
Diet-Induced Obesity Impairs Outcomes and Induces Multi-Factorial Deficiencies in Effector T Cell Responses Following Anti-CTLA-4 Combinatorial Immunotherapy in Renal Tumor-Bearing Mice

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. Murine In Vivo Tumor Modeling
2.3. NanoString Immunogenetic Profiling
2.4. Flow Cytometry
2.5. Tumor-Specific Protein Quantification
2.6. Statistical Analysis
3. Results
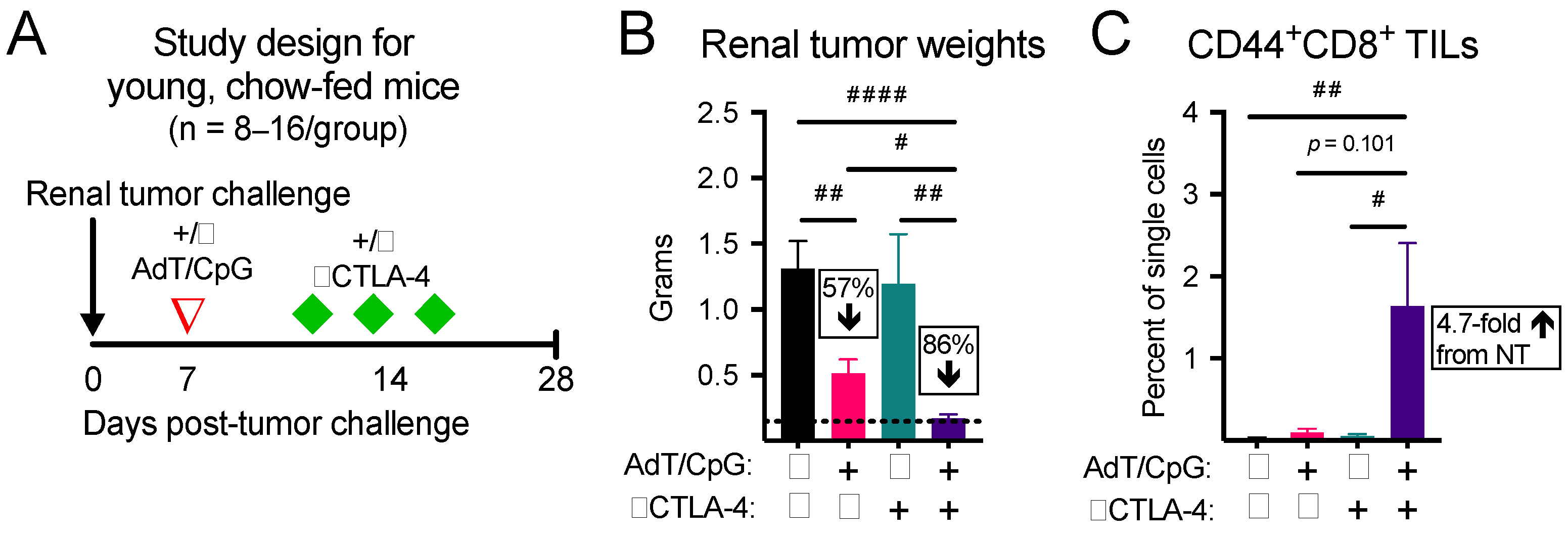
3.1. Combinatorial Therapy Induces the Greatest Reduction in Tumor Growth and Expansion in CD44+CD8+ Tumor-Infiltrating Lymphocytes (TILs) in Young, Chow-Fed Mice
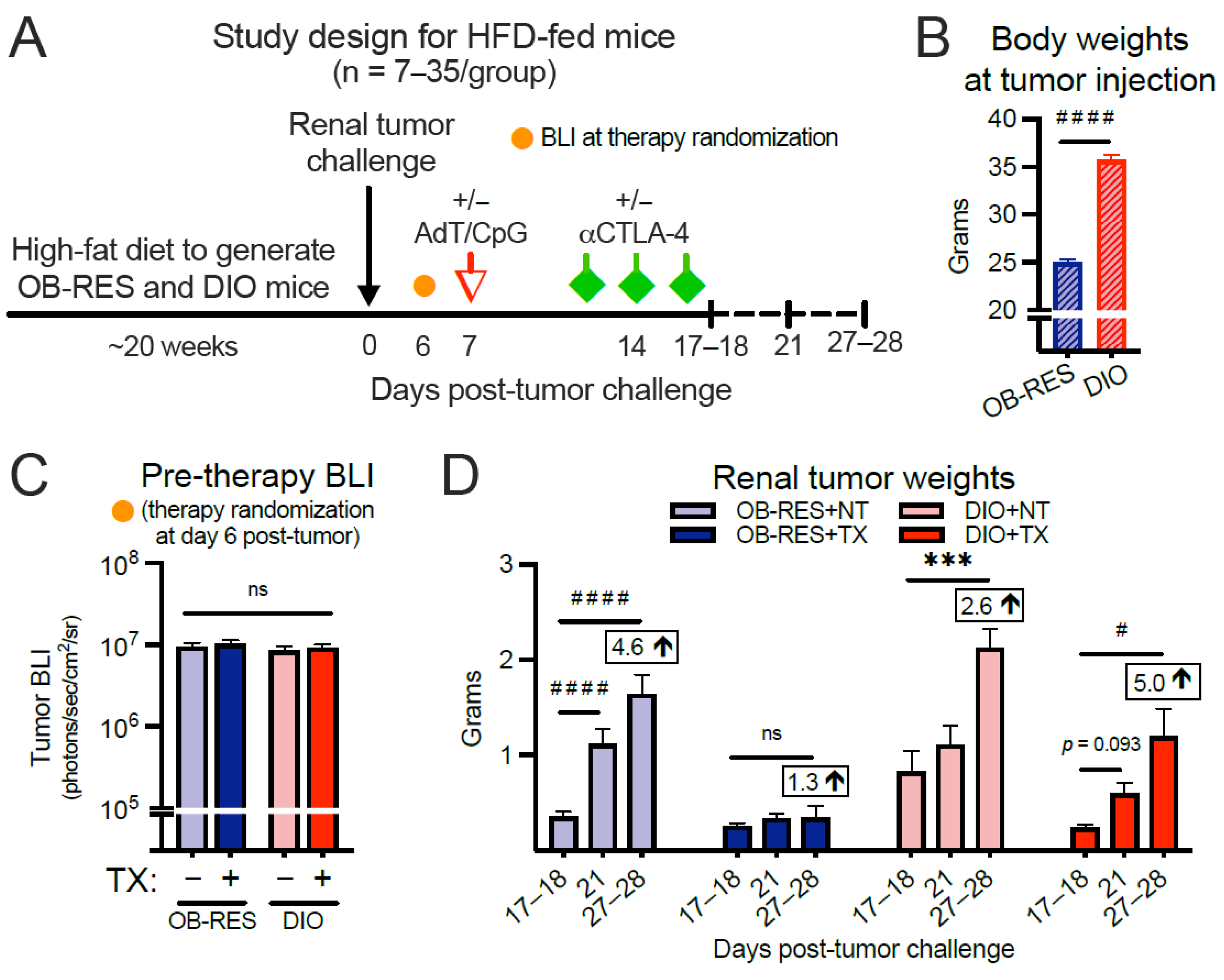
3.2. Therapy-Treated OB-RES Mice Have a Sustained Reduction in Tumor Growth over Time; Therapy-Treated DIO Mice Experience an Acute, Transient Response Followed by Tumor Outgrowth
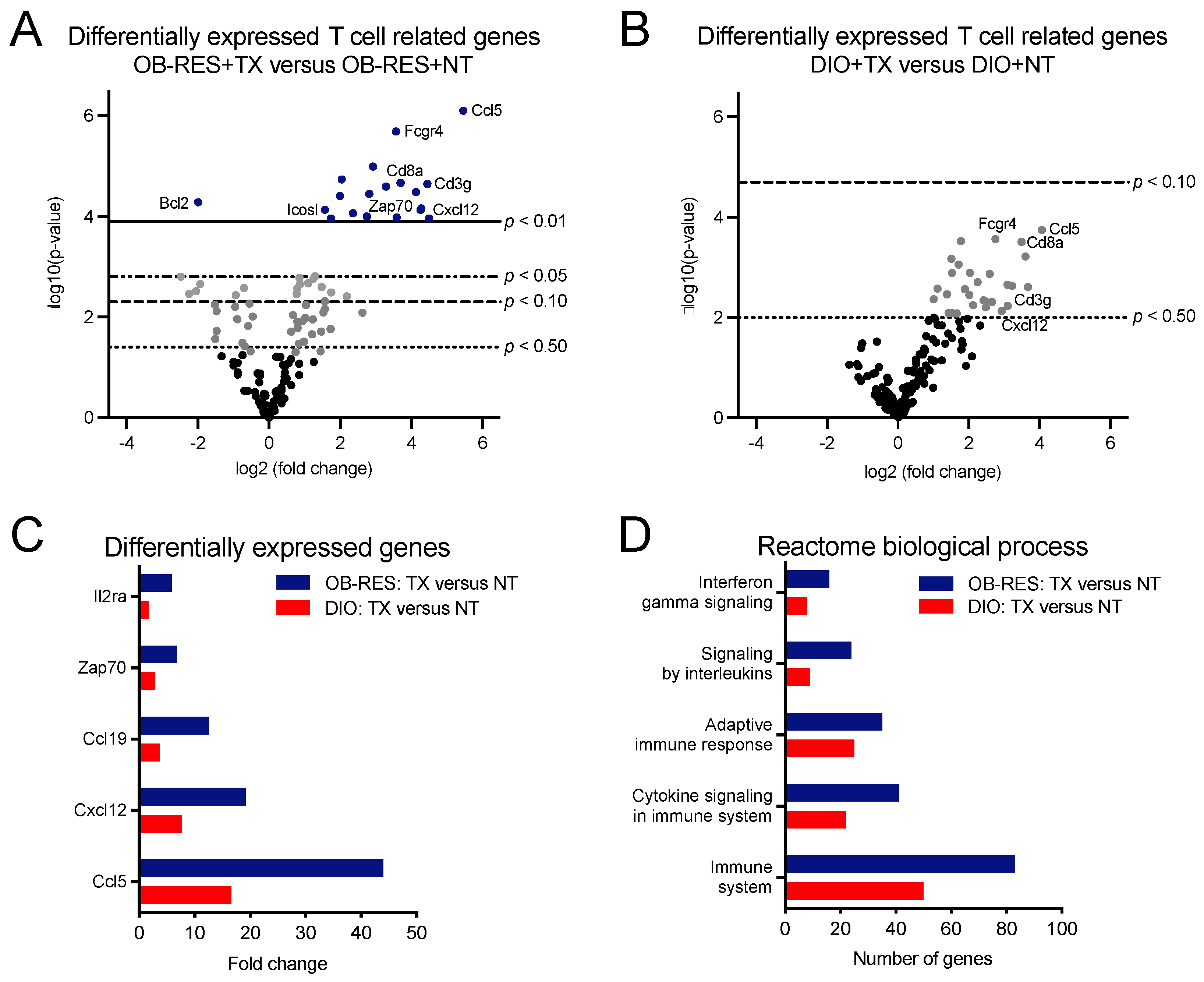
3.3. Therapy-Treated OB-RES Mice Display an Immunogenetic Profile Characterized by Increased Expression of T Cell Related Genes and Biological Processes
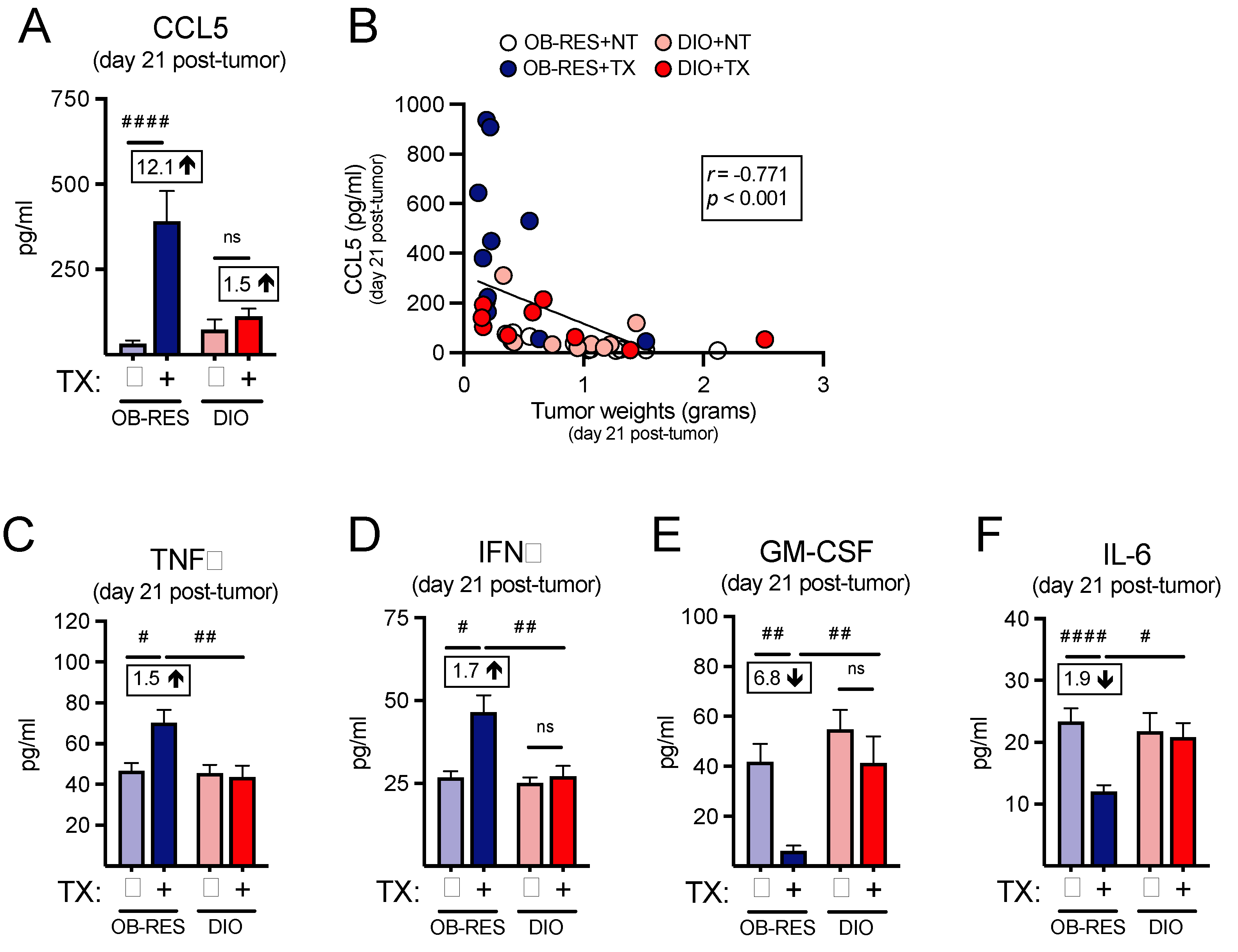
3.4. Therapy-Treated OB-RES Mice Possess an Intratumoral Milieu Favoring T Cell Chemoattractants and Reductions in Pro-Inflammatory Mediators
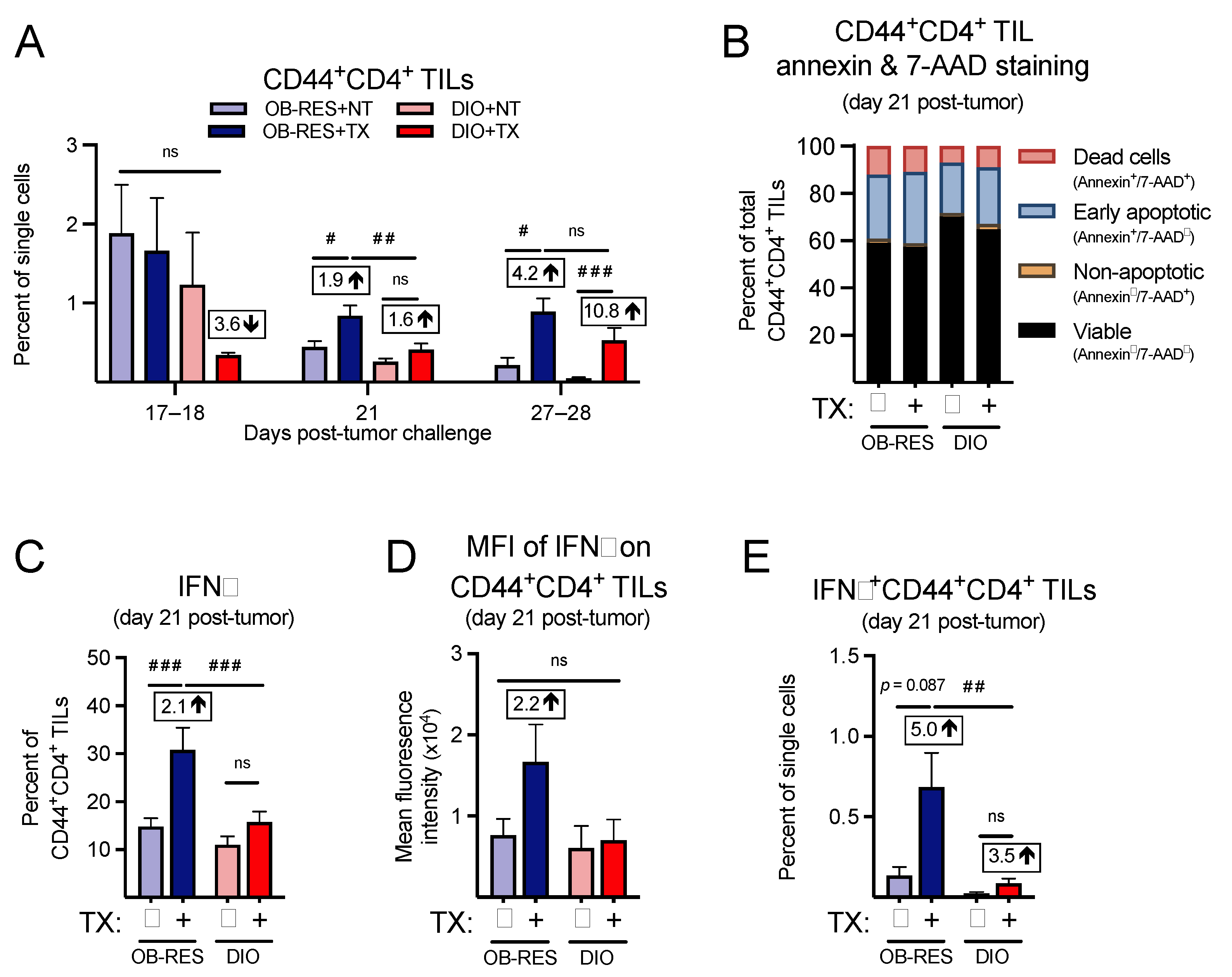
3.5. Therapy-Treated OB-RES Mice Have an Enhanced CD44+CD4+ TIL Response
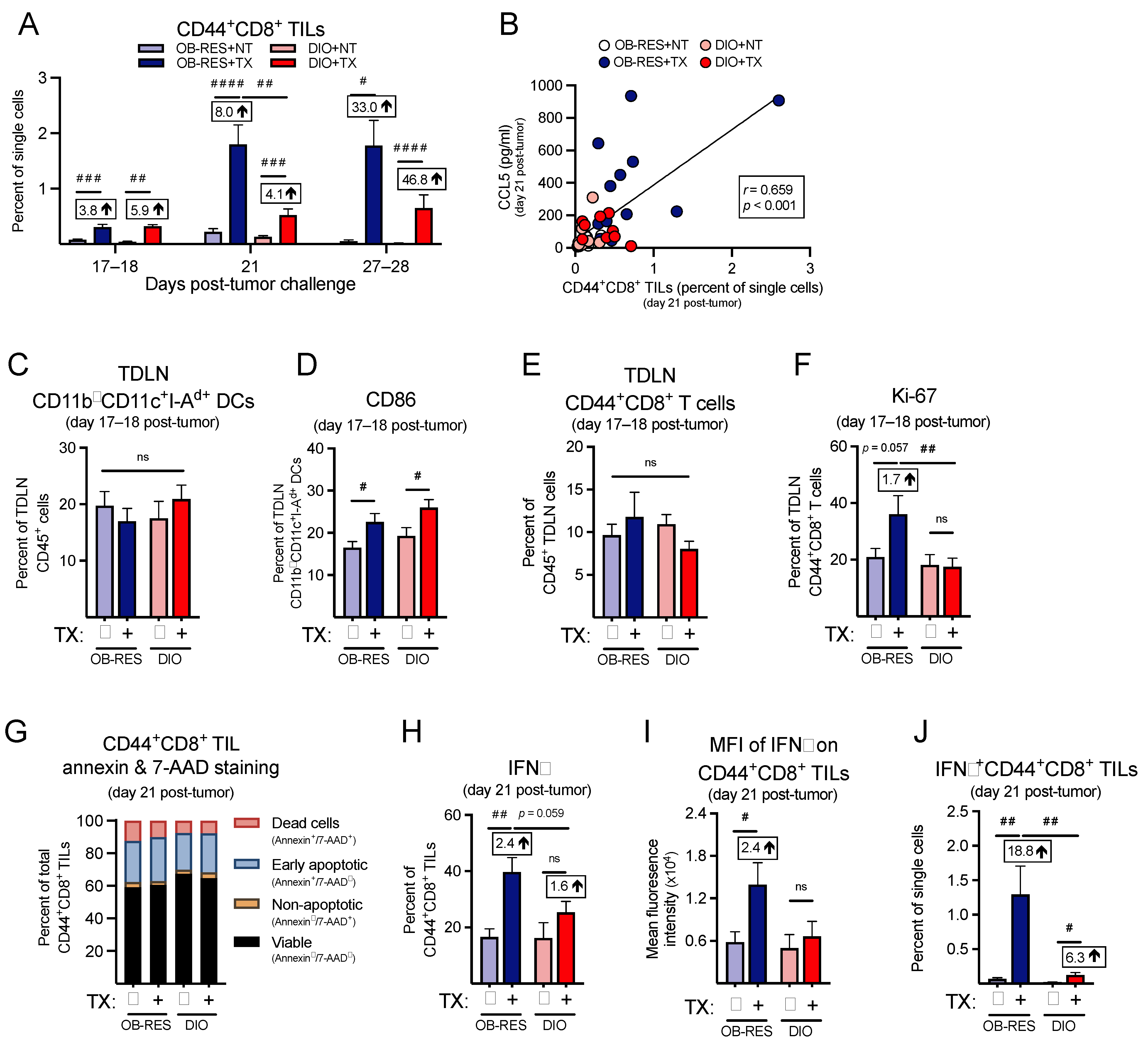
3.6. Therapy-Treated OB-RES Mice Have a Sustained CD44+CD8+ TIL Response over Time
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hales, C.M.; Fryar, C.D.; Carroll, M.D.; Freedman, D.S.; Ogden, C.L. Trends in Obesity and Severe Obesity Prevalence in US Youth and Adults by Sex and Age, 2007–2008 to 2015–2016. JAMA 2018, 319, 1723–1725. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [PubMed]
- James, B.R.; Tomanek-Chalkley, A.; Askeland, E.J.; Kucaba, T.; Griffith, T.S.; Norian, L.A. Diet-Induced Obesity Alters Dendritic Cell Function in the Presence and Absence of Tumor Growth. J. Immunol. 2012, 189, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Hale, M.; Itani, F.; Buchta, C.M.; Wald, G.; Bing, M.; Norian, L.A. Obesity Triggers Enhanced MDSC Accumulation in Murine Renal Tumors via Elevated Local Production of CCL2. PLoS ONE 2015, 10, e0118784. [Google Scholar] [CrossRef]
- Kolb, R.; Phan, L.; Borcherding, N.; Liu, Y.; Yuan, F.; Janowski, A.M.; Xie, Q.; Markan, K.R.; Li, W.; Potthoff, M.J.; et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat. Commun. 2016, 7, 13007. [Google Scholar] [CrossRef]
- Incio, J.; Ligibel, J.A.; McManus, D.T.; Suboj, P.; Jung, K.; Kawaguchi, K.; Pinter, M.; Babykutty, S.; Chin, S.M.; Vardam, T.D.; et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci. Transl. Med. 2018, 10, eaag0945. [Google Scholar] [CrossRef]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef]
- Turbitt, W.J.; Collins, S.D.; Meng, H.; Rogers, C.J. Increased Adiposity Enhances the Accumulation of MDSCs in the Tumor Microenvironment and Adipose Tissue of Pancreatic Tumor-Bearing Mice and in Immune Organs of Tumor-Free Hosts. Nutrients 2019, 11, 3012. [Google Scholar] [CrossRef]
- Boi, S.K.; Orlandella, R.M.; Gibson, J.T.; Turbitt, W.J.; Wald, G.; Thomas, L.; Rosean, C.B.; Norris, K.E.; Bing, M.; Bertrand, L.; et al. Obesity diminishes response to PD-1-based immunotherapies in renal cancer. J. Immunother. Cancer 2020, 8, e000725. [Google Scholar] [CrossRef]
- Gibson, J.T.; Orlandella, R.M.; Turbitt, W.J.; Behring, M.; Manne, U.; Sorge, R.E.; Norian, L.A. Obesity-Associated Myeloid-Derived Suppressor Cells Promote Apoptosis of Tumor-Infiltrating CD8 T Cells and Immunotherapy Resistance in Breast Cancer. Front. Immunol. 2020, 11, 590794. [Google Scholar] [CrossRef]
- Quail, D.F.; Olson, O.C.; Bhardwaj, P.; Walsh, L.A.; Akkari, L.; Quick, M.L.; Chen, I.-C.; Wendel, N.; Ben-Chetrit, N.; Walker, J.; et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat. Cell Biol. 2017, 19, 974–987. [Google Scholar] [CrossRef]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; García-Cañaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866. [Google Scholar] [CrossRef] [PubMed]
- Turbitt, W.J.; Rosean, C.B.; Weber, K.S.; Norian, L.A. Obesity and CD8 T cell metabolism: Implications for anti-tumor immunity and cancer immunotherapy outcomes. Immunol. Rev. 2020, 295, 203–219. [Google Scholar] [CrossRef] [PubMed]
- McCune, J.S. Rapid Advances in Immunotherapy to Treat Cancer. Clin. Pharmacol. Ther. 2018, 103, 540–544. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Rini, B.I.; Battle, D.; Figlin, R.A.; George, D.J.; Hammers, H.; Hutson, T.; Jonasch, E.; Joseph, R.W.; McDermott, D.F.; Motzer, R.J.; et al. The society for immunotherapy of cancer consensus statement on immunotherapy for the treatment of advanced renal cell carcinoma (RCC). J. Immunother. Cancer 2019, 7, 354. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef]
- Canter, R.J.; Le, C.T.; Beerthuijzen, J.M.; Murphy, W.J. Obesity as an immune-modifying factor in cancer immunotherapy. J. Leukoc. Biol. 2018, 104, 487–497. [Google Scholar] [CrossRef]
- Ba, D.J.M.; Ba, M.R.K.; Liu, Y.; Ba, J.M.S.; Williams, M.A.; Khan, A.I.; Lewis, C.; Np, H.C.; Akce, M.; Kissick, H.T.; et al. Adiposity may predict survival in patients with advanced stage cancer treated with immunotherapy in phase 1 clinical trials. Cancer 2020, 126, 575–582. [Google Scholar] [CrossRef]
- Turbitt, W.J.; Demark-Wahnefried, W.; Peterson, C.M.; Norian, L.A. Targeting Glucose Metabolism to Enhance Immunotherapy: Emerging Evidence on Intermittent Fasting and Calorie Restriction Mimetics. Front. Immunol. 2019, 10, 1402. [Google Scholar] [CrossRef]
- Warner, A.B.; McQuade, J.L. Modifiable Host Factors in Melanoma: Emerging Evidence for Obesity, Diet, Exercise, and the Microbiome. Curr. Oncol. Rep. 2019, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Young, A.C.; Quach, H.T.; Song, H.; Davis, E.J.; Moslehi, J.J.; Ye, F.; Williams, G.R.; Johnson, D.B. Impact of body composition on outcomes from anti-PD1+/− anti-CTLA-4 treatment in melanoma. J. Immunother. Cancer 2012, 8, e000821. [Google Scholar] [CrossRef]
- Woodall, M.J.; Neumann, S.; Campbell, K.; Pattison, S.T.; Young, S.L. The Effects of Obesity on Anti-Cancer Immunity and Cancer Immunotherapy. Cancers 2020, 12, 1230. [Google Scholar] [CrossRef]
- Li, M.; Bu, R. Biological Support to Obesity Paradox in Renal Cell Carcinoma: A Review. Urol. Int. 2020, 104, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cao, D.; He, A.; Ge, W. The prognostic role of obesity is independent of sex in cancer patients treated with immune checkpoint inhibitors: A pooled analysis of 4090 cancer patients. Int. Immunopharmacol. 2019, 74, 105745. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Furberg, H.; Kuo, F.; Vuong, L.; Ged, Y.; Patil, S.; Ostrovnaya, I.; Petruzella, S.; Reising, A.; Patel, P.; et al. Transcriptomic signatures related to the obesity paradox in patients with clear cell renal cell carcinoma: A cohort study. Lancet Oncol. 2020, 21, 283–293. [Google Scholar] [CrossRef]
- James, B.R.; Anderson, K.G.; Brincks, E.L.; Kucaba, T.A.; Norian, L.A.; Masopust, D.; Griffith, T.S. CpG-mediated modulation of MDSC contributes to the efficacy of Ad5-TRAIL therapy against renal cell carcinoma. Cancer Immunol. Immunother. 2014, 63, 1213–1227. [Google Scholar] [CrossRef]
- Murphy, K.A.; James, B.R.; Sjaastad, F.V.; Kucaba, T.A.; Kim, H.; Brincks, E.L.; Chua, S.C.; Wilber, A.; Griffith, T.S. Cutting Edge: Elevated Leptin during Diet-Induced Obesity Reduces the Efficacy of Tumor Immunotherapy. J. Immunol. 2018, 201, 1837–1841. [Google Scholar] [CrossRef]
- Boi, S.K.; Buchta, C.M.; Pearson, N.A.; Francis, M.B.; Meyerholz, D.K.; Grobe, J.L.; Norian, L.A. Obesity alters immune and metabolic profiles: New insight from obese-resistant mice on high-fat diet. Obesisty 2016, 24, 2140–2149. [Google Scholar] [CrossRef]
- Norian, L.A.; Kresowik, T.P.; Rosevear, H.M.; James, B.R.; Rosean, T.R.; Lightfoot, A.J.; Kucaba, T.A.; Schwarz, C.; Weydert, C.J.; Henry, M.D.; et al. Eradication of Metastatic Renal Cell Carcinoma after Adenovirus-Encoded TNF-Related Apoptosis-Inducing Ligand (TRAIL)/CpG Immunotherapy. PLoS ONE 2012, 7, e31085. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Sheng, I.Y.; Ornstein, M.C. Ipilimumab and Nivolumab as First-Line Treatment of Patients with Renal Cell Carcinoma: The Evidence to Date. Cancer Manag. Res. 2020, 12, 4871–4881. [Google Scholar] [CrossRef]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Richmond, A. Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Front. Immunol. 2019, 10, 333. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S. Myeloid derived-suppressor cells: Their role in cancer and obesity. Curr. Opin. Immunol. 2018, 51, 68–75. [Google Scholar] [CrossRef]
- Bähr, I.; Goritz, V.; Doberstein, H.; Hiller, G.G.R.; Rosenstock, P.; Jahn, J.; Pörtner, O.; Berreis, T.; Mueller, T.; Spielmann, J.; et al. Diet-Induced Obesity Is Associated with an Impaired NK Cell Function and an Increased Colon Cancer Incidence. J. Nutr. Metab. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2020, 31, 148–161. [Google Scholar] [CrossRef]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef]
- Tenold, M.; Ravi, P.; Kumar, M.; Bowman, A.; Hammers, H.; Choueiri, T.K.; Lara, P.N. Current Approaches to the Treatment of Advanced or Metastatic Renal Cell Carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.-L.; Shen, K.-Y.; Tien, C.-Y.; Chen, Y.-A.; Liu, S.-J. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017, 9, 347–360. [Google Scholar] [CrossRef]
- Poeta, V.M.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Cachot, A.; Bilous, M.; Liu, Y.-C.; Li, X.; Saillard, M.; Cenerenti, M.; Rockinger, G.A.; Wyss, T.; Guillaume, P.; Schmidt, J.; et al. Tumor-specific cytolytic CD4 T cells mediate immunity against human cancer. Sci. Adv. 2021, 7, eabe3348. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—New insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef] [PubMed]
- Neel, N.F.; Schutyser, E.; Sai, J.; Fan, G.-H.; Richmond, A. Chemokine receptor internalization and intracellular trafficking. Cytokine Growth Factor Rev. 2005, 16, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Turbitt, W.J.; Orlandella, R.M.; Gibson, J.T.; Peterson, C.M.; Norian, L.A. Therapeutic Time-restricted Feeding Reduces Renal Tumor Bioluminescence in Mice but Fails to Improve Anti-CTLA-4 Efficacy. Anticancer Res. 2020, 40, 5445–5456. [Google Scholar] [CrossRef]
- Orlandella, R.M.; Turbitt, W.J.; Gibson, J.T.; Boi, S.K.; Li, P.; Smith, J.D.L.; Norian, L.A. The Antidiabetic Agent Acarbose Improves Anti-PD-1 and Rapamycin Efficacy in Preclinical Renal Cancer. Cancers 2020, 12, 2872. [Google Scholar] [CrossRef] [PubMed]
- Klevorn, L.E.; Teague, R.M. Adapting Cancer Immunotherapy Models for the Real World. Trends Immunol. 2016, 37, 354–363. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turbitt, W.J.; Boi, S.K.; Gibson, J.T.; Orlandella, R.M.; Norian, L.A. Diet-Induced Obesity Impairs Outcomes and Induces Multi-Factorial Deficiencies in Effector T Cell Responses Following Anti-CTLA-4 Combinatorial Immunotherapy in Renal Tumor-Bearing Mice. Cancers 2021, 13, 2295. https://doi.org/10.3390/cancers13102295
Turbitt WJ, Boi SK, Gibson JT, Orlandella RM, Norian LA. Diet-Induced Obesity Impairs Outcomes and Induces Multi-Factorial Deficiencies in Effector T Cell Responses Following Anti-CTLA-4 Combinatorial Immunotherapy in Renal Tumor-Bearing Mice. Cancers. 2021; 13(10):2295. https://doi.org/10.3390/cancers13102295
Chicago/Turabian StyleTurbitt, William J., Shannon K. Boi, Justin T. Gibson, Rachael M. Orlandella, and Lyse A. Norian. 2021. "Diet-Induced Obesity Impairs Outcomes and Induces Multi-Factorial Deficiencies in Effector T Cell Responses Following Anti-CTLA-4 Combinatorial Immunotherapy in Renal Tumor-Bearing Mice" Cancers 13, no. 10: 2295. https://doi.org/10.3390/cancers13102295
APA StyleTurbitt, W. J., Boi, S. K., Gibson, J. T., Orlandella, R. M., & Norian, L. A. (2021). Diet-Induced Obesity Impairs Outcomes and Induces Multi-Factorial Deficiencies in Effector T Cell Responses Following Anti-CTLA-4 Combinatorial Immunotherapy in Renal Tumor-Bearing Mice. Cancers, 13(10), 2295. https://doi.org/10.3390/cancers13102295