogeneity
5.2. Neoantigen Selection
5.3. Vaccine Formulation
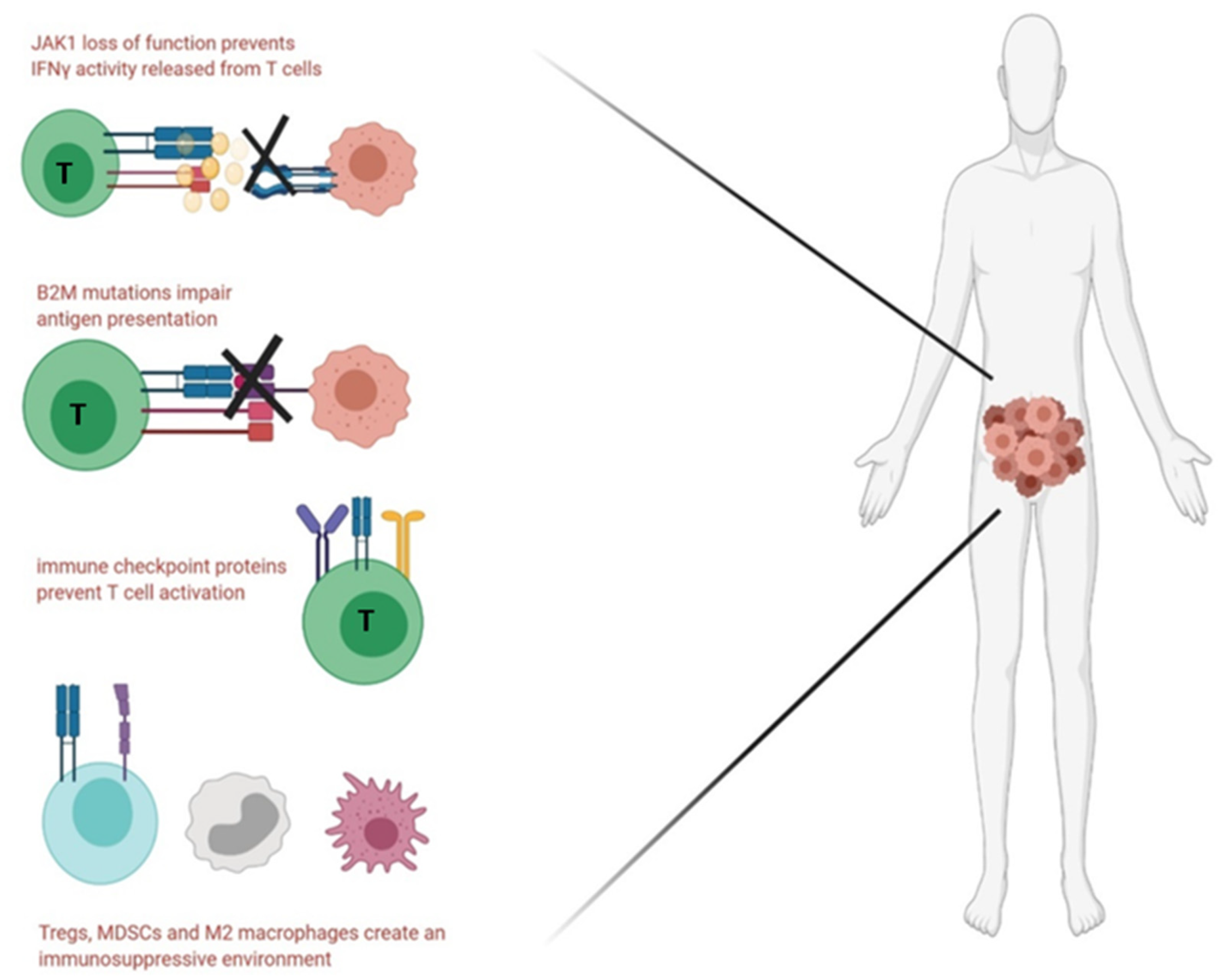
5.4. Immune Evasion and Immunosuppression
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hampel, H.; Hoffner, B. Lynch Syndrome and Immunotherapy. J. Adv. Pract. Oncol. 2017, 8, 7–21. [Google Scholar]
- Lynch, H.T.; Lanspa, S.; Shaw, T.; Casey, M.J.; Rendell, M.; Stacey, M.; Townley, T.; Snyder, C.; Hitchins, M.; Bailey-Wilson, J. Phenotypic and genotypic heterogeneity of Lynch syndrome: A complex diagnostic challenge. Fam. Cancer 2018, 17, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef]
- Ricciardone, M.D.; Tayfun, O. Advances in Brief Human MLH1 Deficiency Predisposes to Hematological Malignancy and. Cancer Res. 1999, 59, 290–293. [Google Scholar]
- Wang, Q.; Lasset, C.; Desseigne, F.; Saurin, J.-C.; Maugard, C.; Navarro, C.; Ruano, E.; Descos, L.; Trillet-Lenoir, V.; Bosset, J.-F.; et al. Prevalence of germline mutations of hMLH1, hMSH2, hPMS1, hPMS2, and hMSH6 genes in 75 French kindreds with nonpolyposis colorectal cancer. Hum. Genet. 1999, 105, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, K.; Kratz, C.P.; Vasen, H.F.A.; Caron, O.; Colas, C.; Entz-Werle, N.; Gerdes, A.-M.; Goldberg, Y.; Ilencikova, D.; Muleris, M.; et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: Suggestions of the European consortium ‘Care for CMMRD’ (C4CMMRD). J. Med. Genet. 2014, 51, 355–365. [Google Scholar] [CrossRef]
- Moreira, L.; Balaguer, F.; Lindor, N.; de la Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef]
- Bucksch, K.; Zachariae, S.; Aretz, S.; Büttner, R.; Holinski-Feder, E.; Holzapfel, S.; Hüneburg, R.; Kloor, M.; Von Knebel Doeberitz, M.; Morak, M.; et al. Cancer risks in Lynch syndrome, Lynch-like syndrome, and familial colorectal cancer type X: A prospective cohort study. BMC Cancer 2020, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise management of ovarian cancer: On the cutting edge. J. Pers. Med. 2020, 10, 41. [Google Scholar] [CrossRef]
- Van Der Post, R.S.; Kiemeney, L.A.; Ligtenberg, M.J.L.; Witjes, J.A.; Hulsbergen-Van De Kaa, C.A.; Bodmer, D.; Schaap, L.; Kets, C.M.; Van Krieken, J.H.J.M.; Hoogerbrugge, N. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J. Med. Genet. 2010, 47, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Mankaney, G.; Macaron, C.; Burke, C.A. Refining Risk Factors for Gastric Cancer in Patients With Lynch Syndrome to Optimize Surveillance Esophagogastroduodenoscopy. Clin. Gastroenterol. Hepatol. 2020, 18, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Abedalthagafi, M. Constitutional mismatch repair-deficiency: Current problems and emerging therapeutic strategies. Oncotarget 2018, 9, 35458–35469. [Google Scholar] [CrossRef] [PubMed]
- Lavoine, N.; Colas, C.; Muleris, M.; Bodo, S.; Duval, A.; Entz-Werle, N.; Coulet, F.; Cabaret, O.; Andreiuolo, F.; Charpy, C.; et al. Constitutional mismatch repair deficiency syndrome: Clinical description in a French cohort. J. Med. Genet. 2015, 52, 770–778. [Google Scholar] [CrossRef]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Westdorp, H.; Kolders, S.; Hoogerbrugge, N.; de Vries, I.J.M.; Jongmans, M.C.J.; Schreibelt, G. Immunotherapy holds the key to cancer treatment and prevention in constitutional mismatch repair deficiency (CMMRD) syndrome. Cancer Lett. 2017, 403, 159–164. [Google Scholar] [CrossRef]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef]
- George, J.T.; Kessler, D.A.; Levine, H. Effects of thymic selection on T cell recognition of foreign and tumor antigenic peptides. Proc. Natl. Acad. Sci. USA 2017, 114, E7875–E7881. [Google Scholar] [CrossRef]
- Zhou, J.; Dudley, M.E.; Rosenberg, S.A.; Robbins, P.F. Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J. Immunother. 2005, 28, 53–62. [Google Scholar] [CrossRef]
- Burgess, J.T.; Rose, M.; Boucher, D.; Plowman, J.; Molloy, C.; Fisher, M.; O’Leary, C.; Richard, D.J.; O’Byrne, K.J.; Bolderson, E. The Therapeutic Potential of DNA Damage Repair Pathways and Genomic Stability in Lung Cancer. Front. Oncol. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Boussios, S.; Moschetta, M.; Karihtala, P.; Samartzis, E.P.; Sheriff, M.; Pappas-Gogos, G.; Ozturk, M.A.; Uccello, M.; Karathanasi, A.; Tringos, M.; et al. Development of new poly(ADP-ribose) polymerase (PARP) inhibitors in ovarian cancer: Quo Vadis? Ann. Transl. Med. 2020, 8, 1706. [Google Scholar] [CrossRef]
- Colle, R.; Cohen, R.; Cochereau, D.; Duval, A.; Lascols, O.; Lopez-Trabada, D.; Afchain, P.; Trouilloud, I.; Parc, Y.; Lefevre, J.H.; et al. Immunotherapy and patients treated for cancer with microsatellite instability. Bull. Cancer 2017, 104, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Arnold, C.N.; Niedzwiecki, D.; Carethers, J.M.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; Bertagnolli, M.M.; Boland, C.R. Frequent Inactivation of PTEN by Promoter Hypermethylation in Microsatellite Instability-High Sporadic Colorectal Cancers. Cancer Res. 2004, 64, 3014–3021. [Google Scholar] [CrossRef] [PubMed]
- Vos, J.R.; Fakkert, I.E.; Spruijt, L.; Willems, R.W.; Langenveld, S.; Mensenkamp, A.R.; Leter, E.M.; Nagtegaal, I.D.; Ligtenberg, M.J.L.; Hoogerbrugge, N. Evaluation of yield and experiences of age-related molecular investigation for heritable and nonheritable causes of mismatch repair deficient colorectal cancer to identify Lynch syndrome. Int. J. Cancer 2020, 147, 2150–2158. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Elze, L.; Mensenkamp, A.R.; Nagtegaal, I.D.; van Zelst-Stams, W.; de Voer, R.M.; Ligtenberg, M.J.L. Somatic Nonepigenetic Mismatch Repair Gene Aberrations Underly Most Mismatch Repair-Deficient Lynch-Like Tumors. Gastroenterology 2021, 160, 1414–1416.e3. [Google Scholar] [CrossRef]
- van Leerdam, M.E.; Roos, V.H.; van Hooft, J.E.; Balaguer, F.; Dekker, E.; Kaminski, M.F.; Latchford, A.; Neumann, H.; Ricciardiello, L.; Rupinska, M.; et al. Endoscopic management of Lynch syndrome and of familial risk of colorectal cancer: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy 2019, 51, 1082–1093. [Google Scholar] [CrossRef]
- Umar, A. Faculty Opinions recommendation of Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Fac. Opin. 2011. [Google Scholar] [CrossRef]
- Leenders, E.K.S.M.; Westdorp, H.; Brüggemann, R.J.; Loeffen, J.; Kratz, C.; Burn, J.; Hoogerbrugge, N.; Jongmans, M.C.J. Cancer prevention by aspirin in children with Constitutional Mismatch Repair Deficiency (CMMRD). Eur. J. Hum. Genet. 2018, 26, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Jo, W.-S.; Carethers, J.M. Chemotherapeutic implications in microsatellite unstable colorectal cancer. Cancer Biomark. 2006, 2, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef] [PubMed]
- Stojic, L.; Brun, R.; Jiricny, J. Mismatch repair and DNA damage signalling. DNA Repair 2004, 3, 1091–1101. [Google Scholar] [CrossRef]
- Sawant, A.; Kothandapani, A.; Zhitkovich, A.; Sobol, R.W.; Patrick, S.M. Role of mismatch repair proteins in the processing of cisplatin interstrand cross-links. DNA Repair 2015, 35, 126–136. [Google Scholar] [CrossRef]
- Seth, S.; Ager, A.; Arends, M.J.; Frayling, I.M. Lynch syndrome—Cancer pathways, heterogeneity and immune escape. J. Pathol. 2018, 246, 129–133. [Google Scholar] [CrossRef]
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001, 91, 2417–2422. [Google Scholar] [CrossRef]
- Bohaumilitzky, L.; von Knebel Doeberitz, M.; Kloor, M.; Ahadova, A. Implications of Hereditary Origin on the Immune Phenotype of Mismatch Repair-Deficient Cancers: Systematic Literature Review. J. Clin. Med. 2020, 9, 1741. [Google Scholar] [CrossRef]
- Ferris, R.L.; Lu, B.; Kane, L.P. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J. Immunol. 2014, 193, 1525–1530. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Le, D.T. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 373, 1979. [Google Scholar] [CrossRef] [PubMed]
- Gurjao, C.; Liu, D.; Hofree, M.; AlDubayan, S.H.; Wakiro, I.; Su, M.-J.; Felt, K.; Gjini, E.; Brais, L.K.; Rotem, A.; et al. Intrinsic Resistance to Immune Checkpoint Blockade in a Mismatch Repair-Deficient Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Salman, P.; Panay, S.; Fernández, R.; Mahave, M.; Soza-Ried, C. Evidence of response to pembrolizumab in a patient with lynch syndrome-related metastatic colon cancer. Onco Targets Ther. 2018, 11, 7295–7300. [Google Scholar] [CrossRef]
- Patil, N.R.; Khan, G.N. Exceptional response to a single cycle of immunotherapy in a lynch syndrome patient with metastatic pancreatic adenocarcinoma. Am. J. Case Rep. 2020, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef]
- AlHarbi, M.; Ali Mobark, N.; AlMubarak, L.; Aljelaify, R.; AlSaeed, M.; Almutairi, A.; Alqubaishi, F.; Hussain, M.E.; Balbaid, A.A.O.; Said Marie, A.; et al. Durable Response to Nivolumab in a Pediatric Patient with Refractory Glioblastoma and Constitutional Biallelic Mismatch Repair Deficiency. Oncologist 2018, 23, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Austin, F.; Richard, H.; Idowu, M.; Williamson, V.; Sabato, F.; Ferreira-Gonzalez, A.; Turner, S.A. Lynch syndrome-associated ultra-hypermutated pediatric glioblastoma mimicking a constitutional mismatch repair deficiency syndrome. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003863. [Google Scholar] [CrossRef]
- Shlien, A.; Campbell, B.B.; De Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef]
- Kloor, M.; von Knebel Doeberitz, M. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef]
- Bakarurraini, N.A.A.R.; Mutalib, N.S.A.; Jamal, R.; Abu, N. The landscape of tumor-specific antigens in colorectal cancer. Vaccines 2020, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- Saeterdal, I.; Bjørheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Møller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 13255–13260. [Google Scholar] [CrossRef] [PubMed]
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune Response Against Frameshift-Induced Neopeptides in HNPCC Patients and Healthy HNPCC Mutation Carriers. Gastroenterology 2008, 134, 988–997. [Google Scholar] [CrossRef]
- Saeterdal, I.; Gjertsen, M.K.; Straten, P.; Eriksen, J.A.; Gaudernack, G. A TGF betaRII frameshift-mutation-derived CTL epitope recognised by HLA-A2-restricted CD8+ T cells. Cancer Immunol. Immunother. 2001, 50, 469–476. [Google Scholar]
- Ripberger, E.; Linnebacher, M.; Schwitalle, Y.; Gebert, J.; Von Knebel Doeberitz, M. Identification of an HLA-A0201-restricted CTL epitope generated by a tumor-specific frameshift mutation in a coding microsatellite of the OGT gene. J. Clin. Immunol. 2003, 23, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Leoni, G.; D’Alise, A.M.; Cotugno, G.; Langone, F.; Garzia, I.; De Lucia, M.; Fichera, I.; Vitale, R.; Bignone, V.; Tucci, F.G.; et al. A Genetic Vaccine Encoding Shared Cancer Neoantigens to Treat Tumors with Microsatellite Instability. Cancer Res. 2020, 80, 3972–3982. [Google Scholar] [CrossRef] [PubMed]
- Westdorp, H.; Gorris, M.A.J.; Boudewijns, S.; Bisseling, T.; de Goede, A.L.; van Rossum, M.M.; Ligtenberg, M.J.L.; Schreibelt, G.; Nagtegaal, I.D.; Figdor, C.G.; et al. Preventive dendritic cell vaccination in healthy Lynch syndrome mutation carriers. Ann. Oncol. 2016, 27, vi362. [Google Scholar] [CrossRef]
- Kloor, M.; Reuschenbach, M.; Karbach, J.; Rafiyan, M.; Al-Batran, S.-E.; Pauligk, C.; Jaeger, E.; von Knebel Doeberitz, M. Vaccination of MSI-H colorectal cancer patients with frameshift peptide antigens: A phase I/IIa clinical trial. J. Clin. Oncol. 2015, 33, 3020. [Google Scholar] [CrossRef]
- Pavelka, Z.; Zitterbart, K.; Nosková, H.; Bajčiová, V.; Slabý, O.; Štěrba, J. Effective Immunotherapy of Glioblastoma in an Adolescent with Constitutional Mismatch Repair-Deficiency Syndrome. Klin. Onkol. 2019, 32. [Google Scholar] [CrossRef]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Inderberg, E.M.; Wälchli, S.; Myhre, M.R.; Trachsel, S.; Almåsbak, H.; Kvalheim, G.; Gaudernack, G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology 2017, 6, e1302631. [Google Scholar] [CrossRef]
- Li, L.; Goedegebuure, S.P.; Gillanders, W.E. Preclinical and clinical development of neoantigen vaccines. Ann. Oncol. 2017, 28, xii11–xii17. [Google Scholar] [CrossRef]
- Jamal-hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational Implications of Tumor Heterogeneity. Clin. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective Lynch syndrome database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef]
- Lynch, H.T.; Boland, C.R.; Gong, G.; Shaw, T.G.; Lynch, P.M.; Fodde, R.; Lynch, J.F.; de la Chapelle, A. Phenotypic and genotypic heterogeneity in the Lynch syndrome: Diagnostic, surveillance and management implications. Eur. J. Hum. Genet. 2006, 14, 390–402. [Google Scholar] [CrossRef]
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Bläker, H.; et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int. J. Cancer 2018, 143, 139–150. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Chang, K.; Taggart, M.W.; Reyes-Uribe, L.; Borras, E.; Riquelme, E.; Barnett, R.M.; Leoni, G.; San Lucas, F.A.; Catanese, M.T.; Mori, F.; et al. Immune Profiling of Premalignant Lesions in Patients With Lynch Syndrome. JAMA Oncol. 2018, 4, 1085–1092. [Google Scholar] [CrossRef]
- Binder, H.; Hopp, L.; Schweiger, M.R.; Hoffmann, S.; Jühling, F.; Kerick, M.; Timmermann, B.; Siebert, S.; Grimm, C.; Nersisyan, L.; et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. J. Pathol. 2017, 243, 242–254. [Google Scholar] [CrossRef]
- Chung, J.; Maruvka, Y.E.; Sudhaman, S.; Kelly, J.; Haradhvala, N.J.; Bianchi, V.; Edwards, M.; Forster, V.J.; Nunes, N.M.; Galati, M.A.; et al. DNA polymerase and mismatch repair exert distinct microsatellite instability signatures in normal and malignant human cells. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Müller, A.; Giuffre, G.; Edmonston, T.B.; Mathiak, M.; Roggendorf, B.; Heinmöller, E.; Brodegger, T.; Tuccari, G.; Mangold, E.; Buettner, R.; et al. Challenges and pitfalls in HNPCC screening by microsatellite analysis and immunohistochemistry. J. Mol. Diagn. 2004, 6, 308–315. [Google Scholar] [CrossRef]
- Sylvester, B.E.; Vakiani, E. Tumor evolution and intratumor heterogeneity in colorectal carcinoma: Insights from comparative genomic profiling of primary tumors and matched metastases. J. Gastrointest. Oncol. 2015, 6, 668–675. [Google Scholar]
- Tougeron, D.; Fauquembergue, E.; Rouquette, A.; Le Pessot, F.; Sesboüé, R.; Laurent, M.; Berthet, P.; Mauillon, J.; Di Fiore, F.; Sabourin, J.C.; et al. Tumor-infiltrating lymphocytes in colorectal cancers with microsatellite instability are correlated with the number and spectrum of frameshift mutations. Mod. Pathol. 2009, 22, 1186–1195. [Google Scholar] [CrossRef]
- Maletzki, C.; Huehns, M.; Bauer, I.; Ripperger, T.; Mork, M.M.; Vilar, E.; Klöcking, S.; Zettl, H.; Prall, F.; Linnebacher, M. Frameshift mutational target gene analysis identifies similarities and differences in constitutional mismatch repair-deficiency and Lynch syndrome. Mol. Carcinog. 2017, 56, 1753–1764. [Google Scholar] [CrossRef]
- Aguadé-Gorgorió, G.; Solé, R. Tumor neoantigen heterogeneity thresholds provide a time window for combination immunotherapy. bioRxiv 2019. [Google Scholar] [CrossRef]
- Bauer, K.; Michel, S.; Reuschenbach, M.; Nelius, N.; von Knebel Doeberitz, M.; Kloor, M. Dendritic cell and macrophage infiltration in microsatellite-unstable and microsatellite-stable colorectal cancer. Fam. Cancer 2011, 10, 557–565. [Google Scholar] [CrossRef]
- Lampis, A.; Ghidini, M.; Ratti, M.; Mirchev, M.B.; Okuducu, A.F.; Valeri, N.; Hahne, J.C. Circulating Tumour DNAs and Non-Coding RNAs as Liquid Biopsies for the Management of Colorectal Cancer Patients. Gastrointest. Disord. 2020, 2, 212–235. [Google Scholar] [CrossRef]
- Arnaud, M.; Duchamp, M.; Bobisse, S.; Renaud, P.; Coukos, G.; Harari, A. Biotechnologies to tackle the challenge of neoantigen identification. Curr. Opin. Biotechnol. 2020, 65, 52–59. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Zhang, X.; Sharma, P.K.; Peter Goedegebuure, S.; Gillanders, W.E. Personalized cancer vaccines: Targeting the cancer mutanome. Vaccine 2017, 35, 1094–1100. [Google Scholar] [CrossRef]
- Williams, D.S.; Bird, M.J.; Jorissen, R.N.; Yu, Y.L.; Walker, F.; Zhang, H.H.; Nice, E.C.; Burgess, A.W. Nonsense mediated decay resistant mutations are a source of expressed mutant proteins in colon cancer cell lines with microsatellite instability. PLoS ONE 2010, 5, e16012. [Google Scholar] [CrossRef]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An Immunogenic Personal Neoantigen Vaccine for Melanoma Patients. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef]
- Majumder, S.; Shah, R.; Elias, J.; Manoharan, M.; Shah, P.; Kumari, A.; Chakraborty, P.; Kode, V.; Mistry, Y.; Coral, K.; et al. A cancer vaccine approach for personalized treatment of Lynch Syndrome. Sci. Rep. 2018, 8, 1–14. [Google Scholar]
- Roudko, V.; Bozkus, C.C.; Orfanelli, T.; McClain, C.B.; Carr, C.; O’Donnell, T.; Chakraborty, L.; Samstein, R.; Huang, K.L.; Blank, S.V.; et al. Shared Immunogenic Poly-Epitope Frameshift Mutations in Microsatellite Unstable Tumors. Cell. 2020, 183, 1634–1649. [Google Scholar] [CrossRef]
- Ballhausen, A.; Przybilla, M.J.; Jendrusch, M.; Haupt, S.; Pfaffendorf, E.; Seidler, F.; Witt, J.; Sanchez, A.H.; Urban, K.; Draxlbauer, M.; et al. The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S.; Kim, S. Neopepsee: Accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. 2018, 29, 1030–1036. [Google Scholar] [CrossRef]
- Guo, Y.; Lei, K.; Tang, L. Neoantigen vaccine delivery for personalized anticancer immunotherapy. Front. Immunol. 2018, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.A.; He, N.; Li, Z.; Ali, Z.; Zhang, L. Nanoparticles for DNA vaccine delivery. J. Biomed. Nanotechnol. 2014, 10, 2332–2349. [Google Scholar] [CrossRef] [PubMed]
- Vormehr, M.; Schrörs, B.; Boegel, S.; Löwer, M.; Türeci, Ö.; Sahin, U. Mutanome Engineered RNA Immunotherapy: Towards Patient-Centered Tumor Vaccination. J. Immunol. Res. 2015, 2015, 595363. [Google Scholar] [CrossRef] [PubMed]
- Dillen, L.; Cuyckens, F. Quantitative Analysis of Peptides with Mass Spectrometry: Selected Reaction Monitoring or High-Resolution Full Scan? In Mass Spectrometry for Drug Discovery and Drug Development; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 403–425. [Google Scholar]
- Reits, E.; Griekspoor, A.; Neijssen, J.; Groothuis, T.; Jalink, K.; van Veelen, P.; Janssen, H.; Calafat, J.; Drijfhout, J.W.; Neefjes, J. Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity 2003, 18, 97–108. [Google Scholar] [CrossRef]
- Kim, J.H.; Kang, G.H. Molecular and prognostic heterogeneity of microsatellite-unstable colorectal cancer. World J. Gastroenterol. 2014, 20, 4230–4243. [Google Scholar] [CrossRef]
- Kloor, M.; Michel, S.; Buckowitz, B.; Rüschoff, J.; Büttner, R.; Holinski-Feder, E.; Dippold, W.; Wagner, R.; Tariverdian, M.; Benner, A.; et al. Beta2-microglobulin mutations in microsatellite unstable colorectal tumors. Int. J. Cancer 2007, 121, 454–458. [Google Scholar] [CrossRef]
- Tikidzhieva, A.; Benner, A.; Michel, S.; Formentini, A.; Link, K.-H.; Dippold, W.; von Knebel Doeberitz, M.; Kornmann, M.; Kloor, M. Microsatellite instability and Beta2-Microglobulin mutations as prognostic markers in colon cancer: Results of the FOGT-4 trial. Br. J. Cancer 2012, 106, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, M.; Janikovits, J.; von Knebel Doeberitz, M.; Kloor, M. Complex pattern of immune evasion in MSI colorectal cancer. Oncoimmunology 2018, 7, 1–10. [Google Scholar] [CrossRef]
- Satoh, A.; Toyota, M.; Ikeda, H.; Morimoto, Y.; Akino, K.; Mita, H.; Suzuki, H.; Sasaki, Y.; Kanaseki, T.; Takamura, Y.; et al. Epigenetic inactivation of class II transactivator (CIITA) is associated with the absence of interferon-gamma-induced HLA-DR expression in colorectal and gastric cancer cells. Oncogene 2004, 23, 8876–8886. [Google Scholar] [CrossRef]
- Michel, S.; Linnebacher, M.; Alcaniz, J.; Voss, M.; Wagner, R.; Dippold, W.; Becker, C.; von Knebel Doeberitz, M.; Ferrone, S.; Kloor, M. Lack of HLA class II antigen expression in microsatellite unstable colorectal carcinomas is caused by mutations in HLA class II regulatory genes. Int. J. Cancer 2010, 127, 889–898. [Google Scholar] [CrossRef]
- Boland, P.M.; Yurgelun, M.B.; Boland, C.R. Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA. Cancer J. Clin. 2018, 68, 217–231. [Google Scholar] [CrossRef]
- Echterdiek, F.; Janikovits, J.; Staffa, L.; Müller, M.; Lahrmann, B.; Frühschütz, M.; Hartog, B.; Nelius, N.; Benner, A.; Tariverdian, M.; et al. Low density of FOXP3-positive T cells in normal colonic mucosa is related to the presence of beta2-microglobulin mutations in Lynch syndrome-associated colorectal cancer. Oncoimmunology 2016, 5, e1075692. [Google Scholar] [CrossRef]
- Janikovits, J.; Müller, M.; Krzykalla, J.; Körner, S.; Echterdiek, F.; Lahrmann, B.; Grabe, N.; Schneider, M.; Benner, A.; Doeberitz, M.; et al. High numbers of PDCD1 (PD-1)-positive T cells and B2M mutations in microsatellite-unstable colorectal cancer. Oncoimmunology 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Cohen, R.; Pellat, A.; Boussion, H.; Svrcek, M.; Lopez-Trabada, D.; Trouilloud, I.; Afchain, P.; André, T. Immunotherapy and metastatic colorectal cancers with microsatellite instability or mismatch repair deficiency. Bull. Cancer 2019, 106, 137–142. [Google Scholar] [CrossRef]
- Sveen, A.; Johannessen, B.; Tengs, T.; Danielsen, S.A.; Eilertsen, I.A.; Lind, G.E.; Berg, K.C.G.; Leithe, E.; Meza-Zepeda, L.A.; Domingo, E.; et al. Multilevel genomics of colorectal cancers with microsatellite instability-clinical impact of JAK1 mutations and consensus molecular subtype 1. Genome Med. 2017, 9, 1–16. [Google Scholar] [CrossRef]
- Feng, Y.; Cao, Y.; Yuan, M.; Chen, R.; Ji, X.; Hu, X. Different responses to anti-programmed cell death protein 1 (PD-1) immunotherapy in a patient with Lynch syndrome and metachronous urothelial and colon cancer: A case report. Oncol. Lett. 2019, 18, 5085–5090. [Google Scholar] [CrossRef]
- Sahin, I.H.; Akce, M.; Alese, O.; Shaib, W.; Lesinski, G.B.; El-Rayes, B.; Wu, C. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br. J. Cancer 2019, 121, 809–818. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abidi, A.; Gorris, M.A.J.; Brennan, E.; Jongmans, M.C.J.; Weijers, D.D.; Kuiper, R.P.; de Voer, R.M.; Hoogerbrugge, N.; Schreibelt, G.; de Vries, I.J.M. Challenges of Neoantigen Targeting in Lynch Syndrome and Constitutional Mismatch Repair Deficiency Syndrome. Cancers 2021, 13, 2345. https://doi.org/10.3390/cancers13102345
Abidi A, Gorris MAJ, Brennan E, Jongmans MCJ, Weijers DD, Kuiper RP, de Voer RM, Hoogerbrugge N, Schreibelt G, de Vries IJM. Challenges of Neoantigen Targeting in Lynch Syndrome and Constitutional Mismatch Repair Deficiency Syndrome. Cancers. 2021; 13(10):2345. https://doi.org/10.3390/cancers13102345
Chicago/Turabian StyleAbidi, Asima, Mark A. J. Gorris, Evan Brennan, Marjolijn C. J. Jongmans, Dilys D. Weijers, Roland P. Kuiper, Richarda M. de Voer, Nicoline Hoogerbrugge, Gerty Schreibelt, and I. Jolanda M. de Vries. 2021. "Challenges of Neoantigen Targeting in Lynch Syndrome and Constitutional Mismatch Repair Deficiency Syndrome" Cancers 13, no. 10: 2345. https://doi.org/10.3390/cancers13102345
APA StyleAbidi, A., Gorris, M. A. J., Brennan, E., Jongmans, M. C. J., Weijers, D. D., Kuiper, R. P., de Voer, R. M., Hoogerbrugge, N., Schreibelt, G., & de Vries, I. J. M. (2021). Challenges of Neoantigen Targeting in Lynch Syndrome and Constitutional Mismatch Repair Deficiency Syndrome. Cancers, 13(10), 2345. https://doi.org/10.3390/cancers13102345