Immunotherapy in Solid Tumors and Gut Microbiota: The Correlation—A Special Reference to Colorectal Cancer

 , , and
, , and 
Abstract
Simple Summary
Abstract
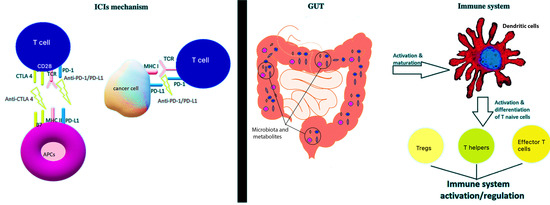
1. Introduction
2. Immunotherapy in Solid Tumors
3. Gut Microbiota and Immunotherapy
3.1. Interference of Gut Microbiota and Immunity
3.2. Correlation of Gut Microbiota and ICIs Efficacy: The Proofs
3.3. Gut Microbiota Modulators and Their Impact on ICIs Efficacy
4. Immunotherapy and Microbiota in CRC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Decker, W.K.; da Silva, R.F.; Sanabria, M.H.; Angelo, L.S.; Guimaraes, F.; Burt, B.M.; Kheradmand, F.; Paust, S. Cancer Immunotherapy: Historical Perspective of a Clinical Revolution and Emerging Preclinical Animal Models. Front. Immunol. 2017, 8, 829. [Google Scholar] [CrossRef]
- Pio, R.; Ajona, D.; Ortiz-Espinosa, S.; Mantovani, A.; Lambris, J.D. Complementing the Cancer-Immunity Cycle. Front. Immunol. 2019, 10, 774. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70 (Suppl. 1), S38–S44. [Google Scholar] [CrossRef] [PubMed]
- Messaritakis, I.; Vogiatzoglou, K.; Tsantaki, K.; Ntretaki, A.; Sfakianaki, M.; Koulouridi, A.; Tsiaoussis, J.; Mavroudis, D.; Souglakos, J. The Prognostic Value of the Detection of Microbial Translocation in the Blood of Colorectal Cancer Patients. Cancers 2020, 12, 1058. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Derosa, L.; Routy, B.; Kroemer, G.; Zitvogel, L. The intestinal microbiota determines the clinical efficacy of immune checkpoint blockers targeting PD-1/PD-L1. Oncoimmunology 2018, 7, e1434468. [Google Scholar] [CrossRef]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Dubin, K.; Callahan, M.K.; Ren, B.; Khanin, R.; Viale, A.; Ling, L.; No, D.; Gobourne, A.; Littmann, E.; Huttenhower, C.; et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat. Commun. 2016, 7, 10391. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 2017, 28, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Mimura, K.; Ashizawa, M.; Okayama, H.; Endo, E.; Saito, K.; Sakamoto, W.; Fujita, S.; Endo, H.; Saito, M.; et al. Characterization of tumor-infiltrating immune cells in relation to microbiota in colorectal cancers. Cancer Immunol. Immunother. 2020, 69, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, P.; Ahmed, S. Current status of immunotherapy in metastatic colorectal cancer. Int. J. Colorectal Dis. 2019, 34, 13–25. [Google Scholar] [CrossRef]
- Santhanam, S.; Alvarado, D.M.; Ciorba, M.A. Therapeutic targeting of inflammation and tryptophan metabolism in colon and gastrointestinal cancer. Transl. Res. 2016, 167, 67–79. [Google Scholar] [CrossRef]
- Zhuo, Q.; Yu, B.; Zhou, J.; Zhang, J.; Zhang, R.; Xie, J.; Wang, Q.; Zhao, S. Lysates of Lactobacillus acidophilus combined with CTLA-4-blocking antibodies enhance antitumor immunity in a mouse colon cancer model. Sci. Rep. 2019, 9, 20128. [Google Scholar] [CrossRef]
- Xu, X.; Lv, J.; Guo, F.; Li, J.; Jia, Y.; Jiang, D.; Wang, N.; Zhang, C.; Kong, L.; Liu, Y.; et al. Gut Microbiome Influences the Efficacy of PD-1 Antibody Immunotherapy on MSS-Type Colorectal Cancer via Metabolic Pathway. Front. Microbiol. 2020, 11, 814. [Google Scholar] [CrossRef]
- Mariotto, A.B.; Yabroff, K.R.; Shao, Y.; Feuer, E.J.; Brown, M.L. Projections of the cost of cancer care in the United States: 2010–2020. J. Natl. Cancer Inst. 2011, 103, 117–128. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.R.; Blomberg, O.S.; Rashidian, M.; Ali, L.; Garforth, S.; Fedorov, E.; Fedorov, A.A.; Bonanno, J.B.; Le Gall, C.; Crowley, S.; et al. Anti-CTLA-4 therapy requires an Fc domain for efficacy. Proc. Natl. Acad. Sci. USA 2018, 115, 3912–3917. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Deshmukh, S.; Reddy, A.; Lightcap, J.; Hayes, M.; McClellan, S.; Singh, S.; Rabideau, B.; Glover, T.G.; Roberts, B.; et al. Cancer Immune Checkpoint Inhibitor Therapy and the Gut Microbiota. Integr. Cancer Ther. 2019, 18, 1534735419846379. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 2018, 49, 1148–1161. [Google Scholar] [CrossRef]
- Li, W.; Deng, Y.; Chu, Q.; Zhang, P. Gut microbiome and cancer immunotherapy. Cancer Lett. 2019, 447, 41–47. [Google Scholar] [CrossRef]
- Gandini, S.; Massi, D.; Mandala, M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2016, 100, 88–98. [Google Scholar] [CrossRef]
- Koustas, E.; Sarantis, P.; Papavassiliou, A.G.; Karamouzis, M.V. The Resistance Mechanisms of Checkpoint Inhibitors in Solid Tumors. Biomolecules 2020, 10, 666. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, B.S.; Lee, S.K. Regulatory T Cells in Tumor Microenvironment and Approach for Anticancer Immunotherapy. Immune Netw. 2020, 20, e4. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Matsushita, T.; Tedder, T.F. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann. N. Y. Acad. Sci. 2010, 1183, 38–57. [Google Scholar] [CrossRef]
- Dysthe, M.; Parihar, R. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 117–140. [Google Scholar] [CrossRef] [PubMed]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Maciel, T.T.; Moura, I.C.; Hermine, O. The role of mast cells in cancers. F1000Prime Rep. 2015, 7, 09. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed]
- Crotzer, V.L.; Blum, J.S. Autophagy and its role in MHC-mediated antigen presentation. J. Immunol. 2009, 182, 3335–3341. [Google Scholar] [CrossRef]
- Castle, J.C.; Uduman, M.; Pabla, S.; Stein, R.B.; Buell, J.S. Mutation-Derived Neoantigens for Cancer Immunotherapy. Front. Immunol. 2019, 10, 1856. [Google Scholar] [CrossRef]
- Johnson, C.H.; Spilker, M.E.; Goetz, L.; Peterson, S.N.; Siuzdak, G. Metabolite and Microbiome Interplay in Cancer Immunotherapy. Cancer Res. 2016, 76, 6146–6152. [Google Scholar] [CrossRef]
- Alegre, M.L.; Bromberg, J.S. Commensal microbiota determine intestinal iTreg. Am. J. Transplant. 2012, 12, 1967. [Google Scholar] [CrossRef]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between drugs and the gut microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef]
- Zeromski, J.; Kaczmarek, M.; Boruczkowski, M.; Kierepa, A.; Kowala-Piaskowska, A.; Mozer-Lisewska, I. Significance and Role of Pattern Recognition Receptors in Malignancy. Arch. Immunol. Ther. Exp. 2019, 67, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zhang, J.; Wu, Q.; Fang, H.; Shi, C.; Li, Z.; Lin, C.; Tang, D.; Wang, D. Intestinal microbiota: A new force in cancer immunotherapy. Cell Commun. Signal. 2020, 18, 90. [Google Scholar] [CrossRef] [PubMed]
- Shcheblyakov, D.V.; Logunov, D.Y.; Tukhvatulin, A.I.; Shmarov, M.M.; Naroditsky, B.S.; Gintsburg, A.L. Toll-Like Receptors (TLRs): The Role in Tumor Progression. Acta Nat. 2010, 2, 21–29. [Google Scholar] [CrossRef]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fahraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Wang, L.; Yu, K.; Zhang, X.; Yu, S. Dual functional roles of the MyD88 signaling in colorectal cancer development. Biomed. Pharmacother. 2018, 107, 177–184. [Google Scholar] [CrossRef]
- Lavelle, E.C.; Murphy, C.; O’Neill, L.A.; Creagh, E.M. The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol. 2010, 3, 17–28. [Google Scholar] [CrossRef]
- Miao, E.A.; Andersen-Nissen, E.; Warren, S.E.; Aderem, A. TLR5 and Ipaf: Dual sensors of bacterial flagellin in the innate immune system. Semin. Immunopathol. 2007, 29, 275–288. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef]
- Monie, T.P.; Bryant, C.E.; Gay, N.J. Activating immunity: Lessons from the TLRs and NLRs. Trends Biochem. Sci. 2009, 34, 553–561. [Google Scholar] [CrossRef]
- Haabeth, O.A.W.; Fauskanger, M.; Manzke, M.; Lundin, K.U.; Corthay, A.; Bogen, B.; Tveita, A.A. CD4(+) T-cell-Mediated Rejection of MHC Class II-Positive Tumor Cells Is Dependent on Antigen Secretion and Indirect Presentation on Host APCs. Cancer Res. 2018, 78, 4573–4585. [Google Scholar] [CrossRef] [PubMed]
- Grosserichter-Wagener, C.; Radjabzadeh, D.; van der Weide, H.; Smit, K.N.; Kraaij, R.; Hays, J.P.; van Zelm, M.C. Differences in Systemic IgA Reactivity and Circulating Th Subsets in Healthy Volunteers With Specific Microbiota Enterotypes. Front. Immunol. 2019, 10, 341. [Google Scholar] [CrossRef] [PubMed]
- Maruya, M.; Kawamoto, S.; Kato, L.M.; Fagarasan, S. Impaired selection of IgA and intestinal dysbiosis associated with PD-1-deficiency. Gut Microbes 2013, 4, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, S.; Tran, T.H.; Maruya, M.; Suzuki, K.; Doi, Y.; Tsutsui, Y.; Kato, L.M.; Fagarasan, S. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science 2012, 336, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Kuen, D.S.; Kim, B.S.; Chung, Y. IL-17-Producing Cells in Tumor Immunity: Friends or Foes? Immune Netw. 2020, 20, e6. [Google Scholar] [CrossRef]
- Razi, S.; Baradaran Noveiry, B.; Keshavarz-Fathi, M.; Rezaei, N. IL-17 and colorectal cancer: From carcinogenesis to treatment. Cytokine 2019, 116, 7–12. [Google Scholar] [CrossRef]
- Khazaie, K.; von Boehmer, H. The impact of CD4+CD25+ Treg on tumor specific CD8+ T cell cytotoxicity and cancer. Semin. Cancer Biol. 2006, 16, 124–136. [Google Scholar] [CrossRef]
- Kindlund, B.; Sjoling, A.; Yakkala, C.; Adamsson, J.; Janzon, A.; Hansson, L.E.; Hermansson, M.; Janson, P.; Winqvist, O.; Lundin, S.B. CD4(+) regulatory T cells in gastric cancer mucosa are proliferating and express high levels of IL-10 but little TGF-beta. Gastric. Cancer 2017, 20, 116–125. [Google Scholar] [CrossRef]
- Sun, M.; Wu, W.; Chen, L.; Yang, W.; Huang, X.; Ma, C.; Chen, F.; Xiao, Y.; Zhao, Y.; Yao, S.; et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat. Commun. 2018, 9, 3555. [Google Scholar] [CrossRef]
- Cottrez, F.; Groux, H. Regulation of TGF-beta response during T cell activation is modulated by IL-10. J. Immunol. 2001, 167, 773–778. [Google Scholar] [CrossRef]
- Zhao, R.; Song, Y.; Wang, Y.; Huang, Y.; Li, Z.; Cui, Y.; Yi, M.; Xia, L.; Zhuang, W.; Wu, X.; et al. PD-1/PD-L1 blockade rescue exhausted CD8+ T cells in gastrointestinal stromal tumours via the PI3K/Akt/mTOR signalling pathway. Cell Prolif. 2019, 52, e12571. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The Role Of PD-1/PD-L1 Axis In Treg Development And Function: Implications For Cancer Immunotherapy. Onco Targets Ther. 2019, 12, 8437–8445. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Limeta, A.; Ji, B.; Levin, M.; Gatto, F.; Nielsen, J. Meta-analysis of the gut microbiota in predicting response to cancer immunotherapy in metastatic melanoma. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Cong, J.; Zhang, X. Roles of intestinal microbiota in response to cancer immunotherapy. Eur. J. Clin. Microbiol. Infect Dis. 2018, 37, 2235–2240. [Google Scholar] [CrossRef]
- Ma, N.; Xing, C.; Xiao, H.; Wang, Y.; Wang, K.; Hou, C.; Han, G.; Chen, G.; Marrero, B.; Shen, B.; et al. C5a regulates IL-12+ DC migration to induce pathogenic Th1 and Th17 cells in sepsis. PLoS ONE 2013, 8, e69779. [Google Scholar] [CrossRef] [PubMed]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Li, A.; Wu, K. Manipulating Gut Microbiota Composition to Enhance the Therapeutic Effect of Cancer Immunotherapy. Integr. Cancer Ther. 2019, 18, 1534735419876351. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Roles of microbiota in response to cancer immunotherapy. Semin. Cancer Biol. 2020, 65, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.Y.; Yin, T.L.; Zhou, J.; Xu, J.; Lu, X.J. Gut microbiome and cancer immunotherapy. J. Cell Physiol. 2020, 235, 4082–4088. [Google Scholar] [CrossRef]
- Soldati, L.; Di Renzo, L.; Jirillo, E.; Ascierto, P.A.; Marincola, F.M.; De Lorenzo, A. The influence of diet on anti-cancer immune responsiveness. J. Transl. Med. 2018, 16, 75. [Google Scholar] [CrossRef]
- Inamura, K. Gut microbiota contributes towards immunomodulation against cancer: New frontiers in precision cancer therapeutics. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Spakowicz, D.; Hoyd, R.; Muniak, M.; Husain, M.; Bassett, J.S.; Wang, L.; Tinoco, G.; Patel, S.H.; Burkart, J.; Miah, A.; et al. Inferring the role of the microbiome on survival in patients treated with immune checkpoint inhibitors: Causal modeling, timing, and classes of concomitant medications. BMC Cancer 2020, 20, 383. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Pezzuto, A.; Sini, C.; Tuzi, A.; Citarella, F.; McCusker, M.G.; Nigro, O.; Tanda, E.; Russo, A. Concomitant medications during immune checkpoint blockage in cancer patients: Novel insights in this emerging clinical scenario. Crit. Rev. Oncol. Hematol. 2019, 142, 26–34. [Google Scholar] [CrossRef]
- Bruno, G.; Zaccari, P.; Rocco, G.; Scalese, G.; Panetta, C.; Porowska, B.; Pontone, S.; Severi, C. Proton pump inhibitors and dysbiosis: Current knowledge and aspects to be clarified. World J. Gastroenterol. 2019, 25, 2706–2719. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyas, E.; Martin-Castillo, B.; Menendez, J.A. Metformin as an archetype immuno-metabolic adjuvant for cancer immunotherapy. Oncoimmunology 2019, 8, e1633235. [Google Scholar] [CrossRef]
- Chalabi, M.; Cardona, A.; Nagarkar, D.R.; Dhawahir Scala, A.; Gandara, D.R.; Rittmeyer, A.; Albert, M.L.; Powles, T.; Kok, M.; Herrera, F.G. Efficacy of chemotherapy and atezolizumab in patients with non-small-cell lung cancer receiving antibiotics and proton pump inhibitors: Pooled post hoc analyses of the OAK and POPLAR trials. Ann. Oncol. 2020, 31, 525–531. [Google Scholar] [CrossRef]
- Hopkins, A.M.; Kichenadasse, G.; Karapetis, C.S.; Rowland, A.; Sorich, M.J. Concomitant Proton Pump Inhibitor Use and Survival in Urothelial Carcinoma Treated with Atezolizumab. Clin. Cancer Res. 2020, 26, 5487–5493. [Google Scholar] [CrossRef]
- Robertson, D.J.; Larsson, H.; Friis, S.; Pedersen, L.; Baron, J.A.; Sorensen, H.T. Proton pump inhibitor use and risk of colorectal cancer: A population-based, case-control study. Gastroenterology 2007, 133, 755–760. [Google Scholar] [CrossRef]
- Yang, Y.X.; Hennessy, S.; Propert, K.; Hwang, W.T.; Sedarat, A.; Lewis, J.D. Chronic proton pump inhibitor therapy and the risk of colorectal cancer. Gastroenterology 2007, 133, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.Y.; Wang, J.H.; Yi, C.H.; Liu, T.T.; Hung, J.S.; Wong, M.W.; Bair, M.J.; Vaezi, M.F.; Orr, W.C.; Chen, C.L. Association between use of proton pump inhibitors and colorectal cancer: A nationwide population-based study. Clin. Res. Hepatol. Gastroenterol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.H.; Chang, H.W.; Lin, I.H.; Chien, L.N.; Wu, M.J.; Liu, Y.R.; Chu, P.G.; Xie, G.; Dong, F.; Jia, W.; et al. Long-term Proton Pump Inhibitor Administration Caused Physiological and Microbiota Changes in Rats. Sci. Rep. 2020, 10, 866. [Google Scholar] [CrossRef] [PubMed]
- Imhann, F.; Bonder, M.J.; Vich Vila, A.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef]
- Jackson, M.A.; Goodrich, J.K.; Maxan, M.E.; Freedberg, D.E.; Abrams, J.A.; Poole, A.C.; Sutter, J.L.; Welter, D.; Ley, R.E.; Bell, J.T.; et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut 2016, 65, 749–756. [Google Scholar] [CrossRef]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Ma, W.; Chen, J.; Meng, Y.; Yang, J.; Cui, Q.; Zhou, Y. Metformin Alters Gut Microbiota of Healthy Mice: Implication for Its Potential Role in Gut Microbiota Homeostasis. Front. Microbiol. 2018, 9, 1336. [Google Scholar] [CrossRef]
- Kyriachenko, Y.; Falalyeyeva, T.; Korotkyi, O.; Molochek, N.; Kobyliak, N. Crosstalk between gut microbiota and antidiabetic drug action. World J. Diabetes 2019, 10, 154–168. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Huang, X.; Hong, X.; Wang, J.; Sun, T.; Yu, T.; Yu, Y.; Fang, J.; Xiong, H. Metformin elicits antitumour effect by modulation of the gut microbiota and rescues Fusobacterium nucleatum-induced colorectal tumourigenesis. EBioMedicine 2020, 61, 103037. [Google Scholar] [CrossRef]
- Jones, G.R.; Molloy, M.P. Metformin, Microbiome and Protection Against Colorectal Cancer. Dig. Dis. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Higurashi, T.; Hosono, K.; Takahashi, H.; Komiya, Y.; Umezawa, S.; Sakai, E.; Uchiyama, T.; Taniguchi, L.; Hata, Y.; Uchiyama, S.; et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: A multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016, 17, 475–483. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Provenzale, D.; Gupta, S.; Ahnen, D.J.; Markowitz, A.J.; Chung, D.C.; Mayer, R.J.; Regenbogen, S.E.; Blanco, A.M.; Bray, T.; Cooper, G.; et al. NCCN Guidelines Insights: Colorectal Cancer Screening, Version 1.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 939–949. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Coleman, O.I.; Lobner, E.M.; Bierwirth, S.; Sorbie, A.; Waldschmitt, N.; Rath, E.; Berger, E.; Lagkouvardos, I.; Clavel, T.; McCoy, K.D.; et al. Activated ATF6 Induces Intestinal Dysbiosis and Innate Immune Response to Promote Colorectal Tumorigenesis. Gastroenterology 2018, 155, 1539–1552. [Google Scholar] [CrossRef]
- Li, S.K.H.; Martin, A. Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends Mol. Med. 2016, 22, 274–289. [Google Scholar] [CrossRef]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef]
- Lee, J.A.; Yoo, S.Y.; Oh, H.J.; Jeong, S.; Cho, N.Y.; Kang, G.H.; Kim, J.H. Differential immune microenvironmental features of microsatellite-unstable colorectal cancers according to Fusobacterium nucleatum status. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Wu, S.; Yan, C.; Zhao, C.; Jin, H.; Yan, N.; Xu, J.; Wu, Y.; Li, C.; Shao, Q.; et al. Butyrate upregulates the TLR4 expression and the phosphorylation of MAPKs and NK-kappaB in colon cancer cell in vitro. Oncol. Lett. 2018, 16, 4439–4447. [Google Scholar] [CrossRef] [PubMed]
- Ciorba, M.A.; Bettonville, E.E.; McDonald, K.G.; Metz, R.; Prendergast, G.C.; Newberry, R.D.; Stenson, W.F. Induction of IDO-1 by immunostimulatory DNA limits severity of experimental colitis. J. Immunol. 2010, 184, 3907–3916. [Google Scholar] [CrossRef]
- Sommariva, M.; De Cecco, L.; De Cesare, M.; Sfondrini, L.; Menard, S.; Melani, C.; Delia, D.; Zaffaroni, N.; Pratesi, G.; Uva, V.; et al. TLR9 agonists oppositely modulate DNA repair genes in tumor versus immune cells and enhance chemotherapy effects. Cancer Res. 2011, 71, 6382–6390. [Google Scholar] [CrossRef]
- He, Y.W.; Wang, H.S.; Zeng, J.; Fang, X.; Chen, H.Y.; Du, J.; Yang, X.Y. Sodium butyrate inhibits interferon-gamma induced indoleamine 2,3-dioxygenase expression via STAT1 in nasopharyngeal carcinoma cells. Life Sci. 2013, 93, 509–515. [Google Scholar] [CrossRef]
- Jiang, G.M.; He, Y.W.; Fang, R.; Zhang, G.; Zeng, J.; Yi, Y.M.; Zhang, S.; Bu, X.Z.; Cai, S.H.; Du, J. Sodium butyrate down-regulation of indoleamine 2, 3-dioxygenase at the transcriptional and post-transcriptional levels. Int. J. Biochem. Cell Biol. 2010, 42, 1840–1846. [Google Scholar] [CrossRef]
- Phan, T.; Nguyen, V.H.; D’Alincourt, M.S.; Manuel, E.R.; Kaltcheva, T.; Tsai, W.; Blazar, B.R.; Diamond, D.J.; Melstrom, L.G. Salmonella-mediated therapy targeting indoleamine 2, 3-dioxygenase 1 (IDO) activates innate immunity and mitigates colorectal cancer growth. Cancer Gene Ther. 2020, 27, 235–245. [Google Scholar] [CrossRef]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Tanoue, T.; Morita, S.; Plichta, D.R.; Skelly, A.N.; Suda, W.; Sugiura, Y.; Narushima, S.; Vlamakis, H.; Motoo, I.; Sugita, K.; et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 2019, 565, 600–605. [Google Scholar] [CrossRef]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillere, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Tumor | Beneficial Microbiota | Immune Response | Immunotherapy | Reference |
|---|---|---|---|---|
| Melanoma | Bifidobacterium spp. | Increase of dendritic cells function and enhancement of CD8+ T cells priming | anti-PD-L1 | [7] |
| NSCLC, Renal cell or urothelial carcinoma | A. muciniphila | CD4+, CD8+ T cells memory towards A. mucinipilla | anti-PD-1/anti-PD-L1 | [8] |
| Melanoma | F. prausnitzii, B. thetaiotamicron, H. filiformis and B. caccae | CD8+ T cells | anti-PD-1/anti-CTLA-4 or combination | [9] |
| Melanoma | F. prausnitzii, R. bromii, P. pasteri, C. hungati and P. faecium | Increased antigen presentation, elevation of CD4+ and CD8+ T cells | anti-PD-1 | [10] |
| Melanoma | F. prausnitzii L2-6, G. formicilitis ATCC27749, butyrate- producing bacteria SS2-1, Ruminococcus, Lachnospiraceae, Clostridium XIVa and Blautia spp. | Low Tregs in peripheral blood, increase in CD4+ cells and serum CD25 | anti-CTLA-4 | [12] |
| Melanoma | Bacteroides spp. | Activation of Th cells, mobilization of CD11b+ DC to lamina propria | anti-CTLA-4 | [26] |
| Gut Microbiota Component/Agent Influencing Microbiota | Correlation with CRC and/or Immunotherapy | Immune Reaction | Reference |
|---|---|---|---|
| Dysbiotic microbiota due to ATF6 activation | Tumorigenesis | MyD88/TRIF- dependent activation | [96] |
| Fusobacterium nucleatum | tumorigenesis, dMMR CRC | binding to TIGIT on NK and T cells | [93,97,98,99,100] |
| Bacteroides spp. and Faecalibacterium sp. | immunosurveillance | Raising of Tregs and M1 TAMs | [13] |
| Butyrate producing bacteria | dMMR CRC mice models: suspends tumor growth, reserves microbioal homeostasis | upregulation of TLR4, phosphorylation of MAPKs and NF-κΒ | [102,103] |
| Lysates of Lactobacillus acidophilus | improvement of antitumor immune response | Increase of CD8+ Tcells, effector memory T cells, decrease of Tregs and M2 TAMs | [16] |
| shRNA targeting IDO delivered via attenuated Salmonella typhimurium | supresses tumor growth | Increase of intratumoral neutrophil concetration | [108] |
| Bacteroidales S24-7 in Control group, A. municiphila in Vanc group, Bacteroides in Coli group | enhace antitumor antiPD-1 activity in MMR-proficient CRC | changes in the expression of INF-γ and IL-2 in tumor microenvironment | [17] |
| Alistipes shahii and Ruminococcus spp. | Enhacement of antitumor response of immunotherapy | TLR4, TNF production | [109] |
| Ruthenibacterium lactatiformans, Eubacterium limosum, F. ulcerans, Phascolarctobacterium succinatutens, Bacteroides uniformis, B. dorei, Paraprevotella xylaniphila, Parabacteroides johnsonii, P. gordonii and Alistipes senegalensis | enhancement of antitumor ICI’s effect | Induce interferon-γ+ CD8 T cells | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koulouridi, A.; Messaritakis, I.; Gouvas, N.; Tsiaoussis, J.; Souglakos, J. Immunotherapy in Solid Tumors and Gut Microbiota: The Correlation—A Special Reference to Colorectal Cancer. Cancers 2021, 13, 43. https://doi.org/10.3390/cancers13010043
Koulouridi A, Messaritakis I, Gouvas N, Tsiaoussis J, Souglakos J. Immunotherapy in Solid Tumors and Gut Microbiota: The Correlation—A Special Reference to Colorectal Cancer. Cancers. 2021; 13(1):43. https://doi.org/10.3390/cancers13010043
Chicago/Turabian StyleKoulouridi, Asimina, Ippokratis Messaritakis, Nikolaos Gouvas, John Tsiaoussis, and John Souglakos. 2021. "Immunotherapy in Solid Tumors and Gut Microbiota: The Correlation—A Special Reference to Colorectal Cancer" Cancers 13, no. 1: 43. https://doi.org/10.3390/cancers13010043
APA StyleKoulouridi, A., Messaritakis, I., Gouvas, N., Tsiaoussis, J., & Souglakos, J. (2021). Immunotherapy in Solid Tumors and Gut Microbiota: The Correlation—A Special Reference to Colorectal Cancer. Cancers, 13(1), 43. https://doi.org/10.3390/cancers13010043