RNA-Binding Proteins in Cancer: Functional and Therapeutic Perspectives

Simple Summary
Abstract
1. Introduction
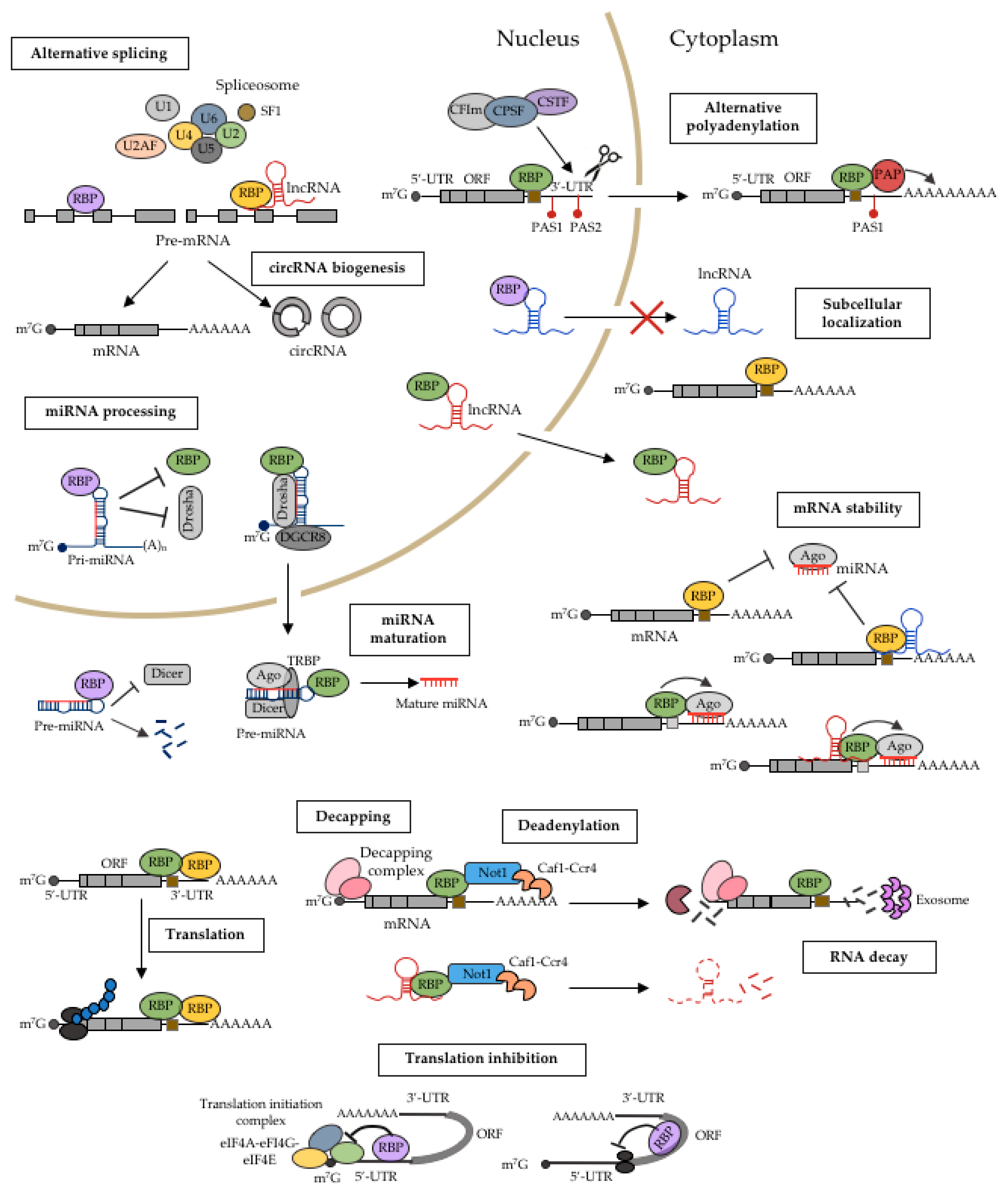
2. Mechanistic Roles of RBPs in Cancer
2.1. miRNA Processing
2.2. Alternative Splicing
2.3. Alternative Polyadenylation
2.4. RNA Localization
2.5. RNA Stability
2.6. Translational Regulation
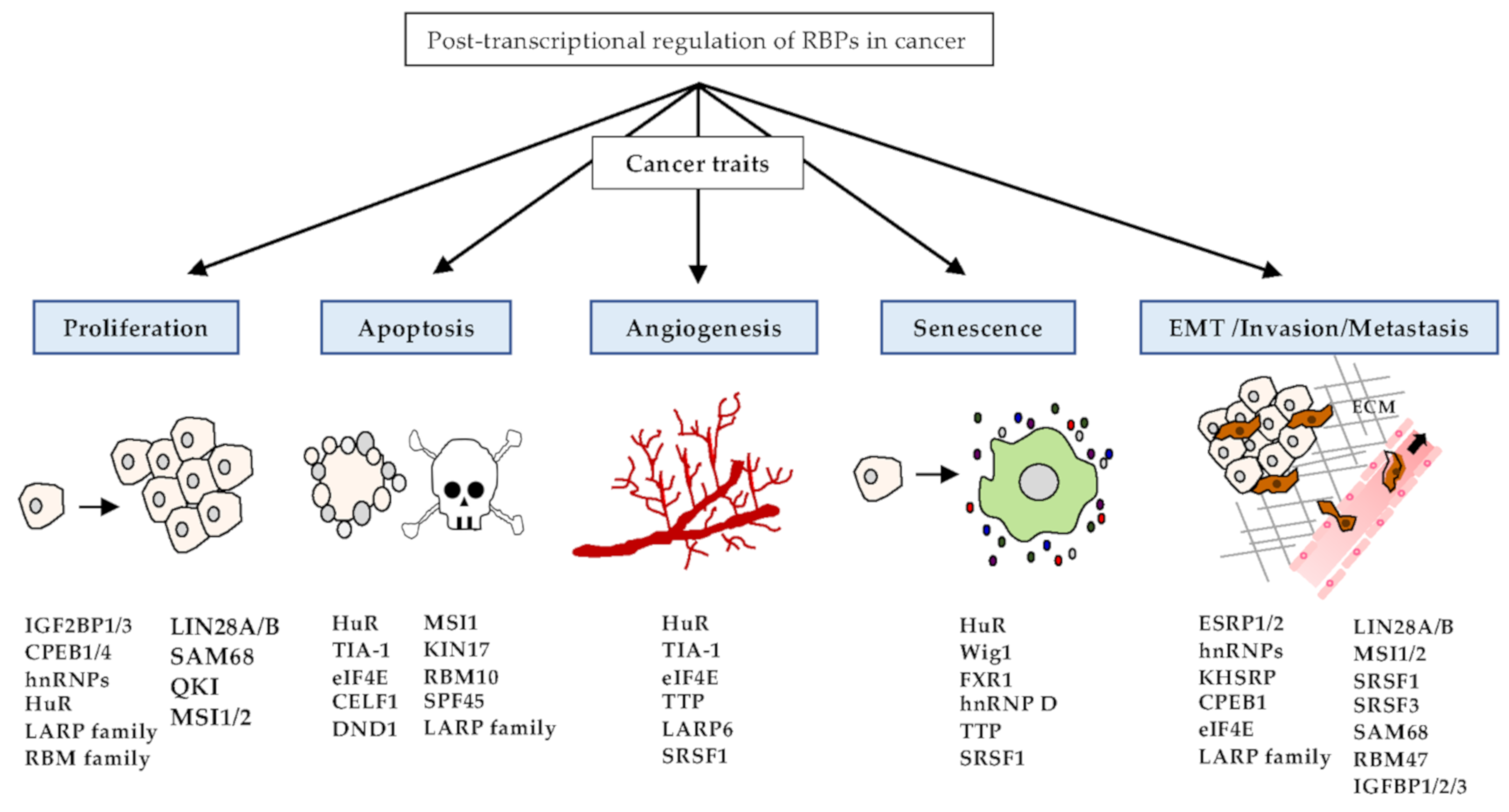
3. Implications of RBPs in Cancer Phenotypes
3.1. Proliferation
3.2. Apoptosis
3.3. Angiogenesis
3.4. Senescence
3.5. EMT, Invasion, and Metastasis
4. RBPs as Therapeutic Targets in Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Hong, S. RNA binding proteins as an emerging therapeutic target for cancer prevention and treatment. J. Cancer Prev. 2017, 22, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Järvelin, A.I.; Davis, I.; Bond, G.L.; Castello, A. Expanding horizons: New roles for non-canonical RNA-binding proteins in cancer. Curr. Opin. Genet. Dev. 2018, 48, 112–120. [Google Scholar] [CrossRef]
- Lukong, K.E.; Chang, K.; Khandjian, E.W.; Richard, S. RNA-binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-binding proteins in cancer: Old players and new actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef]
- Van Kouwenhove, M.; Kedde, M.; Agami, R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer 2011, 11, 644–656. [Google Scholar] [CrossRef]
- Loughlin, F.E.; Gebert, L.F.R.; Towbin, H.; Brunschweiger, A.; Hall, J.; Allain, F.H. Structural basis of pre-let-7 miRNA recognition by the zinc knuckles of pluripotency factor Lin28. Nat. Stuct. Mol. Biol. 2011, 11, 84–89. [Google Scholar] [CrossRef]
- Piskounoa, E.; Viswanathan, S.R.; Janas, M.; LaPierre, R.J.; Daley, G.Q.; Sliz, P.; Gregory, R.I. Determinants of microRNA processing inhibition by the developmentally regulated RNA-binding protein Lin28. J. Biol. Chem. 2008, 283, 21310–21314. [Google Scholar] [CrossRef]
- Heo, I.; Joo, C.; Cho, J.; Ha, M.; Han, J.; Kim, V.N. Lin28 mediates the terminal uridylation of let-7 precursor microRNA. Mol. Cell 2008, 32, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Heo, I.; Joo, C.; Kim, Y.K.; Ha, M.; Yoon, M.J.; Cho, J.; Yeom, K.H.; Han, J.; Kim, V.N. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 2009, 138, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Ustianenko, D.; Hrossova, D.; Potesil, D.; Chalupnikova, K.; Hrazdilova, K.; Pachernik, J.; Cetkobska, K.; Uldrijan, S.; Zdrahal, Z.; Vanacova, S. Mammalian DIS3L2 exoribonuclease targets the uridylated precursors of let-7 miRNAs. RNA 2013, 19, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Viswananthan, S.R.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O’Sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet. 2009, 41, 843–848. [Google Scholar] [CrossRef]
- Balzeau, Z.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 pathway in cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef]
- Trabucchi, M.; Briata, P.; Garcia-Mayoral, M.; Haase, A.D.; Filipowicz, W.; Ramos, A.; Gherzi, R.; Rosenfeld, M.G. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 2009, 459, 1010–1014. [Google Scholar] [CrossRef]
- Tong, L.; Luo, Y.; Wei, T.; Guo, L.; Wang, H.; Zhu, W.; Zhang, J. KH-type splicing regulatory protein (KHSRP) contributes to tumorigenesis by promoting miR-26a maturation in small cell lung cancer. Mol. Cell Biochem. 2016, 422, 61–74. [Google Scholar] [CrossRef]
- Chien, M.H.; Lee, W.J.; Yang, Y.C.; Li, Y.L.; Chen, B.R.; Cheng, T.Y.; Yang, P.W.; Wang, M.Y.; Jan, Y.H.; Lin, Y.K.; et al. KSRP suppresses cell invasion and metastasis through miR-23a-mediated EGR3 mRNA degradation in non-small cell lung cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 1013–1024. [Google Scholar] [CrossRef]
- Puppo, M.; Bucci, G.; Rossi, M.; Giovarelli, M.; Bordo, D.; Moshiri, A.; Gorlero, F.; Gherzi, R.; Briata, P. miRNA-mediated KHSRP silencing rewires distinct post-transcriptional programs during TGF-β-induce epithelial-to-mesenchymal transition. Cell Rep. 2016, 16, 967–978. [Google Scholar] [CrossRef]
- Dhamija, S.; Keuhne, N.; Winzen, R.; Doerrie, A.; Dittrich-Breiholz, O.; Thakur, B.K.; Holtmann, H. Interleukin-1 activates synthesis of interleuin-6 by interfering with a KH-type splicing regulatory protein (KSRP)-dependent translational silencing mechanism. J. Biol. Chem. 2011, 286, 33279–33299. [Google Scholar] [CrossRef]
- Fujita, Y.; Masuda, K.; Hamada, J.; Shoda, K.; Naruto, T.; Hmamda, S.; Miyakami, Y.; Kohmoto, T.; Watanabe, M.; Takahashi, R.; et al. KH-type splicing regulatory protein is involved in esophageal squamous cell carcinoma progression. Oncotarget 2017, 8, 101130–101145. [Google Scholar] [CrossRef] [PubMed]
- Kooshapur, H.; Choudhury, N.R.; Simon, B.; Mühlbauer, M.; Jussupow, A.; Fernandez, N.; Jones, A.N.; Dallmann, A.; Gabel, F.; Camilloni, C.; et al. Structural basis for terminal loop recognition and stimulation of pri-miRNA-18a processing by hnRNP A1. Nat. Commun. 2018, 9, 2479. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Cao, Z.; Zhu, R.; You, L.; Zhang, T. The dual functional role of MicroRNA-18a (miR-18a) in cancer development. Clin. Transl. Med. 2019, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Michlewski, G.; Cáceres, J.F. Antagonistic role of hnRNP A1 and KSRP in the regulation of let-7a biogenesis. Nat. Struct. Mol. Biol. 2010, 17, 1018–1101. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.N.; Hiyard, A.C.; Nguyen, P.H.; Lagna, G.; Hata, A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol. Cell 2010, 39, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Rasheed, S.A.K.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, H.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2018, 27, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef]
- Biamonti, G.; Catillo, M.; Pignataro, D.; Montecucco, A.; Ghigna, C. The alternative splicing side of cancer. Semin. Cell Dev. Biol. 2014, 32, 30–36. [Google Scholar] [CrossRef]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumor suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 2005, 20, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; Stanchina, E.D.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a protooncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An alternative splicing network links cell-cycle control to apoptosis. Cell 2010, 142, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Manley, J.L. Loss of splicing factor ASF/SF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes Dev. 2005, 19, 2705–2714. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef]
- Malakar, P.; Shilo, A.; Mogilevsky, A.; Stein, I.; Pikarsky, E.; Nevo, Y.; Benyamini, H.; Elgavish, S.; Zong, X.; Prasanth, K.V.; et al. Long noncoding MALAT1 promotes hepatocellular carcinoma development by SRSF1 upregulation and mTOR activation. Cancer Res. 2017, 77, 1155–1167. [Google Scholar] [CrossRef]
- Barbagallo, D.; Caponnetto, A.; Brex, D.; Mirabella, F.; Barbagallo, C.; Lauretta, G.; Morrone, A.; Certo, F.; Broggi, G.; Caltabiano, R.; et al. CircSMARCA5 regulates VEGFA mRNA splicing and angiogenesis in glioblastoma multiforme through the binding of SRSF1. Cancers 2019, 11, 194. [Google Scholar] [CrossRef]
- Jia, R.; Li, C.; McCoy, J.P.; Deng, C.X.; Zheng, Z.M. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int. J. Biol. Sci. 2010, 6, 806–826. [Google Scholar] [CrossRef]
- Kurokawa, K.; Akaike, Y.; Masuda, K.; Kuwano, Y.; Nishida, K.; Yamagishi, N.; Kajita, K.; Tanahashi, T.; Rokutan, K. Downregulation of serine/arginine-rich splicing factor 3 induces G1 cell cycle arrest and apoptosis in colon cancer cells. Oncogene 2014, 33, 1407–1417. [Google Scholar] [CrossRef]
- Song, X.; Wan, X.; Huang, T.; Zeng, C.; Sastry, N.; Wu, B.; James, C.D.; Horbinski, C.; Nakano, I.; Zhang, W.; et al. SRSF3-regualed RNA alternative splicing promotes glioblastoma tumorigenicity by affecting multiple cellular processes. Cancer Res. 2019, 79, 5288–5301. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Jumaa, H.; Webster, N.J.G. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun. 2013, 4, 1336. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Langiewicz, M.; Jumaa, H.; Webster, N.J.G. Depletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology 2015, 61, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Meng, N.; Chen, M.; Chen, D.; Chen, X.H.; Wang, J.Z.; Zhu, S.; He, Y.T.; Zhang, X.L.; Lu, R.X.; Yan, G.R. Small protein hidden in lncRNA LOC90024 promotes “Cancerous” RNA splicing and tumorigenesis. Adv. Sci. 2020, 7, 1903233. [Google Scholar] [CrossRef]
- Cohen-Eliav, M.; Golan-Gerstl, R.; Siegfried, Z.; Anderson, C.L.; Thorsen, K.; Ørntoft, T.F.; Mu, D.; Karni, R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J. Pathol. 2013, 229, 630–639. [Google Scholar] [CrossRef]
- Villamizar, O.; Chambers, C.B.; Riberdy, J.M.; Persons, D.A.; Wilber, A. Long noncoding RNA Saf and splicing factor 45 increase soluble Fas and resistance to apoptosis. Oncotarget 2016, 7, 13810–13826. [Google Scholar] [CrossRef]
- Chaudhury, A.; Chander, P.; Howe, P.H. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1’s multifunctional regulatory roles. RNA 2010, 16, 1449–1462. [Google Scholar] [CrossRef]
- Clower, C.V.; Chatterjee, D.; Wang, Z.; Cantley, L.C.; Heiden, M.G.V.; Krainer, A.R. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 1894–1899. [Google Scholar] [CrossRef]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Zhou, Z.J.; Dai, Z.; Zhou, S.L.; Fu, X.T.; Zhao, Y.M.; Shi, Y.H.; Zhou, J.; Fan, J. Overexpression of hnRNP A1 promotes tumor invasion through regulating CD44v6 and indicates poor prognosis for hepatocellular carcinoma. Int. J. Cancer 2013, 132, 1080–1089. [Google Scholar] [CrossRef]
- Golan-Gerstl, R.; Cohen, M.; Shilo, A.; Suh, S.S.; Bakàcs, A.; Coppola, L.; Karni, R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011, 71, 4464–4472. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gao, X.D.; Lee, J.H.; Huang, H.; Tan, H.; Ahn, J.; Reinke, L.M.; Peter, M.E.; Feng, Y.; Gius, D.; et al. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev. 2014, 28, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Lefave, C.V.; Squatrito, M.; Vorlova, S.; Rocco, G.L.; Brennan, C.W.; Holland, E.C.; Pan, Y.X.; Cartegni, L. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011, 30, 4084–4097. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhou, Y.; Wu, T.; Zhu, T.; Ji, X.; Kwon, Y.S.; Zhang, C.; Yeo, G.; Black, D.L.; Sun, H.; et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol. Cell 2010, 36, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; McCutcheon, I.E.; Fuller, M.; Huang, E.S.; Cote, G.J. Fibroblast growth factor receptor-1 alpha-exon exclusion and polypyrimidine tract-binding protein in glioblastoma multiforme tumors. Cancer Res. 2000, 60, 1221–1224. [Google Scholar] [PubMed]
- Yamaguchi, F.; Saya, H.; Bruner, J.M.; Morrison, R.S. Differential expression of two fibroblast growth factor-receptor genes Is associated with malignant progression in human astrocytomas. Proc. Natl. Acad. Sci. USA 1994, 91, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Izaguirre, D.I.; Zhu, W.; Hai, T.; Cheung, H.C.; Krahe, R.; Cote, G.J. PTBP1-dependent regulation of USP5 alternative RNA splicing plays a role in glioblastoma tumorigenesis. Mol. Carcinog. 2012, 51, 895–906. [Google Scholar] [CrossRef]
- Warzecha, C.C.; Sato, T.K.; Nabet, B.; Hogenesch, J.B.; Carstens, R.P. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell 2009, 33, 591–601. [Google Scholar] [CrossRef]
- Warzecha, C.C.; Shen, S.; Xing, Y.; Carstens, R.P. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol. 2009, 6, 536–562. [Google Scholar] [CrossRef]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.J.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H.; et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun. 2012, 3, 883. [Google Scholar] [CrossRef]
- García-Cárdenas, J.M.; Guerrero, S.; López-Cortés, A.; Armendáriz-Castillo, I.; Guevara-Ramírez, P.; Pérez-Villa, A.; Yumiceba, V.; Zambrano, A.K.; Leone, P.E.; Paz-y-Miño, C. Post-transcriptional regulation of colorectal cancer: A focus on RNA-binding proteins. Front. Mol. Biosci. 2019, 6, 65. [Google Scholar]
- Dong, W.; Dai, Z.H.; Liu, F.C.; Guo, X.G.; Ge, C.M.; Ding, J.; Liu, H.; Yang, F. The RNA-binding protein RBM3 promotes cell proliferation in hepatocellular carcinoma by regulating circular RNA SCD-circRNA 2 production. EBioMedicine 2019, 45, 155–167. [Google Scholar] [CrossRef]
- Bielli, P.; Busà, R.; Paronetto, M.P.; Sette, C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr. Relat. Cancer 2011, 18, R91–R102. [Google Scholar] [CrossRef] [PubMed]
- Matter, N.; Herrlich, P.; König, H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002, 420, 691–695. [Google Scholar] [CrossRef]
- Paronetto, M.P.; Cappellari, M.; Busà, R.; Pedrotti, S.; Vitali, R.; Comstock, C.; Hyslop, T.; Knudsen, K.E.; Sette, C. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res. 2010, 70, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Achsel, T.; Massiello, A.; Chalfant, C.E.; Sette, C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 2007, 176, 929–939. [Google Scholar] [CrossRef]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Screiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA binding protein quaking regulates formation of circRNA. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef]
- Erson-Bensan, A.E.E.; Can, T. Alternative polyadenylation: Another foe in cancer. Mol. Cancer Res. 2016, 14, 507–517. [Google Scholar] [CrossRef]
- Fernández-Miranda, G.; Méndez, R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res. Rev. 2012, 11, 460–472. [Google Scholar] [CrossRef]
- Nagaoka, K.; Fujii, K.; Zhang, H.; Usuda, K.; Watanabe, G.; Ivshina, M.; Richter, J.D. CPEB1 mediates epithelial-to-mesenchyme transition and breast cancer metastasis. Oncogene 2016, 35, 2893–2901. [Google Scholar] [CrossRef]
- Hägele, S.; Kühn, U.; Böning, M.; Katschinski, D.M. Cytoplasmic polyadenylation-element-binding protein (CPEB)1 and 2 bind to the HIF-1alpha mRNA 3’-UTR and modulate HIF-1alpha protein expression. Biochem. J. 2009, 417, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Zapater, E.; Pineda, D.; Martínez-Bosch, N.; Fernández-Miranda, G.; Iglesias, M.; Alameda, F.; Moreno, M.; Eliscovich, C.; Eyras, E.; Real, F.X.; et al. Key contribution of CPEB4-mediated translational control to cancer progression. Nat. Med. 2011, 18, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Guijarro, E.; Karras, P.; Cifdaloz, M.; Martínez-Herranz, R.; Cañón, E.; Graña, O.; Horcajada-Reales, C.; Alonso-Curbelo, D.; Calvo, T.G.; Gómez-López, G.; et al. Lineage-specific roles of the cytoplasmic polyadenylation factor CPEB4 in the regulation of melanoma drivers. Nat. Commun. 2016, 7, 13418. [Google Scholar] [CrossRef] [PubMed]
- Calderone, V.; Gallego, J.; Fernandez-Miranda, G.; Garcia-Pras, E.; Maillo, C.; Berzigotti, A.; Mejias, M.; Bava, F.A.; Angulo-Urarte, A.; Graupera, G.; et al. Sequential functions of CPEB1 and CPEB4 regulate pathologic expression of vascular endothelial growth factor and angiogenesis in chronic liver disease. Gastroenterology 2016, 150, 982–997. [Google Scholar] [CrossRef]
- Degrauwe, N.; Suvà, M.L.; Janiszewska, M.; Riggi, N.; Stamenkovic, I. IMPs: An RNA-binding brotein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016, 30, 2459–2474. [Google Scholar] [CrossRef]
- Jonas, K.; Calin, G.A.; Pichler, A. RNA-binding proteins as important regulators of long non-coding RNAs in cancer. Int. J. Mol. Sci. 2020, 21, 2969. [Google Scholar] [CrossRef]
- Nagaoka, K.; Udagawa, T.; Richter, J.D. CPEB-mediated ZO-1 mRNA localization is required for epithelial tight-junction assembly and cell polarity. Nat. Commun. 2012, 3, 675. [Google Scholar] [CrossRef]
- Farina, K.L.; Huttelmaier, S.; Musunuru, K.; Darnell, R.; Singer, R.H. Two ZBP1 KH domains facilitate beta-actin mRNA localization granule formation, and cytoskeletal attachment. J. Cell Biol. 2003, 160, 77–87. [Google Scholar] [CrossRef]
- Gu, W.; Katz, Z.; Wu, B.; Park, H.Y.; Li, D.; Lin, S.; Wells, A.L.; Singer, R.H. Regulation of local expression of cell adhesion and motility-related mRNAs in breast cancer cells by IMP1/ZBP1. J. Cell Sci. 2012, 125, 81–91. [Google Scholar] [CrossRef]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef]
- Noh, J.H.; Kim, K.M.; Abdelmohsen, L.; Yoon, J.H.; Panda, A.C.; Munk, R.; Kim, J.; Curtis, J.; Moad, C.A.; Wohler, C.M.; et al. HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev. 2016, 30, 1224–1239. [Google Scholar]
- Lubelsky, Y.; Ultsky, I. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature 2018, 555, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Kabotyanski, E.B.; Reineke, L.C.; Shao, J.; Xiong, F.; Lee, J.H.; Dubrulle, J.; Johnson, H.; Stossi, F.; Tsoi, P.S.; et al. The SINEB1 element in the long non-coding RNA Malat1 is necessary for TDP-43 proteostasis. Nucleic Acids Res. 2020, 48, 2621–2642. [Google Scholar] [CrossRef] [PubMed]
- Garneau, N.L.; Wilusz, J.; Wilusz, C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007, 8, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Perron, G.; Jandaghi, P.; Solanki, S.; Safisamghabadi, M.; Storoz, C.; Karimzadeh, M.; Papadakis, A.I.; Arseneault, M.; Scelo, G.; Banks, R.E.; et al. A general framework for interrogation of mRNA stability programs identifies RNA-binding proteins that govern cancer transcriptomes. Cell Rep. 2018, 23, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.E.; Chenette, D.M.; Larkin, L.C.; Schneider, R.J. Physiological networks and disease functions of RNA-binding protein AUF1. Wiley Interdiscip. Rev. RNA 2014, 5, 549–564. [Google Scholar] [CrossRef]
- Lin, S.; Wang, W.; Wilson, G.M.; Yang, X.; Brewer, G.; Holbrook, N.J.; Gorospe, M. Down-regulation of cyclin D1 expression by prostaglandin A(2) Is mediated by enhanced cyclin D1 mRNA turnover. Mol. Cell Biol. 2000, 20, 7903–7913. [Google Scholar] [CrossRef]
- Lal, A.; Mazan-Mamczarz, K.; Kawai, T.; Yang, X.; Martindale, J.L.; Gorospe, M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004, 23, 3092–3102. [Google Scholar] [CrossRef]
- Chang, N.; Yi, J.; Guo, G.; Liu, X.; Shang, Y.; Tong, T.; Cui, Q.; Ming, Z.; Gorospe, M.; Wang, W. HuR uses AUF1 as a cofactor to promote p16INK4 mRNA decay. Mol. Cell Biol. 2010, 30, 3875–3886. [Google Scholar] [CrossRef]
- Trojanowicz, B.; Brodauf, L.; Sekulla, C.; Lorenz, K.; Finke, R.; Dralle, H.; Hoang-Vu, C. The role of AUF1 in thyroid carcinoma progression. Endocr. Relat. Cancer 2009, 16, 857–871. [Google Scholar] [CrossRef]
- Zucconi, B.E.; Wilson, G.M. Modulation of neoplastic gene regulatory pathways by the RNAbinding factor AUF1. Front. Biosci. 2011, 16, 2307–2325. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; De, S.; Srikantan, S.; Abdelmohsen, K.; Grammatikakis, I.; Kim, J.; Kim, J.M.; Noh, J.H.; White, E.J.F.; Martindale, J.L.; et al. PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat. Commun. 2014, 5, 5248. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, Y.; Chu, H.; Guan, Y.; Bi, J.; Wang, B. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int. J. Mol. Sci. 2013, 14, 10015–10041. [Google Scholar] [CrossRef] [PubMed]
- Young, L.E.; Moore, A.E.; Sokol, L.; Meisner-Kober, N.; Dixon, D.A. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol. Cancer Res. 2012, 10, 167–180. [Google Scholar] [CrossRef]
- Chai, Y.; Liu, J.; Zhang, Z.; Liu, L. HuR-regulated lncRNA NEAT1 stability in tumorigenesis and progression of ovarian cancer. Cancer Med. 2016, 5, 1588–1598. [Google Scholar] [CrossRef]
- Hu, Y.P.; Jin, Y.P.; Wu, X.S.; Yang, Y.; Li, Y.S.; Li, H.F.; Xiang, S.S.; Song, X.L.; Jiang, L.; Zhang, Y.J.; et al. LNCRNA-HGBC stabilized by HuR promotes gallbladder cancer progression by regulating miR-502-3p/SET/AKT axis. Mol. Cancer 2019, 18, 167. [Google Scholar] [CrossRef]
- Kim, J.; Abdelmohsen, K.; Yang, X.; De, S.; Grammatikakis, I.; Noh, H.; Gorospe, M. LncRNA OIP5-AS1/Cyrano sponges RNA-binding protein HuR. Nucleic Acids Res. 2016, 44, 2378–2392. [Google Scholar] [CrossRef]
- Yoon, J.H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhang, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell 2012, 47, 648–655. [Google Scholar] [CrossRef]
- Yoon, J.H.; Abdelmohsen, K.; Kim, J.; Yang, X.; Martindale, J.L.; Tominaga-Yamanaka, K.; White, E.J.; Orjalo, A.V.; Rinn, J.L.; Kreft, S.G.; et al. Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat. Commun. 2014, 4, 2939. [Google Scholar] [CrossRef]
- Fu, M.; Blackshear, P.J. RNA-binding proteins in immune regulation: A focus on CCCH zinc finger proteins. Nat. Rev. Immunol. 2017, 17, 130–143. [Google Scholar] [CrossRef]
- Sandler, H.; Kreth, J.; Timmers, H.T.M.; Stoecklin, G. Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic Acids Res. 2011, 39, 4373–4386. [Google Scholar] [CrossRef] [PubMed]
- Fenger-Grøn, M.; Fillman, C.; Norrild, B.; Lykke-Andersen, J. Multiple processing body factors and the ARE binding protein activate mRNA decapping. Mol. Cell 2005, 20, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Lee, T.H.; Kang, T.H. Roles of tristetraprolin in tumorigenesis. Int. J. Mol. Sci. 2018, 19, 3384. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Qu, H.; Shan, T.; Chen, Y.; Chen, Y.; Xia, J. Tristetraprolin overexpression in gastric cancer cells suppresses PD-L1 expression and inhibits tumor progression by enhancing antitumor immunity. Mol. Cells 2018, 41, 653–664. [Google Scholar]
- Noubissi, F.K.; Elcheva, I.; Bhatia, N.; Shakoori, A.; Ougolkov, A.; Liu, J.; Minamoto, T.; Ross, J.; Fuchs, S.Y.; Spiegelman, V.S. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to b-catenin signalling. Nature 2006, 441, 898–901. [Google Scholar] [CrossRef]
- Weidensdorfer, D.; Stöhr, N.; Baude, A.; Lederer, M.; Köhn, M.; Schierhorn, A.; Buchmeier, S.; Wahle, E.; Hüttelmaier, S. Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA 2009, 15, 104–115. [Google Scholar] [CrossRef]
- Vikesaa, J.; Hansen, T.V.O.; Jønson, L.; Borup, R.; Wewer, U.M.; Christiansen, J.; Nielsen, F.C. RNA-binding IMPs promote cell adhesion and invadopodia formation. EMBO J. 2006, 25, 1456–1468. [Google Scholar] [CrossRef]
- Elcheva, I.; Goswami, S.; Noubissi, F.K.; Spiegelman, V.S. CRD-BP protects the coding region of betaTrCP1 mRNA from miR-183-mediated degradation. Mol. Cell 2009, 35, 240–246. [Google Scholar] [CrossRef]
- Ye, S.; Song, W.; Xu, X.; Zhao, X.; Yang, L. IGF2BP2 promotes colorectal cancer cell proliferation and survival through interfering with RAF-1 degradation by miR-195. FEBS Lett. 2016, 590, 1641–1650. [Google Scholar] [CrossRef]
- Jønson, L.; Christiansen, J.; Hansen, T.V.O.; Vikeså, J.; Yamamoto, Y.; Nielsen, F.C. IMP3 RNP safe houses prevent miRNA-directed HMGA2 mRNA decay in cancer and development. Cell Rep. 2014, 7, 539–551. [Google Scholar] [CrossRef]
- Hämmerle, M.; Gutschner, T.; Uckelmann, H.; Ozgur, S.; Fiskin, E.; Gross, M.; Skawran, B.; Geffers, R.; Longerich, T.; Breuhahn, K.; et al. Posttranscriptional destabilization of the liver-specific long noncoding RNA HULC by the IGF2 mRNA-binding protein 1 (IGF2BP1). Hepatology 2013, 58, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.; et al. Recognition of RNA N6-methyadenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell. Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Hellborg, F.; Qian, W.; Mendez-Vidal, C.; Asker, C.; Kost-Alimova, M.; Wilhelm, M.; Imreh, S.; Wiman, K.G. Human wig-1, a p53 target gene that encodes a growth inhibitory zinc finger protein. Oncogene 2001, 20, 5466–5474. [Google Scholar] [CrossRef] [PubMed]
- Prahl, M.; Vilborg, A.; Palmberg, C.; Jörnvall, H.; Asker, C.; Wiman, K.G. The p53 target protein Wig-1 binds hnRNP A2/B1 and RNA helicase A via RNA. FEBS Lett. 2008, 582, 2173–2177. [Google Scholar] [CrossRef]
- Viborg, A.; Glahder, J.A.; Wilhelm, M.; Bersani, C.; Corcoran, M.; Mahmoudi, S.; Rosenstierne, M.; Grandér, D.; Farnebo, M.; Norrild, B.; et al. The p53 target Wig-1 regulates p53 mRNA stability through an AU-rich element. Proc. Natl. Acad. Sci. USA 2009, 106, 15756–15761. [Google Scholar] [CrossRef]
- Kim, B.C.; Lee, H.C.; Lee, J.J.; Choi, C.M.; Kim, D.K.; Lee, J.C.; Ko, Y.G.; Lee, J.S. Wig1 prevents cellular senescence by regulating p21 mRNA decay through control of RISC recruitment. EMBO J. 2012, 31, 4289–4303. [Google Scholar] [CrossRef]
- Lee, H.C.; Jung, S.H.; Hwang, H.J.; Kang, D.; De, S.; Dudekula, D.B.; Martindale, J.L.; Park, B.; Park, S.K.; Lee, E.K.; et al. Wig1 is crucial for AGO2-mediated ACOT7 mRNA silencing via miRNA-dependent and -independent mechanisms. Nucleic Acids Res. 2017, 45, 6894–6910. [Google Scholar] [CrossRef]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2017, 17, 332. [Google Scholar] [CrossRef]
- Hsieh, A.C.; Ruggero, D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin. Cancer Res. 2010, 16, 4914–4920. [Google Scholar] [CrossRef]
- Mazan-Mamczarz, K.; Galbán, S.; López de Silanes, I.; Martindale, J.L.; Atasoy, U.; Keene, J.D.; Gorospe, M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. USA 2003, 100, 8354–8359. [Google Scholar] [CrossRef]
- Vo, D.T.; Abdelmohsen, K.; Martindale, J.L.; Qiao, M.; Tominaga, K.; Burton, T.L.; Gelfond, J.A.L.; Brenner, A.J.; Patel, V.; Trageser, D.; et al. The oncogenic RNA-binding protein musashi1 is regulated by HuR via mRNA translation and stability in glioblastoma cells. Mol. Cancer Res. 2012, 10, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Galbán, S.; Kuwano, Y.; Pullmann, R., Jr.; Martindale, M.L.; Kim, H.H.; Lal, A.; Abdelmohsen, K.; Yang, X.; Dang, Y.; Liu, J.O.; et al. RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1alpha. Mol. Cell Biol. 2008, 28, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Glorian, V.; Maillot, G.; Polès, S.; Iacovoni, J.S.; Favre, G.; Vagner, S. HuR-dependent loading of miRNA RISC to the mRNA encoding the Ras-related small GTPase RhoB controls its translation during UV-induced apoptosis. Cell Death Differ. 2011, 18, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Lagadec, C.; Vlashi, E.; Frohnen, P.; Alhiyari, Y.; Chan, M.; Pajonk, F. The RNA-binding protein Musashi-1 regulates proteasome subunit expression in breast cancer- and glioma-initiating cells. Stem Cells 2014, 32, 135–144. [Google Scholar] [CrossRef]
- Park, S.-M.; Gönen, M.; Vu, L.; Minuesa, G.; Tivnan, P.; Barlowe, T.S.; Taggart, J.; Lu, Y.; Deering, R.P.; Hacohen, N.; et al. Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. J. Clin. Investig. 2015, 125, 1286–1298. [Google Scholar] [CrossRef]
- Niu, J.; Zhao, X.; Liu, Q.; Yang, J. Knockdown of MSI1 inhibited the cell proliferation of human osteosarcoma cells by targeting p21 and p27. Oncol. Lett. 2017, 14, 5271–5278. [Google Scholar] [CrossRef]
- Fox, R.G.; Lytle, N.K.; Jaquish, D.V.; Park, F.D.; Ito, T.; Bajaj, J.; Koechlein, C.S.; Zimdahl, B.; Yano, M.; Kopp, J.; et al. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature 2016, 534, 407–411. [Google Scholar] [CrossRef]
- Durie, D.; Lewis, S.M.; Liwak, U.; Kisilewicz, M.; Gorospe, M.; Holcik, M. RNA-binding protein HuR mediates cytoprotection through stimulation of XIAP translation. Oncogene 2011, 30, 1460–1469. [Google Scholar] [CrossRef]
- Hussey, G.S.; Chaudhury, A.; Dawson, A.E.; Lindner, D.J.; Knudsen, C.R.; Wilce, M.C.J.; Merrick, M.C.J.; Howe, P.H. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol. Cell 2011, 41, 419–431. [Google Scholar] [CrossRef]
- Chaudhury, A.; Hussey, G.S.; Ray, P.S.; Jin, G.; Fox, P.L.; Howe, P.H. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat. Cell Biol. 2010, 12, 286–293. [Google Scholar] [CrossRef]
- Biyanee, A.; Ohnheiser, J.; Singh, P.; Klempnauer, K.-H. A novel mechanism for the control of translation of specific mRNAs by tumor suppressor protein Pdcd4: Inhibition of translation elongation. Oncogene 2015, 34, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Parsa, N. Environmental Factors Inducing Human Cancers. Iran. J. Public Health 2012, 41, 1–9. [Google Scholar] [PubMed]
- Lewandowska, A.M.; Rudzki, M.; Rudzki, S.; Lewandowski, T.; Laskowska, B. Environmental risk factors for cancer. Ann. Agric. Environ. Med. 2019, 26, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-L.; Li, B.; Luo, Y.-X.; Lin, Q.; Liu, S.-R.; Zhang, X.-Q.; Zhou, H.; Yang, J.-H.; Qu, L.-H. Comprehensive genomic characterization of RNA-binding proteins across human cancers. Cell Rep. 2018, 22, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hämmerle, M.; Pazaitis, N.; Bley, N.; Fiskin, E.; Uckelmann, H.; Heim, A.; Groβ, M.; Hofmann, N.; Geffers, R.; et al. Insulin-Like Growth Factor 2 mRNA-Binding Protein 1 (IGF2BP1) is an Important Protumorigenic Factor in Hepatocellular Carcinoma. Hepatology 2014, 59, 1900–1911. [Google Scholar] [CrossRef]
- Wang, Z.; Tong, D.; Han, C.; Zhao, Z.; Wang, X.; Jiang, T.; Li, Q.; Liu, S.; Chen, L.; Chen, Y.; et al. Blockade of miR-3614 maturation by IGF2BP3 increases TRIM25 expression and promotes breast cancer cell proliferation. EBioMedicine 2019, 41, 357–369. [Google Scholar] [CrossRef]
- Mizutani, R.; Imamachi, N.; Suzuki, Y.; Yoshida, H.; Tochigi, N.; Oonishi, T.; Suzuki, Y.; Akimitsu, N. Oncofetal protein IGF2BP3 facilitates the activity of proto-oncogene protein eIF4E through the destabilization of EIF4E-BP2 mRNA. Oncogene 2016, 35, 3495–3502. [Google Scholar] [CrossRef]
- Palanichamy, J.K.; Tran, T.M.; Howard, J.M.; Contreras, J.R.; Fernando, T.R.; Sterne-Weiler, T.; Katzman, S.; Toloue, M.; Yan, W.; Basso, G.; et al. RNA-binding protein IGF2BP3 targeting of oncogenic transcripts promotes hematopoietic progenitor proliferation. J. Clin. Investig. 2016, 126, 1495–1511. [Google Scholar] [CrossRef]
- Vargas, T.R.; Boudoukha, S.; Simon, A.; Souidi, M.; Cuvellier, S.; Pinna, G.; Polesskaya, A. Post-transcriptional regulation of cyclins D1, D3 and G1 and proliferation of human cancer cells depend on IMP-3 nuclear localization. Oncogene 2014, 33, 2866–2875. [Google Scholar] [CrossRef]
- Bava, F.A.; Eliscovich, C.; Ferreira, P.G.; Minñana, B.; Ben-Dov, C.; Guigõ, R.; Juan Valcárcel, J.; Méndez, R. CPEB1 coordinates alternative 3′-UTR formation with translational regulation. Nature 2013, 495, 121–125. [Google Scholar] [CrossRef]
- Kang, H.; Heo, S.; Shin, J.; Ji, E.; Tak, H.; Ahn, S.; Lee, K.; Lee, E.; Kim, W. A miR-194/PTBP1/CCND3 axis regulates tumor growth in human hepatocellular carcinoma. J. Pathol. 2019, 249, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Hu, Y.; Gary Brewer, G. Competitive binding of AUF1 and TIAR to MYC mRNA controls its translation. Nat. Struct. Biol. 2007, 14, 511–518. [Google Scholar] [CrossRef]
- Li, F.; Su, M.; Zhao, H.; Xie, W.; Cao, S.; Xu, Y.; Chen, W.; Wang, L.; Hou, L.; Tan, W. HnRNP-F promotes cell proliferation by regulating TPX2 in bladder cancer. Am. J. Transl. Res. 2019, 11, 7035–7048. [Google Scholar] [PubMed]
- Guo, X.; Hartley, R.S. HuR Contributes to Cyclin E1 Deregulation in MCF-7 Breast Cancer Cells. Cancer Res. 2006, 66, 7948–7956. [Google Scholar] [CrossRef]
- Wang, W.; Caldwell, M.C.; Lin, S.; Furneaux, H.; Gorospe, M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO Rep. 2000, 19, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Sommer, G.; Dittmann, J.; Kuehnert, J.; Reumann, K.; Schwartz, P.E.; Will, H.; Coulter, B.L.; Smith, M.T.; Heise, T. The RNA-binding protein La contributes to cell proliferation and CCND1 expression. Oncogene 2011, 30, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Tcherkezian, J.; Cargnello, M.; Romeo, Y.; Huttlin, E.L.; Lavoie, G.; Gygi, S.P.; Roux, P.P. Proteomic analysis of cap-dependent translation identifies LARP1 as a key regulator of 5′ TOP mRNA translation. Genes Dev. 2014, 28, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Bechara, E.G.; Sebestyén, E.; Bernardis, I.; Eyras, E.; Valcárcel, J. RBM5, 6, and 10 Differentially Regulate NUMB Alternative Splicing to Control Cancer Cell Proliferation. Mol. Cell 2013, 52, 720–733. [Google Scholar] [CrossRef]
- Feng, C.; Neumeister, V.; Ma, W.; Xu, J.; Lu, L.; Bordeaux, J.; Maihle, N.J.; Rimm, D.L.; Huang, Y. Lin28 regulates HER2 and promotes malignancy through multiple mechanisms. Cell Cycle 2012, 11, 2486–2494. [Google Scholar] [CrossRef]
- Babic, I.; Jakymiw, A.; Fujita, D.J. The RNA binding protein Sam68 is acetylated in tumor cell lines, and its acetylation correlates with enhanced RNA binding activity. Oncogene 2004, 23, 3781–3789. [Google Scholar] [CrossRef]
- Yang, G.; Fu1, H.; Zhang, J.; Lu, X.; Yu, F.; Jin, L.; Bai, L.; Huang, B.; Shen, L.; Feng, Y.; et al. RNA binding protein Quaking, a critical regulator of colon epithelial differentiation and a suppressor of colon cancer. Gastroenterology 2010, 138, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Wong, R. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Lal, A.; Kim, H.; Gorospe, M. Posttranscriptional Orchestration of an Anti-Apoptotic Program by HuR. Cell Cycle 2007, 6, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Kawai, T.; Yang, X.; Mazan-Mamczarz, K.; Gorospe, M. Antiapoptotic function of RNA-binding protein HuR effected through prothymosin α. EMBO Rep. 2005, 24, 1852–1862. [Google Scholar] [CrossRef]
- Wigington, C.P.; Jung, J.; Rye, E.A.; Belauret, S.L.; Philpot, A.M.; Feng, Y.; Santangelo, P.J.; Corbett, A.H. Post-transcriptional Regulation of Programmed Cell Death (PDCD4) mRNA by the RNA-binding Proteins Human Antigen R (HuR) and T-cell Intracellular Antigen 1 (TIA1). J. Biol. Chem. 2015, 290, 3468–3487. [Google Scholar] [CrossRef]
- Izquierdo, J.M.; Majós, N.; Bonnal, S.; Martínez, C.; Castelo, R.; Guigó, R.; Bilbao, D.; Valcárcel, J. Regulation of Fas Alternative Splicing by Antagonistic Effects of TIA-1 and PTB on Exon Definition. Mol. Cell 2005, 19, 475–484. [Google Scholar] [CrossRef]
- Wendel, H.S.; Stanchina, D.; Fridman, J.S.; Malina, A.; Ray, S.; Kogan, S.; Cordon-Cardo, C.; Pelletier, J.; Lowe, S.W. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004, 428, 332–337. [Google Scholar] [CrossRef]
- Koso, H.; Yi, H.; Sheridan, P.; Miyano, S.; Ino, Y.; Todo, T.; Watanabe, S. Identification of RNA-Binding Protein LARP4B as a Tumor Suppressor in Glioma. Cancer Res. 2016, 76, 2254–2264. [Google Scholar] [CrossRef]
- Trotta, R.; Vignudelli, T.; Candini, O.; Intine, R.V.; Pecorari, L.; Guerzoni, C.; Santilli, G.; Byrom, M.W.; Goldoni, S.; Ford, L.P.; et al. BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer Cell 2003, 3, 145–160. [Google Scholar] [CrossRef]
- Talwar, S.; Balasubramanian, S.; Sundaramurthy, S.; House, R.; Wilusz, C.J.; Kuppuswamy, D.; D’Silva, N.; Gillespie, M.B.; Hill, E.G.; Palanisamy, V. Overexpression of RNA-binding protein CELF1 prevents apoptosis and destabilizes pro-apoptotic mRNAs in oral cancer cells. RNA Biol. 2013, 10, 277–286. [Google Scholar] [CrossRef]
- Cheng, F.; Pan, Y.; Lu, Y.M.; Zhu, L.; Chen, S. RNA-Binding Protein Dnd1 Promotes Breast Cancer Apoptosis by Stabilizing the Bim mRNA in a miR-221 Binding Site. Biomed. Res. Int. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; George, R.J.; Dieckgraefe, B.K.; Mcleod, H.L.; Ramalingam, S.; Bishnupuri, K.S.; Natarajan, G.; Anant, S.; Houchen, C.W. Knockdown of RNA binding protein Musashi-1 leads to tumor regression In Vivo. Gastroenterology 2008, 134, 1448–1458. [Google Scholar] [CrossRef]
- Gao, X.; Liu, Z.; Zhong, M.; Wu, K.; Zhang, Y.; Wang, H.; Zeng, T. Knockdown of DNA/RNA-binding protein KIN17 promotes apoptosis of triple-negative breast cancer cells. Oncol. Lett. 2019, 17, 288–293. [Google Scholar] [CrossRef]
- Jung, J.; Lee, H.; Cao, B.; Liao, P.; Zeng, S.X.; Lu, H. RNA-binding motif protein 10 induces apoptosis and suppresses proliferation by activating p53. Oncogene 2020, 39, 1031–1040. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Gillespie, G.Y.; Harkins, L.; King, P.H. HuR, a RNA Stability Factor, Is Expressed in Malignant Brain Tumors and Binds to Adenine- and Uridine-rich Elements within the 3′ Untranslated Regions of Cytokine and Angiogenic Factor mRNAs. Cancer Res. 2001, 61, 2154–2161. [Google Scholar]
- Zadeh, M.; Amin, E.M.; Hoareau-Aveilla, C.; Domingo, E.; Symonds, K.E.; Yea, X.; Heesom, K.J.; Salmon, A.; D’Silva, O.; Betteridge, K.B.; et al. Alternative splicing of TIA-1 in human colon cancer regulates VEGF isoform expression, angiogenesis, tumour growth and bevacizumab resistance. Mol. Oncol. 2015, 9, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.X.; Hewitt, S.M.; Steinberg, S.M.; Liewehr, D.; Swain, S.M. Expression Levels of eIF4E, VEGF, and Cyclin D1, and Correlation of eIF4E With VEGF and Cyclin D1 in Multi-Tumor Tissue Microarray. Oncol. Rep. 2007, 17, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.A.; Carter, P.; Liu, L.; Li, B.D.; Abreo, F.; Tudor, A.; Zimmer, S.G.; Benedetti, A.D. Elevated Expression of eIF4E and FGF-2 Isoforms During Vascularization of Breast Carcinomas. Oncogene 1997, 15, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Son, Y.J.; Lee, W.H.; Park, Y.W.; Cha, S.W.; Cho, W.J.; Kim, Y.M.; Choi, H.J.; Choi, D.H.; Jung, S.W.; et al. Tristetraprolin regulates expression of VEGF and tumorigenesis in human colon cancer. Int. J. Cancer 2010, 126, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Scully, S.J., Jr.; Yan, W.; Bentley, B.; Mueller, J.; Brown, C.; Bigelow, C.; Schwartz, L.M. The novel lupus antigen related protein acheron enhances the development of human breast cancer. Int. J. Cancer. 2012, 130, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Lee, J.-S. Exploiting tumor cell senescence in anticancer therapy. BMB Rep. 2014, 47, 51–59. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, J.-S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef]
- Lee, J.H.; Jung, M.S.; Hong, J.Y.; Kim, M.K.; Chung, I.K. Loss of RNA-binding protein HuR facilitates cellular senescence through posttranscriptional regulation of TIN2 mRNA. Nucleic Acids Res. 2018, 46, 4271–4285. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; House, R.; Palanisamy, N.; Qie, S.; Day, T.A.; Neskey, D.; Diehl, J.A.; Palanisamy, V. RNA-Binding Protein FXR1 Regulates p21 and TERC RNA to Bypass p53-Mediated Cellular Senescence in OSCC. PLoS Genet. 2016, 12, e1006306. [Google Scholar]
- Pont, A.R.; Sadri, N.; Hsiao, S.J.; Smith, S.; Schneider, R.J. mRNA decay factor AUF1 maintains normal aging, telomere maintenance and suppression of senescence by activation of telomerase transcription. Mol. Cell 2012, 47, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Sanduja, S.; Kaza, V.; Dixon, D.A. The mRNA decay factor tristetraprolin (TTP) induces senescence in human papillomavirus-transformed cervical cancer cells by targeting E6-AP ubiquitin ligase. Aging 2009, 9, 803–817. [Google Scholar] [CrossRef]
- Fregoso, O.I.; Das, S.; Akerman, M.; Krainer, A.R. Splicing-Factor Oncoprotein SRSF1 Stabilizes p53 via RPL5 and Induces Cellular Senescence. Mol. Cell 2013, 50, 56–66. [Google Scholar] [CrossRef]
- Bebee, T.W.; Cieply, B.W.; Carstens, R.P. Genome-wide Activities of RNA Binding Proteins That Regulate Cellular Changes in the Epithelial to Mesenchymal Transition (EMT). Adv. Exp. Med. Biol. 2014, 825, 267–302. [Google Scholar]
- Ueda, J.; Matsuda, Y.; Yamahatsu, K.; Uhida, E.; Naito, Z.; Korc, M.; Ishiwata, T. Epithelial splicing regulatory protein 1 is a favorable prognostic factor in pancreatic cancer that attenuates pancreatic metastases. Oncogene 2014, 33, 4485–4495. [Google Scholar] [CrossRef]
- Lu, H.; Liu, J.; Liu, S.; Zeng, J.; Ding, D.; Carstens, R.P.; Cong, Y.; Xu, X.; Guo, W. Exo70 Isoform Switching upon Epithelial-Mesenchymal Transition Mediates Cancer Cell Invasion. Dev. Cell 2013, 27, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Tauler, J.; Zudaire, E.; Liu, H.; Shih, J.; Mulshine, J.L. hnRNP A2/B1 Modulates Epithelial-Mesenchymal Transition in Lung Cancer Cell Lines. Cancer Res. 2010, 70, 7137–7147. [Google Scholar] [CrossRef] [PubMed]
- Moran-Jones, K.; Grindlay, J.; Jones, M.; Smith, R.; Norman, J.C. hnRNP A2 Regulates Alternative mRNA Splicing of TP53INP2 to Control Invasive Cell Migration. Cancer Res. 2009, 69, 9219–9227. [Google Scholar] [CrossRef]
- Wang, H.; Vardy, L.A.; Tan, C.P.; Loo, J.M.; Guo, K.; Li, J.; Lim, S.G.; Zhou, J.; Chng, W.J.; Ng, S.B.; et al. PCBP1 Suppresses the Translation of Metastasis-Associated PRL-3 Phosphatase. Cancer Cell 2010, 18, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Howley, B.V.; Hussey, G.S.; Link, L.A.; Howe, P.H. Translational regulation of inhibin βA by TGFβ via the RNA-binding protein hnRNP E1 enhances the invasiveness of epithelial-to-mesenchymal transitioned cells. Oncogene 2016, 35, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.-B.; Qin, X.; Peng, K.; Li, Q.; Tang, C.; Wei, Y.-C.; Yu, S.; Gan, L.; Liu, T.-S. HnRNPR-CCNB1/CENPF axis contributes to gastric cancer proliferation and metastasis. Aging 2019, 11, 7473–7491. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zong, Y.; Peng, L.; Kong, S.; Zhou, M.; Zou, J.; Liu, J.; Mia, R.; Sun, X.; Li, L. Overexpression of eIF4E in colorectal cancer patients is associated with liver metastasis. OncoTargets Ther. 2016, 9, 815–822. [Google Scholar]
- Gu, W.; Pan, F.; Singer, R.H. Blocking -catenin binding to the ZBP1 promoter represses ZBP1 expression, leading to increased proliferation and migration of metastatic breast cancer cells. J. Cell Sci. 2009, 122, 1895–1905. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Thi, H.T.H.; Hong, S.T. IMP2 and IMP3 cooperate to promote the metastasis of triple-negative breast cancer through destabilization of progesterone receptor. Cancer Lett. 2018, 415, 30–39. [Google Scholar] [CrossRef]
- Petz, M.; Them, N.; Huber, H.; Beug, H.; Mikulits, W. La enhances IRES-mediated translation of laminin B1 during malignant epithelial to mesenchymal transition. Nucleic Acids Res. 2012, 40, 290–302. [Google Scholar] [CrossRef]
- Ji, X.; Lu, H.; Zhou, Q.; Luo, K. LARP7 suppresses P-TEFb activity to inhibit breast cancer progression and metastasis. eLife 2014, 3, e02907. [Google Scholar] [CrossRef]
- King, C.E.; Cuatrecasas, M.; Castells, A.; Sepulveda, A.R.; Lee, J.-S.; Rustgi, A.K. LIN28B Promotes Colon Cancer Progression and Metastasis. Cancer Res. 2011, 71, 4260–4268. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.; Vogel, G.; Huot, M.-É.; Guo, T.; Muller, W.J.; Lukong, K.E. Sam68 haploinsufficiency delays onset of mammary tumorigenesis and metastasis. Oncogene 2008, 27, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Nakka, K.K.; Chaudhary, N.; Joshi, S.; Bhat, J.; Singh, K.; Chatterjee, S.; Malhotra, R.; De, A.; Santra, M.K.; Dilworth, F.J.; et al. Nuclear matrix-associated protein SMAR1 regulates alternative splicing via HDAC6-mediated deacetylation of Sam68. Proc. Natl. Acad. Sci. USA 2015, 112, 3374–3383. [Google Scholar] [CrossRef] [PubMed]
- Vanharanta, S.; Marney, C.B.; Shu, W.; Valiente, M.; Zou, Y.; Mele, A.; Darnell, R.B.; Massagué, J. Loss of the multifunctional RNA-binding protein RBM47 as a source of selectable metastatic traits in breast cancer. eLife 2014, 3, e02734. [Google Scholar] [CrossRef]
- Mohibi, S.; Chen, X.; Zhang, J. Cancer The ‘RBP’ eutics—RNA-Binding Proteins as Therapeutic Targets for Cancer. Pharmacol. Ther. 2019, 203. [Google Scholar]
- Kentsis, A.; Topisirovic, I.; Culjkovic, B.; Shao, L.; Borden, K.L.B. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc. Natl. Acad. Sci. USA 2004, 101, 18105–18110. [Google Scholar] [CrossRef]
- Li, S.; Jia, Y.; Jacobson, B.; McCauley, J.; Kratzke, R.; Bitterman, P.B.; Wagner, C.R. Treatment of breast and lung cancer cells with a N-7 benzyl guanosine monophosphate tryptamine phosphoramidate pronucleotide (4Ei-1) results in chemosensitization to gemcitabine and induced eIF4E proteasomal degradation. Mol. Pharm. 2013, 10, 523–531. [Google Scholar] [CrossRef]
- Moerke, N.J.; Aktas, H.; Chen, H.; Cantel, S.; Reibarkh, M.Y.; Fahmy, A.; Gross, J.D.; Degterev, A.; Yuan, J.; Chorev, M.; et al. Small-Molecule Inhibition of the Interaction between the Translation Initiation Factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [Google Scholar] [CrossRef]
- Cencic, R.; Hall, D.R.; Robert, F.; Du, Y.; Min, J.; Li, L.; Qui, M.; Lewis, L.; Kurtkaya, S.; Dingledine, R.; et al. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc. Natl. Acad. Sci. USA 2011, 108, 1046–1051. [Google Scholar] [CrossRef]
- Cao, J.; He, L.; Lin, G.; Hu, C.; Dong, R.; Zhang, J.; Zhu, H.; Hu, Y.; Wagner, C.R.; He, Q.; et al. Cap-dependent translation initiation factor, eIF4E, is the target for Ouabain-mediated inhibition of HIF-1a. Biochem. Pharmacol. 2014, 89, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Peffley, D.M.; Sharma, C.; Hentosh, P.; Buechler, R.D. Perillyl alcohol and genistein differentially regulate PKB/Akt and 4E-BP1 phosphorylation as well as eIF4E/eIF4G interactions in human tumor cells. Arch. Biochem. Biophys. 2007, 465, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Konicek, B.W.; Stephens, J.R.; McNulty, A.M.; Robichaud, N.; Peery, R.B.; Dumstorf, C.A.; Dowless, M.S.; Iversen, P.W.; Parsons, S.; Ellis, K.E.; et al. Therapeutic Inhibition of MAP Kinase Interacting Kinase Blocks Eukaryotic Initiation Factor 4E Phosphorylation and Suppresses Outgrowth of Experimental Lung Metastases. Cancer Res. 2011, 71, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.Y.; Guo, H.; Barengo, N.; Naora, H. Inhibition of Ovarian Cancer Growth by a Tumor-Targeting Peptide That Binds Eukaryotic Translation Initiation Factor 4E. Clin. Cancer Res. 2009, 15, 4336–4347. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, C.A.; Zhang, J.; Ma, B.; Chen, M.; Chen, X. Disruption of the Rbm38-eIF4E Complex with a Synthetic Peptide Pep8 Increases p53 Expression. Cancer Res. 2019, 79, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Makarova-Rusher, O.V.; Ulhannan, S.V.; Rahma, O.E.; Fioravanti, S.; Walker, M.; Abdullah, S.; Raffeld, M.; Anderson, V.; Abi-Jaoudeh, N.; et al. Modulation of tumor eIF4E by antisense inhibition: A phase I/II translational clinical trial of ISIS 183750—An antisense oligonucleotide against eIF4E—In combination with irinotecan in solid tumors and irinotecan-refractory colorectal cancer. Int. J. Cancer 2016, 139, 1648–1657. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Oh, Y.; Wheler, J.; Naing, A.; Brail, L.; Callies, S.; André, V.; Kadam, S.K.; Nasir, A.; et al. A Phase 1 Dose Escalation, Pharmacokinetic, and Pharmacodynamic Evaluation of eIF-4E Antisense Oligonucleotide LY2275796 in Patients with Advanced Cancer. Clin. Cancer Res. 2011, 17, 6582–6591. [Google Scholar] [CrossRef]
- Dong, K.; Wang, R.; Wang, X.; Lin, F.; Shen, J.-J.; Gao, P.; Zhang, H.-Z. Tumor-specific RNAi targeting eIF4E suppresses tumor growth, induces apoptosis and enhances cisplatin cytotoxicity in human breast carcinoma cells. Breast Cancer Res. Treat. 2009, 113, 443–456. [Google Scholar] [CrossRef]
- Lang, M.; Berry, D.; Passecker, K.; Mesteri, I.; Bhuju, S.; Ebner, F.; Sedlyarov, V.; Evstatiev, R.; Dammann, K.; Loy, A.; et al. HuR small-molecule inhibitor elicits differential effects in adenomatosis polyposis and colorectal carcinogenesis. Cancer Res. 2017, 77, 2424–2438. [Google Scholar] [CrossRef]
- Lal, P.; Cerofolini, L.; D’Agostino, V.G.; Zucal, C.; Fuccio, C.; Bonomo, I.; Dassi, E.; Giuntini, S.; Maio, D.D.; Vishwakarma, V.; et al. Regulation of HuR structure and function by dihydrotanshinone-I. Nucleic Acids Res. 2017, 45, 9514–9527. [Google Scholar] [CrossRef]
- Filippova, N.; Yang, X.; Ananthan, S.; Sorochinsky, A.; Hackney, J.R.; Gentry, Z.; Bae, S.; King, P.; Nabors, L.B. Hu antigen R (HuR) multimerization contributes to glioma disease progression. J. Biol. Chem. 2017, 292, 16999–17010. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Wu, X.; Fields, J.K.; Johnson, D.K.; Lan, L.; Pratt, M.; Somoza, A.D.; Wang, C.C.C.; Karanicolas, J.; Oakley, B.R.; et al. The fungal natural product azaphilone-9 binds to HuR and inhibits HuR-RNA interaction in vitro. PLoS ONE 2017, 12, e0175471. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, R.; Mehta, M.; Ahmed, R.; Roy, S.; Xu, L.; Aubé, J.; Chen, A.; Zhao, Y.D.; Herman, T.; Ramesh, R.; et al. HuR-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci. Rep. 2017, 7, 9684–9694. [Google Scholar] [CrossRef]
- Allegri, L.; Baldan, F.; Roy, S.; Aubé, J.; Russo, D.; Filetti, S.; Damante, G. The HuR CMLD-2 inhibitor exhibits antitumor effects via MAD2 downregulation in thyroid cancer cells. Sci. Rep. 2019, 9, 7374. [Google Scholar] [CrossRef] [PubMed]
- Amreddy, N.; Babu, A.; Paneerselvam, J.; Srivastava, A.; Muralidharna, R.; Chen, A.; Zhao, Y.D.; Munshi, A.; Ramesh, R. Chemo-biologic combinatorial drug delivery using folate receptor-targeted dendrimer nanoparticles for lung cancer treatment. Nanomedicine 2018, 14, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, R.; Babu, A.; Amreddy, N.; Srivastava, A.; Chen, A.; Zhao, Y.D.; Kompella, U.B.; Munshi, A.; Ramesh, R. Tumor-targeted Nanoparticle Delivery of HuR siRNA Inhibits Lung Tumor Growth In Vitro and In Vivo By Disrupting the Oncogenic Activity of the RNA-binding Protein HuR. Mol. Cancer Ther. 2017, 16, 1470–1486. [Google Scholar] [CrossRef] [PubMed]
- Clingman, C.C.; Deveau, L.M.; Hay, S.A.; Genga, R.M.; Shandilya, S.M.D.; Massi, F.; Ryder, S.P. Allosteric inhibition of a stem cell RNA-binding protein by an intermediary metabolite. eLife 2014, 3, e02848. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Appelman, C.; Smith, A.R.; Yu, J.; Larsen, S.; Marquez, R.T.; Liu, H.; Wu, X.; Gao, P.; Roy, A.; et al. Natural product (L)-gossypol inhibits colon cancer cell growth by targeting RNA-binding protein Musashi-1. Mol. Oncol. 2015, 9, 1406–1420. [Google Scholar] [CrossRef]
- Minuesa, G.; Albanese, S.K.; Xie, W.; Kazansky, Y.; Worroll, D.; Chow, A.; Schurer, A.; Park, S.M.; Rotsides, C.Z.; Taggart, J.; et al. Small-molecule targeting of MUSASHI RNA-binding activity in acute myeloid leukemia. Nat. Commun. 2019, 10, 2691. [Google Scholar] [CrossRef]
- Chen, H.; Liu, J.; Wang, H.; Cheng, Q.; Zhou, C.; Chen, X.; Ye, F. Inhibition of RNA-Binding Protein Musashi-1 Suppresses Malignant Properties and Reverses Paclitaxel Resistance in Ovarian Carcinoma. J. Cancer 2019, 10, 1580–1592. [Google Scholar] [CrossRef]
- Roos, M.; Pradére, U.; Ngondo, R.P.; Behera, A.; Allegrini, S.; Civenni, G.; Zagalak, J.A.; Marchand, J.-R.; Menzi, M.; Towbin, H.; et al. A Small-Molecule Inhibitor of Lin28. ACS Chem. Biol. 2016, 11, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rowe, R.G.; Jaimes, A.; Yu, C.; Nam, Y.; Pearson, D.S.; Zhang, J.; Xie, X.; Marion, W.; Heffron, G.J.; et al. Small-molecule inhibitors disrupt let-7 oligouridylation and release the selective blockade of let-7 processing by LIN28. Cell Rep. 2018, 23, 3091–3101. [Google Scholar] [CrossRef] [PubMed]
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the use of therapeutic peptides for cancer treatment. J. Biomed. Sci. 2017, 24, 21–35. [Google Scholar] [CrossRef]
- Watts, J.K.; Corey, D.R. Gene silencing by siRNAs and antisense oligonucleotides in the laboratory and the clinic. J. Pathol. 2012, 226, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Burdelski, C.; Jakani-karimi, N.; Jacobsen, F.; Möller-Koop, C.; Minner, S.; Simon, R.; Sauter, G.; Steurer, S.; Clauditz, T.S.; Wilczak, W. IMP3 overexpression occurs in various important cancer types and is linked to aggressive tumor features: A tissue microarray study on 8877 human cancers and normal tissues. Oncol. Rep. 2018, 39, 3–12. [Google Scholar]
- Morimatsu, K.; Aishima, S.; Yamamoto, H.; Hayashi, A.; Nakata, K.; Oda, Y.; Shindo, K.; Fujino, M.; Tanaka, M.; Oda, Y. Insulin-like growth factor II messenger RNA–binding protein-3 is a valuable diagnostic and prognostic marker of intraductal papillary mucinous neoplasm. Hum. Pathol. 2013, 44, 1714–1721. [Google Scholar] [CrossRef]
- Heinonen, M.; Bono, P.; Narko, K.; Chang, S.-H.; Lundin, J.; Joensuu, H.; Furneaux, H.; Hla, T.; Haglund, C.; Ristimäki, A. Cytoplasmic HuR Expression Is a Prognostic Factor in Invasive Ductal Breast Carcinoma. Cancer Res. 2005, 65, 2157–2161. [Google Scholar] [CrossRef]
- Huang, H.; Han, Y.; Zhang, C.; Wu, J.; Feng, J.; Qu, L.; Shou, C. HNRNPC as a candidate biomarker for chemoresistance in gastric cancer. Tumor Biol. 2016, 37, 3527–3534. [Google Scholar] [CrossRef]
- Wang, K.; Li, L.; Fu, L.; Yuan, Y.; Dai, H.; Zhu, T.; Zhou, Y.; Yuan, F. Integrated Bioinformatics Analysis the Function of RNA Binding Proteins (RBPs) and Their Prognostic Value in Breast Cancer. Front. Pharmacol. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Wei, L.; Gao, L.-N.; Song, P.-P.; You, C.-G. Development and validation of a RNA binding protein-associated prognostic model for lung adenocarcinoma. Aging 2020, 12, 3558–3573. [Google Scholar]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60–75. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| RBP | Cancer Types | Expression | Mechanisms | Targets | Cancer Traits | References |
|---|---|---|---|---|---|---|
| LIN28A/B | Brain, breast, colon, cervical esophageal, head and neck, liver, lung, lymphoma, renal, prostate, ovary | Upregulated | miRNA processing | let-7a | Proliferation, invasion, metastasis, angiogenesis | [9,10,11,12,13,14,15,149,192] |
| SMAD | Colon | Upregulated | miRNA processing | miR-21, miR-199a | Invasion, metastasis | [25,26] |
| SRSF1 (SF2/ASF) | Lung, colon, kidney, liver, pancreas, breast, prostate | Upregulated | Alternative splicing | Ron, BIN1, MKN2, S6K1, Bcl-x, Mcl-1, CASP2, CASP9, ICAD, BIM, TEAD1, VEGF, RPL-MDM2 | Senescence, EMT, invasion, metastasis, proliferation, angiogenesis | [32,33,34,35,36,37,38,178] |
| SRSF3 (SRp20) | Lung, breast, stomach, skin, bladder, colon, thyroid, kidney, brain | Upregulated | Alternative splicing | FoxM2, HIPK2, ETV1, NDE1, HNF1a, ERN1, HMGCS1, DHCR7, SCAP, LIFR, Epb4.1l5, Myo1b, CTNND1, GIT2, SLK, SP4 | Proliferation, apoptosis, EMT, metastasis | [39,40,41,42,43,44] |
| SRSF6 (SRp55) | Lung, colon | Upregulated | Alternative splicing | INSR, DLG1, MKNK2 | Proliferation | [45] |
| SPF45 (RBM17) | Leukemia, cervical | Upregulated | Alternative splicing | Fas | Apoptosis | [46] |
| RBM3 | Liver | Upregulated | circRNA biogenesis | SCD-circRNA | Proliferation | [61,62] |
| RBM5, 6, 10 | Liver, lung | Upregulated or downregulated | Alternative splicing | NUMB, p53, C-PARP | Proliferation, apoptosis | [148,164] |
| RBM47 | Breast, lung | Downregulated | mRNA stability | DKK1 | Metastasis | [195] |
| KHSRP | Brain, breast, esophageal, liver, lung, renal, testis, thyroid | Upregulated or downregulated | miRNA processing | miR-26a/b, let-7a, miR-23a, miR-192-5p, miR-21, miR-130b, miR-301 | EMT, invasion, metastasis | [16,17,18,19,20,21] |
| hnRNPA1 | Brain, breast, colon, liver, lung, pancreas, cervical, gastric | Upregulated | miRNA processing | miR-18a, let-7a | Proliferation | [19,22,24,47,48,49,50] |
| hnRNPA2/B1 | Brain, lung | Upregulated | Alternative splicing | BIN1, WWOX, CFLAR, CASP9, CD44, TP53IP2, E-cadherin, Twist, Snail1 | Proliferation, EMT, metastasis | [47,48,49,51,182,183] |
| hnRNP D (AUF1) | Breast, colon, gastric, liver, lung, pancreas, renal, sarcoma, thyroid | Upregulated | mRNA stability | Cyclin D1, p21, p16, Bax, Bcl-2, Gadd45a, CASP2, MMP9, FGF9, Fos, TYMS, JunD, Myc, lncRNA NEAT1 | Proliferation, Senescence | [86,87,88,89,90,91,92,142,176] |
| hnRNP E1/2 (PCBP1/2) | Breast, lung, colon, pancreas | Upregulated or downregulated | mRNA stability, translation | Dab2, ILEI, PRL-3, Inhibin βA, p73, GLS2 | Senescence, EMT, invasion, metastasis | [129,130,184,185] |
| hnRNP F | Bladder | Upregulated | mRNA stability | TPX2 | Proliferation | [143] |
| hnRNP M | Breast | Upregulated | Alternative splicing | CD44 | EMT, invasion, metastasis | [52] |
| hnRNP H | Brain | Upregulated | Alternative splicing | Ron, MST1R | Invasion, migration | [53] |
| hnRNP I (PTB) | Breast, ovarian, brain, colon | Upregulated | Alternative splicing | FGFR-1, USP5, PKM, Cyclin D3 | Proliferation | [48,49,55,56,57,122,141] |
| hnRNP K | Breast | Upregulated | Subcellular localization | lncRNA MALAT1 | Metastasis | [82,83] |
| hnRNP R | Gastric | Upregulated | mRNA stability | Cyclin B1, CENPF | EMT, invasion, metastasis | [186] |
| CELF1 | Oral | Upregulated | mRNA stability | Bad, Bax, JunD | Apoptosis | [160] |
| DND1 | Breast | Downregulated | mRNA stability | BIM | Apoptosis | [161] |
| FXR1 | Oral | Upregulated | mRNA stability | PI3K/AKT, p53, PTEN, p21, p27, TERC | Senescence | [175] |
| ESRP 1/2 | Breast, colon, head and neck, lung, pancreas, renal, melanoma | Upregulated | Alternative splicing | FGFR2, CD44, CTNND1, ENAH, Snail | EMT, invasion, metastasis | [52,58,59,60,180,181] |
| SAM68 | Prostate, renal, breast, colon, cervical | Upregulated | Alternative splicing | CD44, Cyclin D1, Bcl-x | Proliferation, EMT, invasion, metastasis | [63,64,65,66,150,193,194] |
| CPEB1 | Breast, liver | Upregulated | Alternative polyadenylation | MMP9, VEGF, HIF-1α | Proliferation, angiogenesis, EMT, invasion, metastasis | [70,71,74,77,140] |
| Subcellular localization | ZO-1 | EMT | ||||
| CPEB2 | Breast | Upregulated | Alternative polyadenylation | HIF-1α | Metastasis | [71] |
| CPEB4 | Breast, liver, melanoma, pancreas | Upregulated | Alternative polyadenylation | MIFT, RAB7A, VEGF, tPA | Proliferation, angiogenesis | [72,73,74] |
| IGF2BP1 (IMP1/ZBP1) | Breast, colon, lung, melanoma, ovary, skin, liver | Upregulated | Subcellular localization | β-actin, E-cadherin, α-actinin, Arp-16 | Proliferation, EMT, invasion, metastasis | [78,79,105,106,107,108,111,112,135,188] |
| mRNA stability | β-TrCP1, CD44, lncRNA HULC | |||||
| IGFBP2 (IMP2) | Brain, breast, leukemia, lung, colon | Upregulated | Subcellular localization | NDUFS3, COX7b | EMT, invasion, metastasis | [80,109,112,189] |
| mRNA stability | RAF1, PR | |||||
| IGF2BP3 (IMP3) | Breast, colon, leukemia, lung, ovary, pancreas, renal, liver | Upregulated | mRNA stability | HMGA2, LIN28, Myc, PR | Proliferation, EMT, invasion, metastasis | [110,112,136,137,138,139,189] |
| HuR | Brain, breast, cervical, colon, gastric, liver, leukemia, prostate, ovary, gallbladder | Upregulated in most cancers | mRNA stability | Cyclin (A, B1, D1, and E), IL-1β, IL-2, TNF-α, IL-8, MMP9, HIF-1α, VEGF, SIRT1, Snail, c-Myc, Wnt5a, COX-2, c-fos, p21, p16, p27, TIN2, MSI1, Bcl-2, Bcl-xL, Mcl-1, ProTα , lncRNA NEAT1, lncRNA-HGBC, OIP5-AS1, LincRNA-p21, HOTAIR | Proliferation, apoptosis, angiogenesis, senescence, invasion, metastasis | [93,94,95,96,97,98,99,120,121,122,123,128,144,145,153,154,155,166,174] |
| mRNA translation | ProTα, p53, MSI1, HIF-1α, XIAP | |||||
| eIF4E | B-cell Lymphoma, breast, colon, lymphoma, melanoma | Upregulated | Translation | Bcl-2, Bcl-xL, VEGF, FGF2, MMP2, MMP9 | Apoptosis, angiogenesis, EMT, invasion, metastasis | [119,157,168,169,187] |
| KIN17 | Breast, cervical | Upregulated | mRNA translation | Caspase 3/7 | Apoptosis | [163] |
| La | Cervical, liver | Upregulated | Translation | Cyclin D1, LamB1 | Proliferation | [146,190] |
| LARP 1 | Cervical, lung | Upregulated | mRNA stability, translation | Bax, Bcl-2, Bik, Mdm2, XIAP, 5′ TOP mRNAs | Proliferation, apoptosis | [147,158,159] |
| LARP6 | Breast | Upregulated | Translation | MMP9, VEGF | Angiogenesis | [171] |
| LARP 7 | Breast | Downregulated | Stability | P-TEFb, 7 SK snRNP | EMT, invasion, metastasis | [191] |
| MSI 1/2 | Bone, Liver | Upregulated | mRNA translation | BRD4, c-Met, HMGA2, NUMB, Fos, Fyn, p21, p27, Caspase 3 | Proliferation, apoptosis, EMT, invasion, metastasis | [124,125,126,127,162] |
| QKI | Breast, colon | Upregulated | circRNA biogenesis | SMARCAS5, POLE2, OXNAD1, SHPRH, SMAD2, ATXN2, DOCK1, GNB1 | Invasion, metastasis | [67] |
| Downregulated | mRNA translation | p27, β-catenin | Proliferation | [151] | ||
| PDCD4 | Leukemia, breast, colon | Downregulated | mRNA translation | A-Myb | Proliferation | [131] |
| TIA-1 | Breast, colon, cervical | Upregulated | Alternative splicing, mRNA stability, Translation | Fas, VEGF, PDCD4, GADD45A | Apoptosis, angiogenesis | [155,156,167] |
| TTP | Brain, breast, colon, leukemia, liver, melanoma, pancreas, prostate | Upregulated or downregulated | mRNA stability | Cyclin B1, Cyclin D1, Bcl-2, cIAP, VEGF, Snail, Twist1, ZEB1, SOX9, MACC1, MMP2, MMP9, IL-6, PD-L1 | Angiogenesis, senescence | [100,101,102,103,104,170,177] |
| Wig1 (ZMAT3) | Breast, lung, osteosarcoma | Downregulated | mRNA stability | p53, p21, ACOT7 | Senescence | [113,114,115,116,117] |
| Translation | ACOT7 |
| RBP | Therapeutic Types | Compounds | Functions | Cancer Types | References |
|---|---|---|---|---|---|
| eIF4E | Small molecule inhibitor | Ribavirin | Mimic of 5′ 7-methyl guanosine cap-structure. Interacts with eIF4E and inhibits eIF4E-mediated translation of oncogenes | Human squamous cell carcinoma (HSCC), acute myeloid leukemia (AML) | [197] |
| 4Ei-1 | Converts to 7Bn-GMP. Antagonizes eIF4E cap binding and induces eIF4E proteasomal degradation, inhibiting translation initiation | Breast, lung, mesothelioma | [198] | ||
| 4EGI-1, 4E1RCat, 4E2RCat, Ouabain, Perillyl alcohol, Rapamycin, Torin1, AZD8055 | Interferes with association of eIF4E and eIF4G, inhibiting translation initiation | Lung, melanoma, lymphoma, colon, liver, breast, prostate | [199,200,201,202] | ||
| MnK inhibitors | Inhibits phosphorylation of eIF4E | Melanoma, lymphoma, colon, lung | [203] | ||
| Therapeutic peptides | GnRH-4EBP | Binds to eIF4E and disrupts eIF4E interacting with eIF4G | Ovary | [204,205] | |
| Pep8 | Interrupts RBM38-eIF4E and increases p53 translation | Colon, breast | |||
| ASO | ISIS 183750 | Synergetic effect with chemical drugs, such as irinotecan | Colon | [206,207] | |
| LY2275796 | Combination therapy with chemical drugs or radiation | Prostate, lung | |||
| siRNA | Stimulates the cytotoxic effects of cisplatin | Breast | [208] | ||
| HuR | Small molecule inhibitor | MS-444, okicenone, dehydromutactin, DHTS, AZA-9 | Targets RRM1 and 2 of HuR, and inhibits RNA-binding activities of HuR | Pancreas, colon, melanoma, brain, breast | [209,210,211,212] |
| CMLD-2 | Downregulates the expression of HuR Competitively interacts with HuR and inhibits its interaction with target mRNAs | Lung, thyroid | [213,214] | ||
| siRNA | Enhances the efficiency with chemo-biologic combinatorial drug delivery system | Lung | [215,216] | ||
| MSI | Small molecule inhibitor | Oleic acid | Binds to RRM1 of MSI and induces conformational change of MSI1, inhibiting the interaction between MSI and target RNAs | Brain (CNS) | [217] |
| (-)-gossypol | Interacts with RRM1 of MSI1 and block MSI1 RNA binding | Colon | [218] | ||
| Ro 80-2750 (Ro) | Selectively binds to RRM1 of MSI2 and disrupts MSI2 interacting with RNA | AML | [219] | ||
| ASO | Targets MSI with undruggable approaches | Ovary, pancreas | [127,220] | ||
| siRNA | Targets MSI with liposomal preparation | Colon | [127,220] | ||
| LIN28 | Small molecule inhibitor | Compound 1632 | Inhibits the interaction of LIN28/pre-let-7 and induces the maturation of let-7 | Prostate, liver | [221] |
| Compound 1, KCB3602, LI71 | Binds to cold shock domain (CSD) of LIN28 and inactivates LIN28 binding against let-7 | Ovary | [222] | ||
| TPEN | Binds to and destabilize zinc-knuckle domain (ZKD) of LIN28. TPEN binding disrupts LIN28-mediated oligouridylation and elevates the level of let-7 | Ovary, placenta | [222] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, D.; Lee, Y.; Lee, J.-S. RNA-Binding Proteins in Cancer: Functional and Therapeutic Perspectives. Cancers 2020, 12, 2699. https://doi.org/10.3390/cancers12092699
Kang D, Lee Y, Lee J-S. RNA-Binding Proteins in Cancer: Functional and Therapeutic Perspectives. Cancers. 2020; 12(9):2699. https://doi.org/10.3390/cancers12092699
Chicago/Turabian StyleKang, Donghee, Yerim Lee, and Jae-Seon Lee. 2020. "RNA-Binding Proteins in Cancer: Functional and Therapeutic Perspectives" Cancers 12, no. 9: 2699. https://doi.org/10.3390/cancers12092699
APA StyleKang, D., Lee, Y., & Lee, J.-S. (2020). RNA-Binding Proteins in Cancer: Functional and Therapeutic Perspectives. Cancers, 12(9), 2699. https://doi.org/10.3390/cancers12092699