Randomized Controlled Immunotherapy Clinical Trials for GBM Challenged

 ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Current Anti-Cancer Treatment Strategies for GBM
3. Challenges for RCTs
4. Immunotherapy for GBM
5. Risk Factors and Levels of Personalized Medicine
6. Current Landscape
7. Non-Scientific Challenges
8. Individualized Multimodal Immunotherapy for GBM
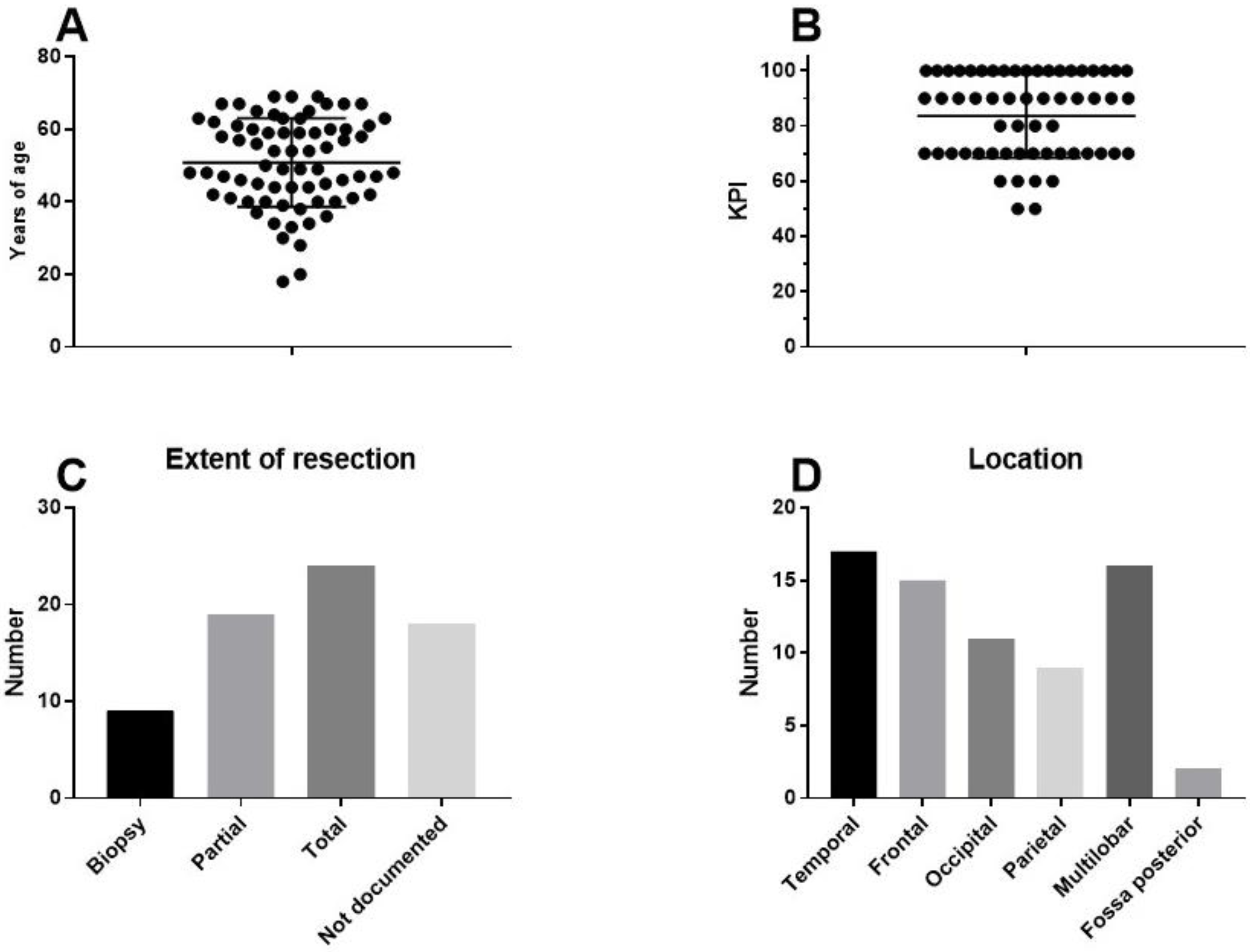
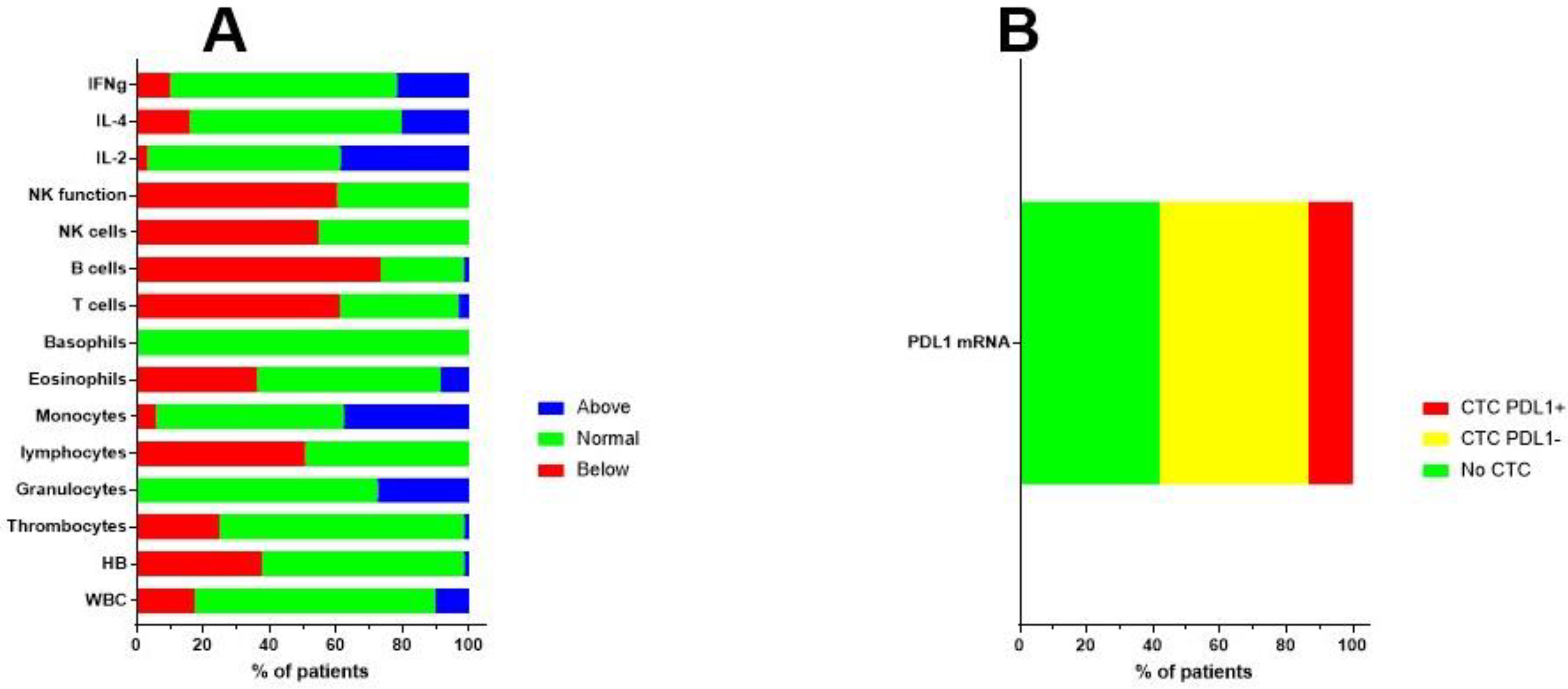
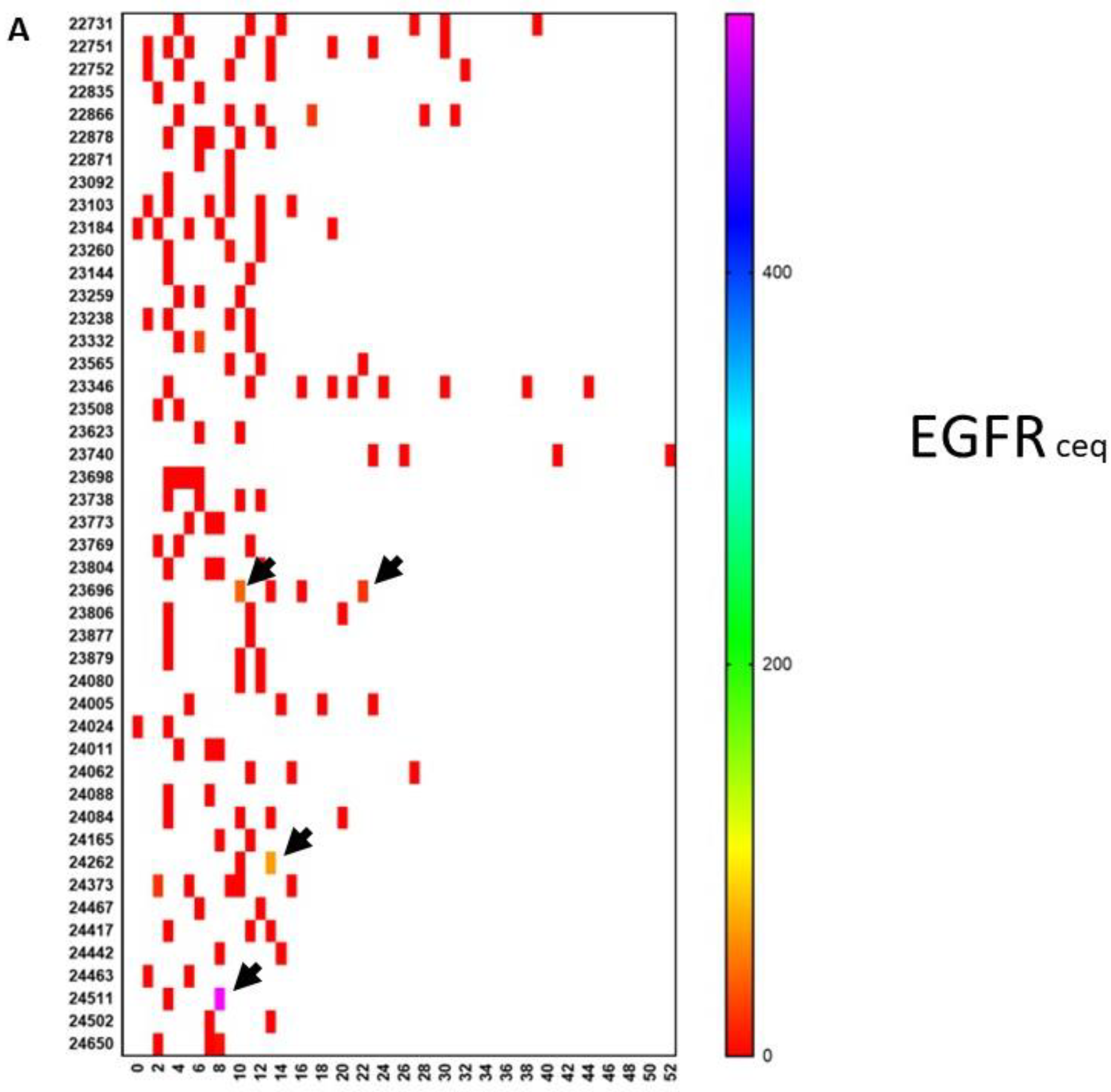
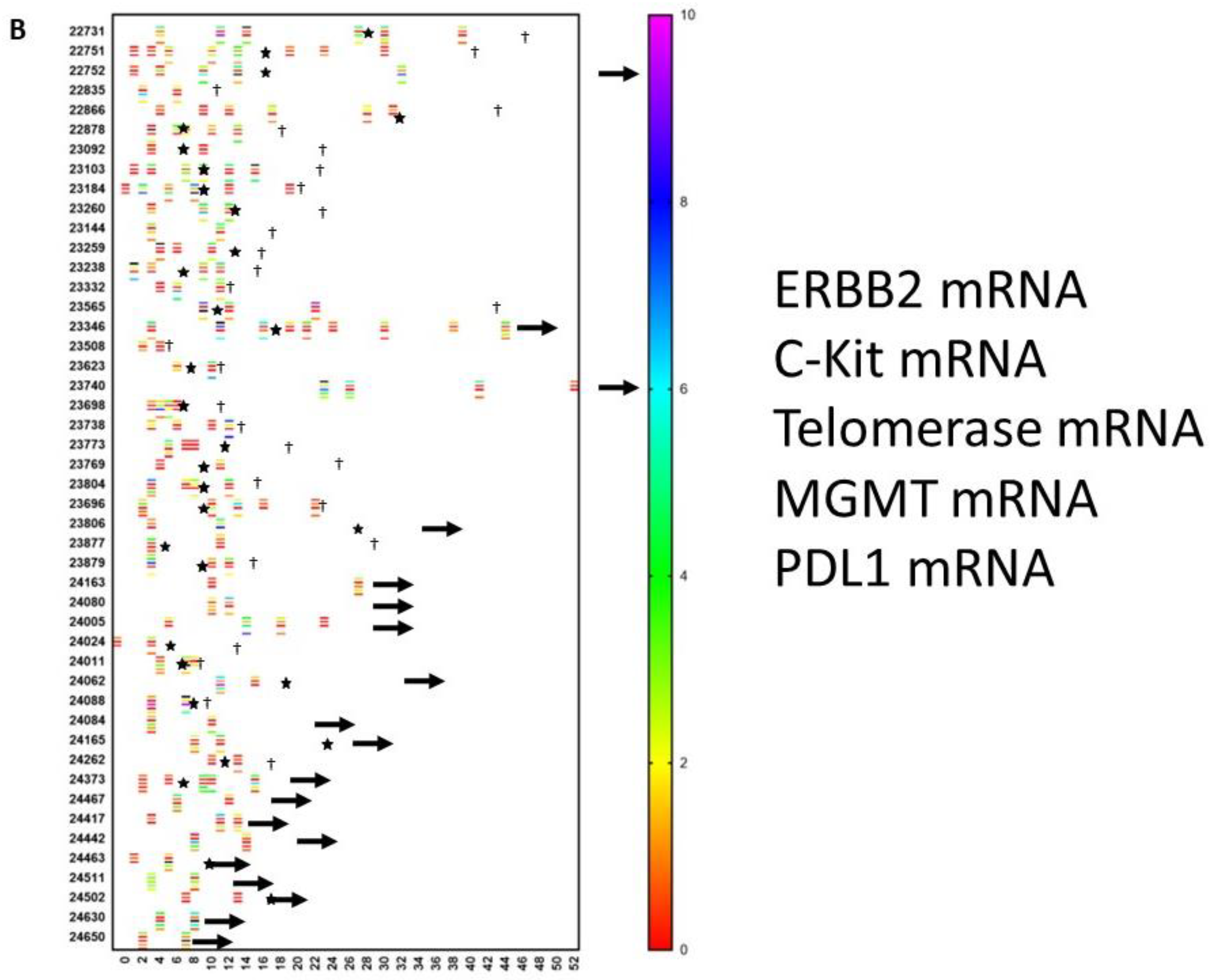
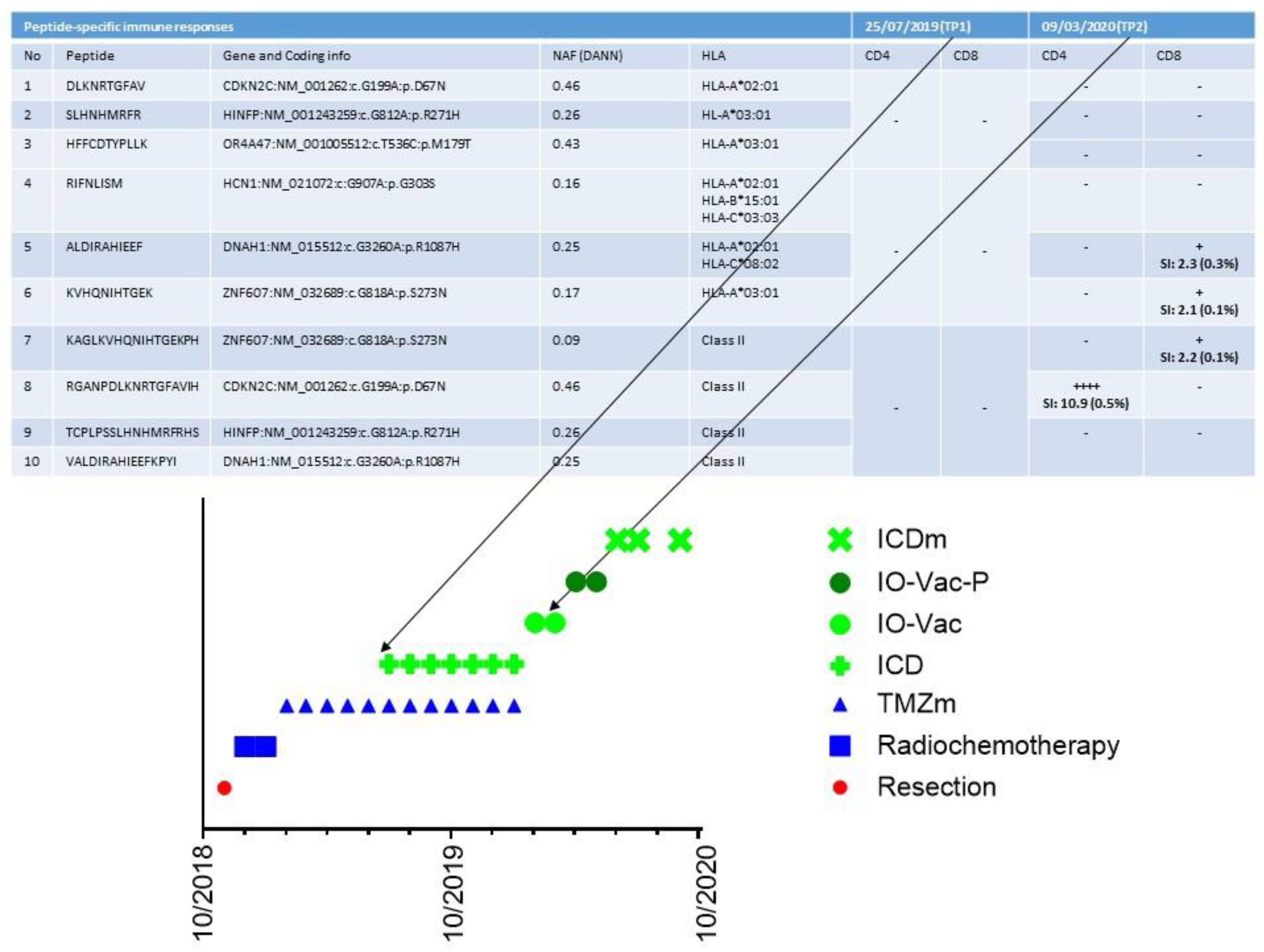
9. A Case of Complete Remission and Specific T-Cell Response
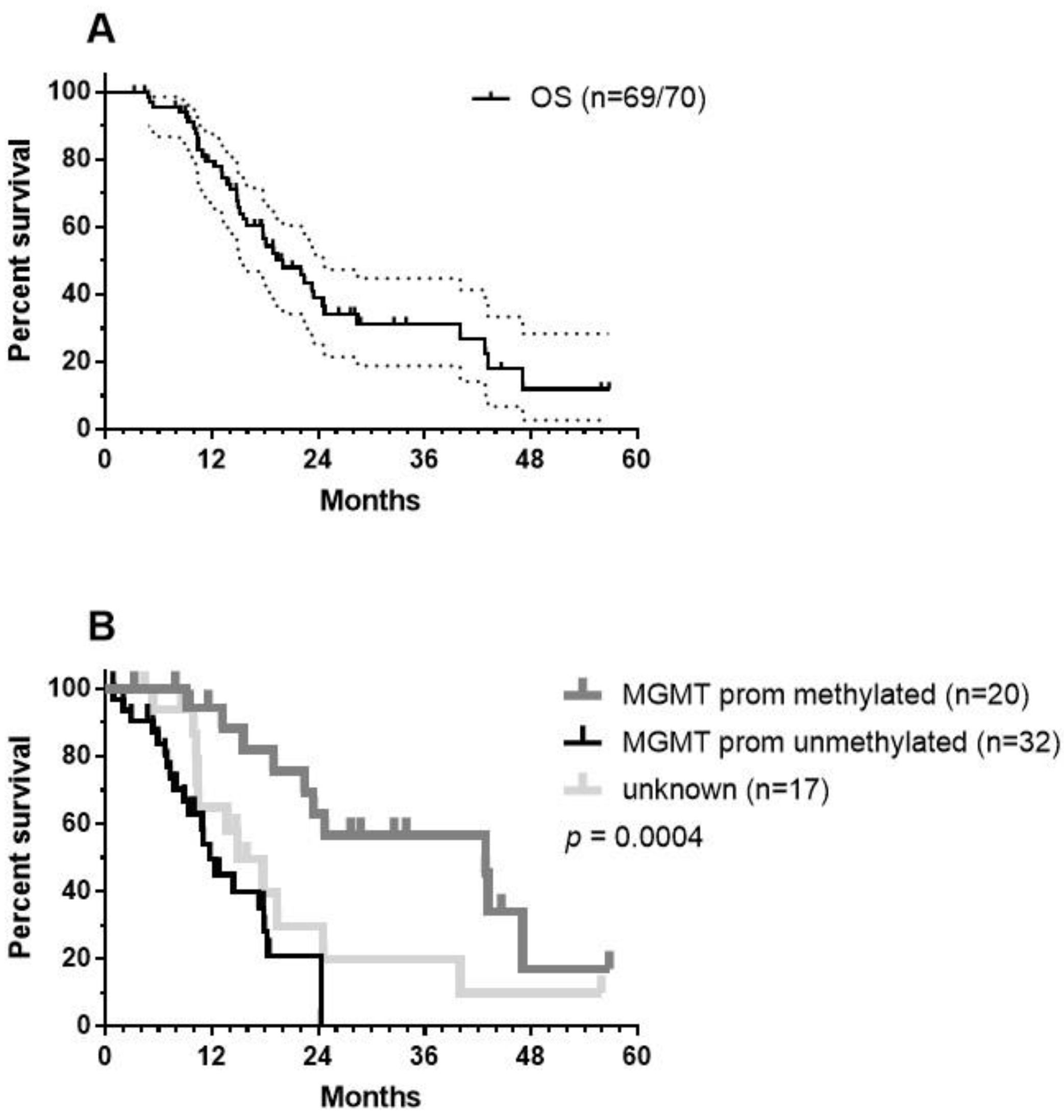
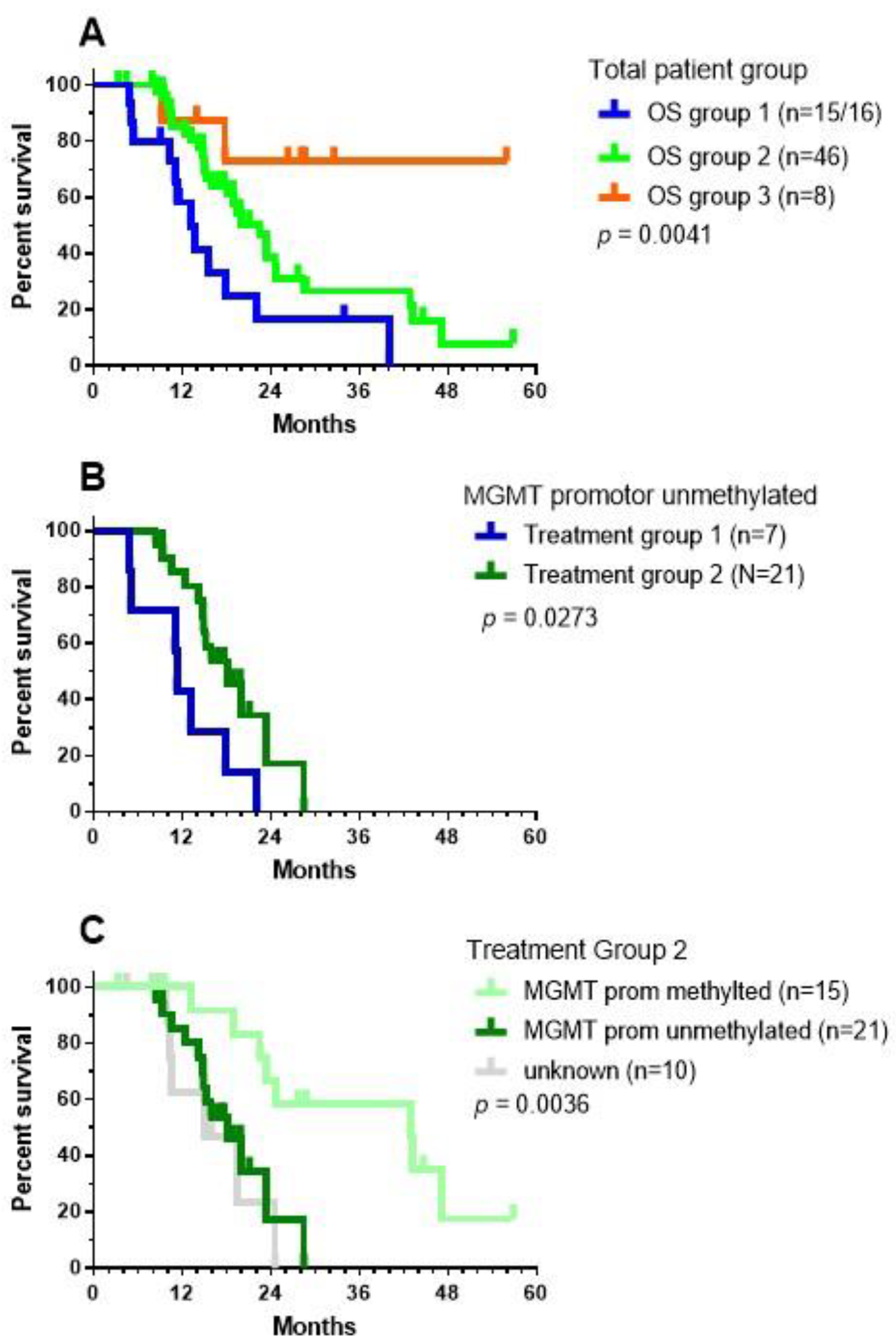
10. Results in Term of OS
11. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 18 December 2020).
- Available online: https://www.cancer.gov (accessed on 18 December 2020).
- Creutzig, U.; Zimmermann, M.; Dworzak, M.N.; Ritter, J.; Schellong, G.; Reinhardt, D. Development of a curative treatment within the AML-BFM studies. Klin. Padiatr. 2013, 225 (Suppl. 1), S79–S86. [Google Scholar] [CrossRef]
- Schrappe, M.; Reiter, A.; Ludwig, W.D.; Harbott, J.; Zimmermann, M.; Hiddemann, W.; Niemeyer, C.; Henze, G.; Feldges, A.; Zintl, F.; et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: Results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 2000, 95, 3310–3322. [Google Scholar]
- Loning, L.; Zimmermann, M.; Reiter, A.; Kaatsch, P.; Henze, G.; Riehm, H.; Schrappe, M. Secondary neoplasms subsequent to Berlin-Frankfurt-Munster therapy of acute lymphoblastic leukemia in childhood: Significantly lower risk without cranial radiotherapy. Blood 2000, 95, 2770–2775. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. Ca Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Ladomersky, E.; Scholtens, D.M.; Kocherginsky, M.; Hibler, E.A.; Bartom, E.T.; Otto-Meyer, S.; Zhai, L.; Lauing, K.L.; Choi, J.; Sosman, J.A.; et al. The Coincidence between Increasing Age, Immunosuppression, and the Incidence of Patients with Glioblastoma. Front. Pharm. 2019, 10, 200. [Google Scholar] [CrossRef] [PubMed]
- Izycka-Swieszewska, E.; Bien, E.; Stefanowicz, J.; Szurowska, E.; Szutowicz-Zielinska, E.; Koczkowska, M.; Sigorski, D.; Kloc, W.; Rogowski, W.; Adamkiewicz-Drozynska, E. Malignant Gliomas as Second Neoplasms in Pediatric Cancer Survivors: Neuropathological Study. Biomed. Res. Int. 2018, 2018, 4596812. [Google Scholar] [CrossRef] [PubMed]
- Hardell, L.; Carlberg, M.; Hansson Mild, K. Use of mobile phones and cordless phones is associated with increased risk for glioma and acoustic neuroma. Pathophysiology 2013, 20, 85–110. [Google Scholar] [CrossRef] [PubMed]
- Soderberg-Naucler, C.; Johnsen, J.I. Cytomegalovirus infection in brain tumors: A potential new target for therapy? Oncoimmunology 2012, 1, 739–740. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoom, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Eng. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the Eortc-Ncic trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- De Vleeschouwer, S.; Van Gool, S.W.; Van Calenbergh, F. Immunotherapy for malignant gliomas: Emphasis on strategies of active specific immunotherapy using autologous dendritic cells. Childs Nerv. Syst. 2005, 21, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.iddi.com/news/news-events/symposium-are-randomized-trials-still-needed-in-2020/ (accessed on 18 December 2020).
- Schirrmacher, V.; Bihari, A.S.; Stucker, W.; Sprenger, T. Long-term remission of prostate cancer with extensive bone metastases upon immuno- and virotherapy: A case report. Oncol. Lett. 2014, 8, 2403–2406. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Stucker, W.; Lulei, M.; Bihari, A.S.; Sprenger, T. Long-term survival of a breast cancer patient with extensive liver metastases upon immune and virotherapy: A case report. Immunotherapy 2015, 7, 855–860. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Feyen, O.; Prix, L.; Schirrmacher, V.; Stuecker, W. The induction of immunogenic cell death (ICD) during maintenance chemotherapy and subsequent multimodal immunotherapy for glioblastoma (GBM). Austin Oncol. Case Rep. 2018, 3, 1010. [Google Scholar]
- Van Gool, S.W.; Makalowski, J.; Bonner, E.R.; Feyen, O.; Domogalla, M.P.; Prix, L.; Schirrmacher, V.; Nazarian, J.; Stuecker, W. Addition of Multimodal Immunotherapy to Combination Treatment Strategies for Children with DIPG: A Single Institution Experience. Medicines 2020, 7, 29. [Google Scholar] [CrossRef]
- Cao, J.X.; Zhang, X.Y.; Liu, J.L.; Li, D.; Li, J.L.; Liu, Y.S.; Wang, M.; Xu, B.L.; Wang, H.B.; Wang, Z.X. Clinical efficacy of tumor antigen-pulsed DC treatment for high-grade glioma patients: Evidence from a meta-analysis. PLoS ONE 2014, 9, e107173. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, H.Y.; Zhang, F.C.; Sun, Y.; Xiong, Z.Y.; Jiang, X.B. Dendritic cell-based vaccine for the treatment of malignant glioma: A systematic review. Cancer Investig. 2014, 32, 451–457. [Google Scholar] [CrossRef]
- Eagles, M.E.; Nassiri, F.; Badhiwala, J.H.; Suppiah, S.; Almenawer, S.A.; Zadeh, G.; Aldape, K.D. Dendritic cell vaccines for high-grade gliomas. Clin. Risk Manag. 2018, 14, 1299–1313. [Google Scholar] [CrossRef]
- Vatu, B.I.; Artene, S.A.; Staicu, A.G.; Turcu-Stiolica, A.; Folcuti, C.; Dragoi, A.; Cioc, C.; Baloi, S.C.; Tataranu, L.G.; Silosi, C.; et al. Assessment of efficacy of dendritic cell therapy and viral therapy in high grade glioma clinical trials. A meta-analytic review. J. Immunoass. Immunochem. 2019, 40, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Huang, J.; Xi, H.; Zhou, X. Efficacy and safety of dendritic cell vaccines for patients with glioblastoma: A meta-analysis of randomized controlled trials. Int. Immunopharmacol. 2020, 83, 106336. [Google Scholar] [CrossRef] [PubMed]
- Burnet, N.G.; Jefferies, S.J.; Benson, R.J.; Hunt, D.P.; Treasure, F.P. Years of life lost (YLL) from cancer is an important measure of population burden--and should be considered when allocating research funds. Br. J. Cancer 2005, 92, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Rouse, C.; Gittleman, H.; Ostrom, Q.T.; Kruchko, C.; Barnholtz-Sloan, J.S. Years of potential life lost for brain and CNS tumors relative to other cancers in adults in the United States, 2010. Neuro Oncol. 2016, 18, 70–77. [Google Scholar] [CrossRef]
- Walker, M.D.; Strike, T.A.; Sheline, G.E. An analysis of dose-effect relationship in the radiotherapy of malignant gliomas. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1725–1731. [Google Scholar] [CrossRef]
- Walker, M.D.; Alexander, E., Jr.; Hunt, W.E.; MacCarty, C.S.; Mahaley, M.S., Jr.; Mealey, J., Jr.; Norrell, H.A.; Owens, G.; Ransohoff, J.; Wilson, C.B.; et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J. Neurosurg. 1978, 49, 333–343. [Google Scholar] [CrossRef]
- Gzell, C.; Back, M.; Wheeler, H.; Bailey, D.; Foote, M. Radiotherapy in Glioblastoma: The Past, the Present and the Future. Clin. Oncol. R Coll. Radiol. 2017, 29, 15–25. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. New Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Dix, A.R.; Brooks, W.H.; Roszman, T.L.; Morford, L.A. Immune defects observed in patients with primary malignant brain tumors. J. Neuroimmunol. 1999, 100, 216–232. [Google Scholar] [CrossRef]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2020. [Google Scholar] [CrossRef]
- North, R.J. Gamma-irradiation facilitates the expression of adoptive immunity against established tumors by eliminating suppressor T cells. Cancer Immunol. Immunother. 1984, 16, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.T.; Sun, W.; Hussain, S.F.; DeAngulo, G.; Prabhu, S.S.; Heimberger, A.B. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol. Immunother. 2008, 57, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Fadul, C.E.; Fisher, J.L.; Gui, J.; Hampton, T.H.; Cote, A.L.; Ernstoff, M.S. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol. 2011, 13, 393–400. [Google Scholar] [CrossRef]
- Dutoit, V.; Philippin, G.; Widmer, V.; Marinari, E.; Vuilleumier, A.; Migliorini, D.; Schaller, K.; Dietrich, P.Y. Impact of Radiochemotherapy on Immune Cell Subtypes in High-Grade Glioma Patients. Front. Oncol. 2020, 10, 89. [Google Scholar] [CrossRef]
- Antonopoulos, M.; Van Gool, S.W.; Dionysiou, D.; Graf, N.; Stamatakos, G. Immune phenotype correlates with survival in patients with GBM treated with standard temozolomide-based therapy and immunotherapy. Anticancer Res. 2019, 39, 2043–2051. [Google Scholar] [CrossRef]
- Sathornsumetee, S.; Rich, J.N. Designer therapies for glioblastoma multiforme. Ann. N. Y. Acad. Sci. 2008, 1142, 108–132. [Google Scholar] [CrossRef]
- Chen, R.; Cohen, A.L.; Colman, H. Targeted Therapeutics in Patients with High-Grade Gliomas: Past, Present, and Future. Curr. Treat. Options Oncol. 2016, 17, 42. [Google Scholar] [CrossRef]
- Dos Santos, M.A.; Pignon, J.P.; Blanchard, P.; Lefeuvre, D.; Levy, A.; Touat, M.; Louvel, G.; Dhermain, F.; Soria, J.C.; Deutsch, E.; et al. Systematic review and meta-analysis of phase I/II targeted therapy combined with radiotherapy in patients with glioblastoma multiforme: Quality of report, toxicity, and survival. J. Neurooncol. 2015, 123, 307–314. [Google Scholar] [CrossRef]
- Su, J.; Cai, M.; Li, W.; Hou, B.; He, H.; Ling, C.; Huang, T.; Liu, H.; Guo, Y. Molecularly Targeted Drugs Plus Radiotherapy and Temozolomide Treatment for Newly Diagnosed Glioblastoma: A Meta-Analysis and Systematic Review. Oncol. Res. 2016, 24, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Cihoric, N.; Tsikkinis, A.; Minniti, G.; Lagerwaard, F.J.; Herrlinger, U.; Mathier, E.; Soldatovic, I.; Jeremic, B.; Ghadjar, P.; Elicin, O.; et al. Current status and perspectives of interventional clinical trials for glioblastoma-analysis of ClinicalTrials.gov. Radiat. Oncol. 2017, 12, 63. [Google Scholar] [CrossRef]
- Sim, H.W.; Morgan, E.R.; Mason, W.P. Contemporary management of high-grade gliomas. Cns Oncol. 2018, 7, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Lavacchi, D.; Roviello, G.; D’Angelo, A. Tumor-Agnostic Treatment for Cancer: When How is Better than Where. Clin. Drug Investig. 2020, 40, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Medical Research Council Streptomycin in Tuberculosis Trials Committee. Streptomycin treatment of pulmonary tuberculosis. Br. Med. J. 1948, 2, 769–782. [Google Scholar] [CrossRef]
- Sorensen, H.T.; Lash, T.L.; Rothman, K.J. Beyond randomized controlled trials: A critical comparison of trials with nonrandomized studies. Hepatology 2006, 44, 1075–1082. [Google Scholar] [CrossRef]
- Shikata, S.; Nakayama, T.; Noguchi, Y.; Taji, Y.; Yamagishi, H. Comparison of effects in randomized controlled trials with observational studies in digestive surgery. Ann. Surg. 2006, 244, 668–676. [Google Scholar] [CrossRef]
- Vincent, J.L. We should abandon randomized controlled trials in the intensive care unit. Crit. Care Med. 2010, 38, S534–S538. [Google Scholar] [CrossRef]
- Jones, D.S.; Podolsky, S.H. The history and fate of the gold standard. Lancet 2015, 385, 1502–1503. [Google Scholar] [CrossRef]
- Trentino, K.; Farmer, S.; Gross, I.; Shander, A.; Isbister, J. Observational studies-should we simply ignore them in assessing transfusion outcomes? BMC Anesth. 2016, 16, 96. [Google Scholar] [CrossRef]
- Sharma, M.; Nazareth, I.; Petersen, I. Observational studies of treatment effectiveness: Worthwhile or worthless? Clin. Epidemiol. 2019, 11, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Halabi, S.; Michiels, S. (Eds.) Textbook of Clinical trials in Oncology. A Statistical Perspective; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Stepanenko, A.A.; Chekhonin, V.P. Recent Advances in Oncolytic Virotherapy and Immunotherapy for Glioblastoma: A Glimmer of Hope in the Search for an Effective Therapy? Cancers 2018, 10, 492. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef] [PubMed]
- Suter, R.; Rodriguez-Blanco, J.; Ayad, N.G. Epigenetic pathways and plasticity in brain tumors. Neurobiol. Dis. 2020, 105060. [Google Scholar] [CrossRef] [PubMed]
- Muller Bark, J.; Kulasinghe, A.; Chua, B.; Day, B.W.; Punyadeera, C. Circulating biomarkers in patients with glioblastoma. Br. J. Cancer 2020, 122, 295–305. [Google Scholar] [CrossRef]
- Fiegl, H.; Millinger, S.; Mueller-Holzner, E.; Marth, C.; Ensinger, C.; Berger, A.; Klocker, H.; Goebel, G.; Widschwendter, M. Circulating tumor-specific DNA: A marker for monitoring efficacy of adjuvant therapy in cancer patients. Cancer Res. 2005, 65, 1141–1145. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Macdonald, D.R.; Cascino, T.L.; Schold, S.C., Jr.; Cairncross, J.G. Response criteria for phase II studies of supratentorial malignant glioma. J. Clin. Oncol. 1990, 8, 1277–1280. [Google Scholar] [CrossRef]
- Jaspan, T.; Morgan, P.S.; Warmuth-Metz, M.; Sanchez Aliaga, E.; Warren, D.; Calmon, R.; Grill, J.; Hargrave, D.; Garcia, J.; Zahlmann, G. Response Assessment in Pediatric Neuro-Oncology: Implementation and Expansion of the RANO Criteria in a Randomized Phase II Trial of Pediatric Patients with Newly Diagnosed High-Grade Gliomas. Am. J. Neuroradiol. 2016, 37, 1581–1587. [Google Scholar] [CrossRef]
- Okada, H.; Weller, M.; Huang, R.; Finocchiaro, G.; Gilbert, M.R.; Wick, W.; Ellingson, B.M.; Hashimoto, N.; Pollack, I.F.; Brandes, A.A.; et al. Immunotherapy response assessment in neuro-oncology: A report of the RANO working group. Lancet Oncol. 2015, 16, e534–e542. [Google Scholar] [CrossRef]
- Aquino, D.; Gioppo, A.; Finocchiaro, G.; Bruzzone, M.G.; Cuccarini, V. MRI in Glioma Immunotherapy: Evidence, Pitfalls, and Perspectives. J. Immunol. Res. 2017, 2017, 5813951. [Google Scholar] [CrossRef]
- Sharma, P.; Debinski, W. Receptor-Targeted Glial Brain Tumor Therapies. Int. J. Mol. Sci. 2018, 19, 3326. [Google Scholar] [CrossRef]
- Huang, J.; Liu, F.; Liu, Z.; Tang, H.; Wu, H.; Gong, Q.; Chen, J. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front. Pharm. 2017, 8, 242. [Google Scholar] [CrossRef]
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 87. [Google Scholar] [CrossRef]
- Ameratunga, M.; Coleman, N.; Welsh, L.; Saran, F.; Lopez, J. CNS cancer immunity cycle and strategies to target this for glioblastoma. Oncotarget 2018, 9, 22802–22816. [Google Scholar] [CrossRef]
- Zhu, C.; Zou, C.; Guan, G.; Guo, Q.; Yan, Z.; Liu, T.; Shen, S.; Xu, X.; Chen, C.; Lin, Z.; et al. Development and validation of an interferon signature predicting prognosis and treatment response for glioblastoma. Oncoimmunology 2019, 8, e1621677. [Google Scholar] [CrossRef]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting from Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef]
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells 2020, 9, 263. [Google Scholar] [CrossRef]
- Wang, B.; Niu, D.; Lai, L.; Ren, E.C. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat. Commun. 2013, 4, 2359. [Google Scholar] [CrossRef]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. Cancer Immunology. The cancer immunogram. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Micro-environment and Host State-Implications for Immunotherapy. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pires-Afonso, Y.; Niclou, S.P.; Michelucci, A. Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shen, R.; Cheng, S.; Feng, L. Immune microenvironments differ in immune characteristics and outcome of glioblastoma multiforme. Cancer Med. 2019, 8, 2897–2907. [Google Scholar] [CrossRef] [PubMed]
- Hallaert, G.; Pinson, H.; Van den Broecke, C.; Vanhauwaert, D.; Van Roost, D.; Boterberg, T.; Kalala, J.P. Subventricular zone contacting glioblastoma: Tumor size, molecular biological factors and patient survival. Acta Oncol. 2020. [Google Scholar] [CrossRef]
- Berendsen, S.; van Bodegraven, E.; Seute, T.; Spliet, W.G.M.; Geurts, M.; Hendrikse, J.; Schoysman, L.; Huiszoon, W.B.; Varkila, M.; Rouss, S.; et al. Adverse prognosis of glioblastoma contacting the subventricular zone: Biological correlates. PLoS ONE 2019, 14, e0222717. [Google Scholar] [CrossRef]
- Wee, C.W.; Kim, I.H.; Park, C.K.; Kim, J.W.; Dho, Y.S.; Ohka, F.; Aoki, K.; Motomura, K.; Natsume, A.; Kim, N.; et al. Validation of a novel molecular RPA classification in glioblastoma (GBM-molRPA) treated with chemoradiation: A multi-institutional collaborative study. Radiother. Oncol. 2018, 129, 347–351. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Golinelli, G.; Grisendi, G.; Prapa, M.; Bestagno, M.; Spano, C.; Rossignoli, F.; Bambi, F.; Sardi, I.; Cellini, M.; Horwitz, E.M.; et al. Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rammensee, H.G.; Singh-Jasuja, H. HLA ligandome tumor antigen discovery for personalized vaccine approach. Expert Rev. Vaccines 2013, 12, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017. [Google Scholar] [CrossRef] [PubMed]
- Johanns, T.M.; Miller, C.A.; Liu, C.J.; Perrin, R.J.; Bender, D.; Kobayashi, D.K.; Campian, J.L.; Chicoine, M.R.; Dacey, R.G.; Huang, J.; et al. Detection of neoantigen-specific T cells following a personalized vaccine in a patient with glioblastoma. Oncoimmunology 2019, 8, e1561106. [Google Scholar] [CrossRef] [PubMed]
- De Vleeschouwer, S.; Arredouani, M.; Ade, M.; Cadot, P.; Vermassen, E.; Ceuppens, J.L.; Van Gool, S.W. Uptake and presentation of malignant glioma tumor cell lysates by monocyte-derived dendritic cells. Cancer Immunol. Immunother. 2005, 54, 372–382. [Google Scholar] [CrossRef] [PubMed]
- De Vleeschouwer, S.; Spencer, L.I.; Ceuppens, J.L.; Van Gool, S.W. Persistent IL-10 production is required for glioma growth suppressive activity by Th1-directed effector cells after stimulation with tumor lysate-loaded dendritic cells. J. Neurooncol. 2007, 84, 131–140. [Google Scholar] [CrossRef]
- Maes, W.; Deroose, C.; Reumers, V.; Krylyshkina, O.; Gijsbers, R.; Ceuppens, J.; Baekelandt, V.; Debyser, Z.; Van Gool, S. In vivo bioluminescence imaging in an experimental mouse model for dendritic cell based immunotherapy against malignant glioma. J. Neuro Oncol. 2009, 91, 127–139. [Google Scholar] [CrossRef]
- Maes, W.; Galicia Rosas, G.; Verbinnen, B.; Boon, L.; De Vleeschouwer, S.; Ceuppens, J.L.; Van Gool, S.W. DC vaccination with anti-CD25 treatment leads to long-term immunity against experimental glioma. Neuro Oncol. 2009, 11, 529–542. [Google Scholar] [CrossRef]
- Ardon, H.; Verbinnen, B.; Maes, W.; Beez, T.; Van Gool, S.; De Vleeschouwer, S. Technical advancement in regulatory T cell isolation and characterization using CD127 expression in patients with malignant glioma treated with autologous dendritic cell vaccination. J. Immunol. Methods 2010, 352, 169–173. [Google Scholar] [CrossRef]
- Maes, W.; Van Gool, S.W. Experimental immunotherapy for malignant glioma: Lessons from two decades of research in the GL261 model. Cancer Immunol. Immunother. 2011, 60, 153–160. [Google Scholar] [CrossRef]
- Vandenberk, L.; Van Gool, S.W. Treg infiltration in glioma: A hurdle for antiglioma immunotherapy. Immunotherapy 2012, 4, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Koks, C.A.E.; Garg, A.D.; Ehrhardt, M.; Riva, M.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2014, 136, e313–e325. [Google Scholar] [CrossRef]
- Koks, C.A.; De Vleeschouwer, S.; Graf, N.; Van Gool, S.W. Immune Suppression during Oncolytic Virotherapy for High-Grade Glioma; Yes or No? J. Cancer 2015, 6, 203–217. [Google Scholar] [CrossRef][Green Version]
- Vandenberk, L.; Belmans, J.; Van Woensel, M.; Riva, M.; Van Gool, S.W. Exploiting the Immunogenic Potential of Cancer Cells for Improved Dendritic Cell Vaccines. Front. Immunol. 2015, 6, 663. [Google Scholar] [CrossRef]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra327. [Google Scholar] [CrossRef] [PubMed]
- Vandenberk, L.; Garg, A.D.; Verschuere, T.; Koks, C.; Belmans, J.; Beullens, M.; Agostinis, P.; De Vleeschouwer, S.; Van Gool, S.W. Irradiation of necrotic cancer cells, employed for pulsing dendritic cells (DCs), potentiates DC vaccine-induced antitumor immunity against high-grade glioma. Oncoimmunology 2016, 5, e1083669. [Google Scholar] [CrossRef] [PubMed]
- Belmans, J.; Van Woensel, M.; Creyns, B.; Dejaegher, J.; Bullens, D.M.; Van Gool, S.W. Immunotherapy with subcutaneous immunogenic autologous tumor lysate increases murine glioblastoma survival. Sci. Rep. 2017, 7, 13902. [Google Scholar] [CrossRef]
- Liau, L.M.; Black, K.L.; Martin, N.A.; Sykes, S.N.; Bronstein, J.M.; Jouben-Steele, L.; Mischel, P.S.; Belldegrun, A.; Cloughesy, T.F. Treatment of a patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I-matched tumor peptides. Case Report. Neurosurg. Focus 2000, 9, e8. [Google Scholar] [CrossRef]
- Yu, J.S.; Wheeler, C.J.; Zeltzer, P.M.; Ying, H.; Finger, D.N.; Lee, P.K.; Yong, W.H.; Incardona, F.; Thompson, R.C.; Riedinger, M.S.; et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001, 61, 842–847. [Google Scholar]
- Kikuchi, T.; Akasaki, Y.; Irie, M.; Homma, S.; Abe, T.; Ohno, T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol. Immunother. 2001, 50, 337–344. [Google Scholar] [CrossRef]
- Wheeler, C.J.; Black, K.L.; Liu, G.; Ying, H.; Yu, J.S.; Zhang, W.; Lee, P.K. Thymic CD8(+) T cell production strongly influences tumor antigen recognition and age-dependent glioma mortality. J. Immunol. 2003, 171, 4927–4933. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Abe, T.; Yajima, N.; Tsuchiya, N.; Homma, J.; Kobayashi, T.; Narita, M.; Takahashi, M.; Tanaka, R. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: Results of a clinical phase I/II trial. Br. J. Cancer 2003, 89, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Caruso, D.A.; Orme, L.M.; Neale, A.M.; Radcliff, F.J.; Amor, G.M.; Maixner, W.; Downie, P.; Hassall, T.E.; Tang, M.L.; Ashley, D.M. Results of a phase 1 study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro. Oncol. 2004, 6, 236–246. [Google Scholar] [CrossRef] [PubMed]
- De Vleeschouwer, S.; Van Calenbergh, F.; Demaerel, P.; Flamen, P.; Rutkowski, S.; Kaempgen, E.; Wolff, J.E.A.; Plets, C.; Sciot, R.; Van Gool, S.W. Transient local response and persistent tumor control of recurrent malignant glioma treated with combination therapy including dendritic cell therapy. J. Neurosurg. 2004, 100, 492–497. [Google Scholar] [PubMed]
- Kikuchi, T.; Akasaki, Y.; Abe, T.; Fukuda, T.; Saotome, H.; Ryan, J.L.; Kufe, D.W.; Ohno, T. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J. Immunother. 2004, 27, 452–459. [Google Scholar] [CrossRef]
- Rutkowski, S.; De Vleeschouwer, S.; Kaempgen, E.; Wolff, J.E.A.; Kuhl, J.; Demaerel, P.; Warmuth-Metz, M.; Flamen, P.; Van Calenbergh, F.; Plets, C.; et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br. J. Cancer 2004, 91, 1656–1662. [Google Scholar] [CrossRef]
- Wheeler, C.J.; Das, A.; Liu, G.; Yu, J.S.; Black, K.L. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin. Cancer Res. 2004, 10, 5316–5326. [Google Scholar] [CrossRef]
- Yu, J.S.; Liu, G.; Ying, H.; Yong, W.H.; Black, K.L.; Wheeler, C.J. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004, 64, 4973–4979. [Google Scholar] [CrossRef]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef]
- Yamanaka, R.; Honma, J.; Tsuchiya, N.; Yajima, N.; Kobayashi, T.; Tanaka, R. Tumor lysate and IL-18 loaded dendritic cells elicits Th1 response, tumor-specific CD8+ cytotoxic T cells in patients with malignant glioma. J. Neurooncol. 2005, 72, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Homma, J.; Yajima, N.; Tsuchiya, N.; Sano, M.; Kobayashi, T.; Yoshida, S.; Abe, T.; Narita, M.; Takahashi, M.; et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: Results of a clinical phase I/II trial. Clin. Cancer Res. 2005, 11, 4160–4167. [Google Scholar] [CrossRef]
- Khan, J.A.; Yaqin, S. Dendritic cell therapy with improved outcome in glioma multiforme—A case report. J. Zhejiang Univ. Sci. B 2006, 7, 114–117. [Google Scholar] [CrossRef]
- Okada, H.; Lieberman, F.S.; Walter, K.A.; Lunsford, L.D.; Kondziolka, D.S.; Bejjani, G.K.; Hamilton, R.L.; Torres-Trejo, A.; Kalinski, P.; Cai, Q.; et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J. Transl. Med. 2007, 5, 67. [Google Scholar] [CrossRef]
- De Vleeschouwer, S.; Fieuws, S.; Rutkowski, S.; Van Calenbergh, F.; Van Loon, J.; Goffin, J.; Sciot, R.; Wilms, G.; Demaerel, P.; Warmuth-Metz, M.; et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 3098–3104. [Google Scholar] [CrossRef]
- Prins, R.M.; Cloughesy, T.F.; Liau, L.M. Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. New Engl. J. Med. 2008, 359, 539–541. [Google Scholar] [CrossRef]
- Walker, D.G.; Laherty, R.; Tomlinson, F.H.; Chuah, T.; Schmidt, C. Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: Potential interaction with adjuvant chemotherapy. J. Clin. Neurosci. 2008, 15, 114–121. [Google Scholar] [CrossRef]
- Wheeler, C.J.; Black, K.L.; Liu, G.; Mazer, M.; Zhang, X.X.; Pepkowitz, S.; Goldfinger, D.; Ng, H.; Irvin, D.; Yu, J.S. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008, 68, 5955–5964. [Google Scholar] [CrossRef]
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Herndon, J.E.; Lally-Goss, D.; McGehee-Norman, S.; Paolino, A.; Reardon, D.A.; Friedman, A.H.; et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol. Cancer 2009, 8, 2773–2779. [Google Scholar] [CrossRef]
- Ardon, H.; Van Gool, S.; Lopes, I.S.; Maes, W.; Sciot, R.; Wilms, G.; Demaerel, P.; Bijttebier, P.; Claes, L.; Goffin, J.; et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: A pilot study. J. Neurooncol. 2010, 99, 261–272. [Google Scholar] [CrossRef]
- Ardon, H.; De Vleeschouwer, S.; Van Calenbergh, F.; Claes, L.; Kramm, C.M.; Rutkowski, S.; Wolff, J.E.; Van Gool, S.W. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr. Blood Cancer 2010, 54, 519–525. [Google Scholar] [CrossRef]
- Chang, C.N.; Huang, Y.C.; Yang, D.M.; Kikuta, K.; Wei, K.J.; Kubota, T.; Yang, W.K. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J. Clin. Neurosci. 2011, 18, 1048–1054. [Google Scholar] [CrossRef]
- Fadul, C.E.; Fisher, J.L.; Hampton, T.H.; Lallana, E.C.; Li, Z.; Gui, J.; Szczepiorkowski, Z.M.; Tosteson, T.D.; Rhodes, C.H.; Wishart, H.A.; et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J. Immunother. 2011, 34, 382–389. [Google Scholar] [CrossRef]
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K.; et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 2011, 29, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Prins, R.M.; Soto, H.; Konkankit, V.; Odesa, S.K.; Eskin, A.; Yong, W.H.; Nelson, S.F.; Liau, L.M. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res. 2011, 17, 1603–1615. [Google Scholar] [CrossRef]
- Akiyama, Y.; Oshita, C.; Kume, A.; Iizuka, A.; Miyata, H.; Komiyama, M.; Ashizawa, T.; Yagoto, M.; Abe, Y.; Mitsuya, K.; et al. alpha-type-1 polarized dendritic cell-based vaccination in recurrent high-grade glioma: A phase I clinical trial. BMC Cancer 2012, 12, 623. [Google Scholar] [CrossRef]
- Ardon, H.; Van Gool, S.W.; Verschuere, T.; Maes, W.; Fieuws, S.; Sciot, R.; Wilms, G.; Demaerel, P.; Goffin, J.; Van Calenbergh, F.; et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: Results of the HGG-2006 phase I/II trial. Cancer Immunol. Immunother. 2012, 61, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.Y.; Yang, W.K.; Lee, H.C.; Hsu, D.M.; Lin, H.L.; Lin, S.Z.; Chen, C.C.; Harn, H.J.; Liu, C.L.; Lee, W.Y.; et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: A phase II clinical trial. World Neurosurg. 2012, 77, 736–744. [Google Scholar] [CrossRef] [PubMed]
- De Vleeschouwer, S.; Ardon, H.; van Calenbergh, F.; Sciot, R.; Wilms, G.; van Loon, J.; Goffin, J.; van Gool, S. Stratification according to Hgg-Immuno Rpa model predicts outcome in a large group of patients with relapsed malignant glioma treated by adjuvant postoperative dendritic cell vaccination. Cancer Immunol. Immunother. 2012, 61, 2105–2112. [Google Scholar] [CrossRef]
- Elens, I.; De Vleeschouwer, S.; Pauwels, F.; Van Gool, S.W. Resection and immunotherapy for recurrent grade III glioma. Isrn Immunol. 2012, 2012, 530179. [Google Scholar] [CrossRef]
- Fong, B.; Jin, R.; Wang, X.; Safaee, M.; Lisiero, D.N.; Yang, I.; Li, G.; Liau, L.M.; Prins, R.M. Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PLoS ONE 2012, 7, e32614. [Google Scholar] [CrossRef]
- Iwami, K.; Shimato, S.; Ohno, M.; Okada, H.; Nakahara, N.; Sato, Y.; Yoshida, J.; Suzuki, S.; Nishikawa, H.; Shiku, H.; et al. Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor alpha2 chain in recurrent malignant glioma patients with HLA-A*24/A*02 allele. Cytotherapy 2012. [Google Scholar] [CrossRef]
- Jie, X.; Hua, L.; Jiang, W.; Feng, F.; Feng, G.; Hua, Z. Clinical application of a dendritic cell vaccine raised against heat-shocked glioblastoma. Cell Biochem. Biophys. 2012, 62, 91–99. [Google Scholar] [CrossRef]
- Qin, K.; Tian, G.; Li, P.; Chen, Q.; Zhang, R.; Ke, Y.Q.; Xiao, Z.C.; Jiang, X.D. Anti-glioma response of autologous T cells stimulated by autologous dendritic cells electrofused with CD133(+) or CD133(-) glioma cells. J. Neuroimmunol. 2012, 242, 9–15. [Google Scholar] [CrossRef]
- Sampson, J.H.; Schmittling, R.J.; Archer, G.E.; Congdon, K.L.; Nair, S.K.; Reap, E.A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E.; et al. A Pilot Study of IL-2Ralpha Blockade during Lymphopenia Depletes Regulatory T-cells and Correlates with Enhanced Immunity in Patients with Glioblastoma. PLoS ONE 2012, 7, e31046. [Google Scholar] [CrossRef]
- Valle, R.D.; de Cerio, A.L.; Inoges, S.; Tejada, S.; Pastor, F.; Villanueva, H.; Gallego, J.; Espinos, J.; Aristu, J.; Idoate, M.A.; et al. Dendritic cell vaccination in glioblastoma after fluorescence-guided resection. World J. Clin. Oncol. 2012, 3, 142–149. [Google Scholar] [CrossRef]
- Lasky, J.L., III; Panosyan, E.H.; Plant, A.; Davidson, T.; Yong, W.H.; Prins, R.M.; Liau, L.M.; Moore, T.B. Autologous Tumor Lysate-pulsed Dendritic Cell Immunotherapy for Pediatric Patients with Newly Diagnosed or Recurrent High-grade Gliomas. Anticancer Res. 2013, 33, 2047–2056. [Google Scholar]
- Pellegatta, S.; Eoli, M.; Frigerio, S.; Antozzi, C.; Bruzzone, M.G.; Cantini, G.; Nava, S.; Anghileri, E.; Cuppini, L.; Cuccarini, V.; et al. The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. Oncoimmunology 2013, 2, e23401. [Google Scholar] [CrossRef] [PubMed]
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuno, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M.; et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135. [Google Scholar] [CrossRef]
- Prins, R.M.; Wang, X.; Soto, H.; Young, E.; Lisiero, D.N.; Fong, B.; Everson, R.; Yong, W.H.; Lai, A.; Li, G.; et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J. Immunother. 2013, 36, 152–157. [Google Scholar] [CrossRef]
- Vik-Mo, E.O.; Nyakas, M.; Mikkelsen, B.V.; Moe, M.C.; Due-Tonnesen, P.; Suso, E.M.; Saeboe-Larssen, S.; Sandberg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013. [Google Scholar] [CrossRef]
- Eyrich, M.; Schreiber, S.C.; Rachor, J.; Krauss, J.; Pauwels, F.; Hain, J.; Wolfl, M.; Lutz, M.B.; De, V.S.; Schlegel, P.G.; et al. Development and validation of a fully GMP-compliant production process of autologous, tumor-lysate-pulsed dendritic cells. Cytotherapy 2014, 16, 946–964. [Google Scholar] [CrossRef]
- Ishikawa, E.; Muragaki, Y.; Yamamoto, T.; Maruyama, T.; Tsuboi, K.; Ikuta, S.; Hashimoto, K.; Uemae, Y.; Ishihara, T.; Matsuda, M.; et al. Phase I/IIa trial of fractionated radiotherapy, temozolomide, and autologous formalin-fixed tumor vaccine for newly diagnosed glioblastoma. J. Neurosurg. 2014, 121, 543–553. [Google Scholar] [CrossRef]
- Hunn, M.K.; Bauer, E.; Wood, C.E.; Gasser, O.; Dzhelali, M.; Ancelet, L.R.; Mester, B.; Sharples, K.J.; Findlay, M.P.; Hamilton, D.A.; et al. Dendritic cell vaccination combined with temozolomide retreatment: Results of a phase I trial in patients with recurrent glioblastoma multiforme. J. Neurooncol. 2015, 121, 319–329. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef]
- Muller, K.; Henke, G.; Pietschmann, S.; van Gool, S.; De Vleeschouwer, S.; von Bueren, A.O.; Compter, I.; Friedrich, C.; Matuschek, C.; Klautke, G.; et al. Re-irradiation or re-operation followed by dendritic cell vaccination? Comparison of two different salvage strategies for relapsed high-grade gliomas by means of a new prognostic model. J. Neurooncol. 2015, 124, 325–332. [Google Scholar] [CrossRef]
- Sakai, K.; Shimodaira, S.; Maejima, S.; Udagawa, N.; Sano, K.; Higuchi, Y.; Koya, T.; Ochiai, T.; Koide, M.; Uehara, S.; et al. Dendritic cell-based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent malignant glioma. J. Neurosurg. 2015, 123, 989–997. [Google Scholar] [CrossRef]
- Van Gool, S.W. Brain tumor immunotherapy: What have we learned so far? Front. Oncol. 2015, 5, 98. [Google Scholar] [CrossRef]
- Akasaki, Y.; Kikuchi, T.; Homma, S.; Koido, S.; Ohkusa, T.; Tasaki, T.; Hayashi, K.; Komita, H.; Watanabe, N.; Suzuki, Y.; et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol. Immunother. 2016, 65, 1499–1509. [Google Scholar] [CrossRef]
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Normolle, D.P.; Connelly, A.K.; Dibridge, S.; Mason, G.; Whiteside, T.L.; et al. Antigen-specific immunoreactivity and clinical outcome following vaccination with glioma-associated antigen peptides in children with recurrent high-grade gliomas: Results of a pilot study. J. Neurooncol. 2016, 130, 517–527. [Google Scholar] [CrossRef]
- Inoges, S.; Tejada, S.; de Cerio, A.L.; Gallego Perez-Larraya, J.; Espinos, J.; Idoate, M.A.; Dominguez, P.D.; de Eulate, R.G.; Aristu, J.; Bendandi, M.; et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J. Transl. Med. 2017, 15, 104. [Google Scholar] [CrossRef]
- Sakai, K.; Shimodaira, S.; Maejima, S.; Sano, K.; Higuchi, Y.; Koya, T.; Sugiyama, H.; Hongo, K. Clinical effect and immunological response in patients with advanced malignant glioma treated with WT1-pulsed dendritic cell-based immunotherapy: A report of two cases. Interdiscip. Neurosurg. Adv. Tech. Case Manag. 2017, 9, 24–29. [Google Scholar] [CrossRef]
- Benitez-Ribas, D.; Cabezon, R.; Florez-Grau, G.; Molero, M.C.; Puerta, P.; Guillen, A.; Paco, S.; Carcaboso, A.M.; Santa-Maria Lopez, V.; Cruz, O.; et al. Immune Response Generated with the Administration of Autologous Dendritic Cells Pulsed with an Allogenic Tumoral Cell-Lines Lysate in Patients with Newly Diagnosed Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2018, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Buchroithner, J.; Erhart, F.; Pichler, J.; Widhalm, G.; Preusser, M.; Stockhammer, G.; Nowosielski, M.; Iglseder, S.; Freyschlag, C.F.; Oberndorfer, S.; et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers 2018, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Jan, C.I.; Tsai, W.C.; Harn, H.J.; Shyu, W.C.; Liu, M.C.; Lu, H.M.; Chiu, S.C.; Cho, D.Y. Predictors of Response to Autologous Dendritic Cell Therapy in Glioblastoma Multiforme. Front. Immunol. 2018, 9, 727. [Google Scholar] [CrossRef] [PubMed]
- Erhart, F.; Buchroithner, J.; Reitermaier, R.; Fischhuber, K.; Klingenbrunner, S.; Sloma, I.; Hibsh, D.; Kozol, R.; Efroni, S.; Ricken, G.; et al. Immunological analysis of phase II glioblastoma dendritic cell vaccine (Audencel) trial: Immune system characteristics influence outcome and Audencel up-regulates Th1-related immunovariables. Acta Neuropathol. Commun. 2018, 6, 135. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Pellegatta, S.; Eoli, M.; Cuccarini, V.; Anghileri, E.; Pollo, B.; Pessina, S.; Frigerio, S.; Servida, M.; Cuppini, L.; Antozzi, C.; et al. Survival gain in glioblastoma patients treated with dendritic cell immunotherapy is associated with increased NK but not CD8(+) T cell activation in the presence of adjuvant temozolomide. Oncoimmunology 2018, 7, e1412901. [Google Scholar] [CrossRef]
- Yao, Y.; Luo, F.; Tang, C.; Chen, D.; Qin, Z.; Hua, W.; Xu, M.; Zhong, P.; Yu, S.; Chen, D.; et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: An exploratory randomized phase II clinical trial. Cancer Immunol. Immunother. 2018. [Google Scholar] [CrossRef]
- Rudnick, J.D.; Sarmiento, J.M.; Uy, B.; Nuno, M.; Wheeler, C.J.; Mazer, M.J.; Wang, H.; Hu, J.L.; Chu, R.M.; Phuphanich, S.; et al. A phase I trial of surgical resection with Gliadel Wafer placement followed by vaccination with dendritic cells pulsed with tumor lysate for patients with malignant glioma. J. Clin. Neurosci. 2020, 74, 187–193. [Google Scholar] [CrossRef]
- Wang, Q.T.; Nie, Y.; Sun, S.N.; Lin, T.; Han, R.J.; Jiang, J.; Li, Z.; Li, J.Q.; Xiao, Y.P.; Fan, Y.Y.; et al. Tumor-associated antigen-based personalized dendritic cell vaccine in solid tumor patients. Cancer Immunol. Immunother. 2020, 69, 1375–1387. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Dejaegher, J.; Solie, L.; Hunin, Z.; Sciot, R.; Capper, D.; Siewert, C.; Van Cauter, S.; Wilms, G.; van Loon, J.; Ectors, N.; et al. Methylation based glioblastoma subclassification is related to tumoral T cell infiltration and survival. Neuro Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dejaegher, J. Local and Systemic Immune Interactions in Malignant Gliomas; KU Leuven: Leuven, Belgium, 2017. [Google Scholar]
- Available online: https://immuno-oncologynews.com/2017/06/26/glioblastoma-potential-therapy-ict-107-trial-suspended-cash-strapped-immunocellular/ (accessed on 18 December 2020).
- Kast, R.E.; Karpel-Massler, G.; Halatsch, M.E. CUSP9* treatment protocol for recurrent glioblastoma: Aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 2014, 5, 8052–8082. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Messina, G.; Lissoni, A.; Franco, R. The psychoneuroendocrine-immunotherapy of cancer: Historical evolution and clinical results. J. Res. Med. Sci. 2017, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.G.; Da Cruz, W.M.S.; Schonthal, A.H.; Salazar, M.D.; Fontes, C.A.P.; Quirico-Santos, T.; Da Fonseca, C.O. Efficacy of a ketogenic diet with concomitant intranasal perillyl alcohol as a novel strategy for the therapy of recurrent glioblastoma. Oncol. Lett. 2018, 15, 1263–1270. [Google Scholar] [CrossRef]
- Lopez-Valero, I.; Torres, S.; Salazar-Roa, M.; Garcia-Taboada, E.; Hernandez-Tiedra, S.; Guzman, M.; Sepulveda, J.M.; Velasco, G.; Lorente, M. Optimization of a preclinical therapy of cannabinoids in combination with temozolomide against glioma. Biochem. Pharm. 2018, 157, 275–284. [Google Scholar] [CrossRef]
- Friesen, C.; Hormann, I.; Roscher, M.; Fichtner, I.; Alt, A.; Hilger, R.; Debatin, K.M.; Miltner, E. Opioid receptor activation triggering downregulation of cAMP improves effectiveness of anti-cancer drugs in treatment of glioblastoma. Cell Cycle 2014, 13, 1560–1570. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Domogalla, M.P.; Marko, M.; Feyen, O.; Sprenger, K.; Schirrmacher, V.; Stuecker, W. Personalised medicine in glioblastoma multiforme. In Challenges and Solutions of Oncological Hyperthermia; Szasz, A., Ed.; Cambridge Scholars Publishing: Newcastle upon Tyne, UK, 2020; pp. 126–158. [Google Scholar]
- Wang, Z.; Gao, L.; Guo, X.; Lian, W.; Deng, K.; Xing, B. Development and Validation of a Novel DNA Methylation-Driven Gene Based Molecular Classification and Predictive Model for Overall Survival and Immunotherapy Response in Patients with Glioblastoma: A Multiomic Analysis. Front. Cell Dev. Biol. 2020, 8, 576996. [Google Scholar] [CrossRef]
- Bertram, M.Y.; Lauer, J.A.; De Joncheere, K.; Edejer, T.; Hutubessy, R.; Kieny, M.P.; Hill, S.R. Cost-effectiveness thresholds: Pros and cons. Bull. World Health Organ. 2016, 94, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Luo, S.; Zhong, L.; Lai, S.; Zeng, D.; Rao, X.; Huang, P.; Weng, X. Cost-effectiveness of atezolizumab plus chemotherapy for advanced non-small-cell lung cancer. Int. J. Clin. Pharm. 2020, 42, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, P.; Barreto, C.M.N.; Bychkovsky, B.L.; de Lima Lopes, G. Cost-effectiveness studies in oncology. In Mehods and Biostatistics in Oncology; Araujo, R., Riechelmann, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Jonsson, B.; Hofmarcher, T.; Lindgren, P.; Wilking, N. The cost and burden of cancer in the European Union 1995-2014. Eur. J. Cancer 2016, 66, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Li, L.; Brooks, G.; Hassett, M.; Schrag, D. Medicare Spending for Breast, Prostate, Lung, and Colorectal Cancer Patients in the Year of Diagnosis and Year of Death. Health Serv. Res. 2018, 53, 2118–2132. [Google Scholar] [CrossRef] [PubMed]
- Blumen, H.; Fitch, K.; Polkus, V. Comparison of Treatment Costs for Breast Cancer, by Tumor Stage and Type of Service. Am. Health Drug Benefits 2016, 9, 23–32. [Google Scholar] [PubMed]
- Vivot, A.; Jacot, J.; Zeitoun, J.D.; Ravaud, P.; Crequit, P.; Porcher, R. Clinical benefit, price and approval characteristics of FDA-approved new drugs for treating advanced solid cancer, 2000–2015. Ann. Oncol. 2017, 28, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; De Jesus, K.; Mailankody, S. The high price of anticancer drugs: Origins, implications, barriers, solutions. Nat. Rev. Clin. Oncol. 2017, 14, 381–390. [Google Scholar] [CrossRef]
- Roussakow, S.V. Clinical and economic evaluation of modulated electrohyperthermia concurrent to dose-dense temozolomide 21/28 days regimen in the treatment of recurrent glioblastoma: A retrospective analysis of a two-centre German cohort trial with systematic comparison and effect-to-treatment analysis. BMJ Open 2017, 7, e017387. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Recursive Partitioning Analysis (RPA) clinical classification
4. Tumor–host immune reaction
|
1. Molecular biology of tumor
6. Immunotherapy components
|
| Label | Phase | Number of Patients | Randomized | Status | Primary Outcome | Estimated Study Completion |
|---|---|---|---|---|---|---|
| NCT00576537 | 2 | 50 | No | Completed | Safety/Toxicity | 10/2011 |
| NCT02649582 | 1 + 2 | 20 | No | Recruiting | OS, Safety/Toxicity Feasibility | 12/2020 |
| NCT00846456 | 1 + 2 | 20 | No | Completed | Toxicity, Immune response | 02/2013 |
| NCT00323115 | 2 | 11 | No | Completed | Immune response, Toxicity, PFS | 07/2013 |
| NCT02366728 | 2 | 100 | RCT | Active, not recruiting | OS, DC migration | 08/2020 |
| NCT01204684 | 2 | 60 | RCT | Active, not recruiting | PFS, OS | 01/2021 |
| NCT03927222 | 2 | 48 | No | Recruiting | OS, DC migration | 12/2023 |
| NCT01006044 | 2 | 26 | No | Completed | PFS, Toxicity | 08/2014 |
| NCT03395587 | 2 | 136 | RCT | Recruiting | OS, PFS | 06/2023 |
| NCT04523688 | 2 | 28 | No | Not recruiting | PFS | 12/2025 |
| NCT03548571 | 2 + 3 | 60 | RCT | Recruiting | PFS, OS | 05/2023 |
| NCT04115761 | 2 | 24 | RCT | Recruiting | PSF | 06/2022 |
| NCT03014804 | 2 | 0 | RCT | Withdrawn | ||
| NCT03879512 | 1 + 2 | 25 | No | Recruiting | OS, PFS | 01/2022 |
| NCT02772094 | 2 | 50 | No | Unknown | OS, Toxicity | 12/2016 |
| NCT01567202 | 2 | 100 | RCT | Recruiting | Response, PFS, OS | 02/2020 |
| NCT01213407 | 2 | 87 | RCT | Completed | PFS, OS | 11/2015 |
| NCT01291420 | 1 + 2 | 10 | No | Unknown | Immune response | Unknown |
| NCT02465268 | 2 | 120 | RCT | Recruiting | OS, Immune response, PFS | 06/2024 |
| NCT04277221 | 3 | 118 | RCT | Recruiting | OS, PFS | 12/2022 |
| NCT00323115 | 2 | 11 | No | Completed | T-cell response | 07/2013 |
| NCT03400917 | 2 | 55 | No | Completed | OS | 02/2023 |
| NCT02546102 | 3 | 414 | RCT | Suspended | OS | 12/2021 |
| NCT01280552 | 2 | 124 | RCT | Completed | OS | 12/2015 |
| NCT00045968 | 3 | 348 | RCT | Unknown | PFS | 11/2016 |
| NCT03688178 | 2 | 112 | RCT | Recruiting | OS, Varlimumab safety, Treg level | 03/2025 |
| NCT01759810 | 2 + 3 | 60 | No | Enrolling by invitation | OS | 12/2020 |
| NCT02754362 | 2 | No | Withdrawn | Immune response | 06/2019 | |
| NCT04388033 | 1 + 2 | 10 | No | Recruiting | Safety, PFS | 12/2023 |
| Vaccine | Local Hyperthermia | NDV | DC Total | DC/Vaccine | |
|---|---|---|---|---|---|
| N | 69 | 70 | 69 | 70 | 112 |
| Minimum | 0 | 0 | 0 | 0 | 2,400,000 |
| 25%P | 1 | 13 | 17 | 6,950,000 | 8,075,000 |
| Median | 2 | 25 | 30 | 20,115,000 | 12,200,000 |
| 75% | 2 | 42 | 42 | 34,200,000 | 19,145,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Gool, S.W.; Makalowski, J.; Fiore, S.; Sprenger, T.; Prix, L.; Schirrmacher, V.; Stuecker, W. Randomized Controlled Immunotherapy Clinical Trials for GBM Challenged. Cancers 2021, 13, 32. https://doi.org/10.3390/cancers13010032
Van Gool SW, Makalowski J, Fiore S, Sprenger T, Prix L, Schirrmacher V, Stuecker W. Randomized Controlled Immunotherapy Clinical Trials for GBM Challenged. Cancers. 2021; 13(1):32. https://doi.org/10.3390/cancers13010032
Chicago/Turabian StyleVan Gool, Stefaan W., Jennifer Makalowski, Simon Fiore, Tobias Sprenger, Lothar Prix, Volker Schirrmacher, and Wilfried Stuecker. 2021. "Randomized Controlled Immunotherapy Clinical Trials for GBM Challenged" Cancers 13, no. 1: 32. https://doi.org/10.3390/cancers13010032
APA StyleVan Gool, S. W., Makalowski, J., Fiore, S., Sprenger, T., Prix, L., Schirrmacher, V., & Stuecker, W. (2021). Randomized Controlled Immunotherapy Clinical Trials for GBM Challenged. Cancers, 13(1), 32. https://doi.org/10.3390/cancers13010032